
Synthesis and Evaluation of ePSMA-DM1: A New Theranostic Small-Molecule Drug Conjugate (T-SMDC) for Prostate Cancer
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. General Methods
4.1.1. Chemistry
4.1.2. Radiochemistry
4.2. Chemical Synthesis
4.2.1. Synthesis of EuK(Ahx-Sta-Phe-Asp-Lys(C(O)-(CH2)2-SH)-DOTA) (1)
4.2.2. Synthesis of EuK(Ahx-Sta-Phe-Asp-Lys(C(O)-(CH2)2-S-S-Py)-DOTA) (2)
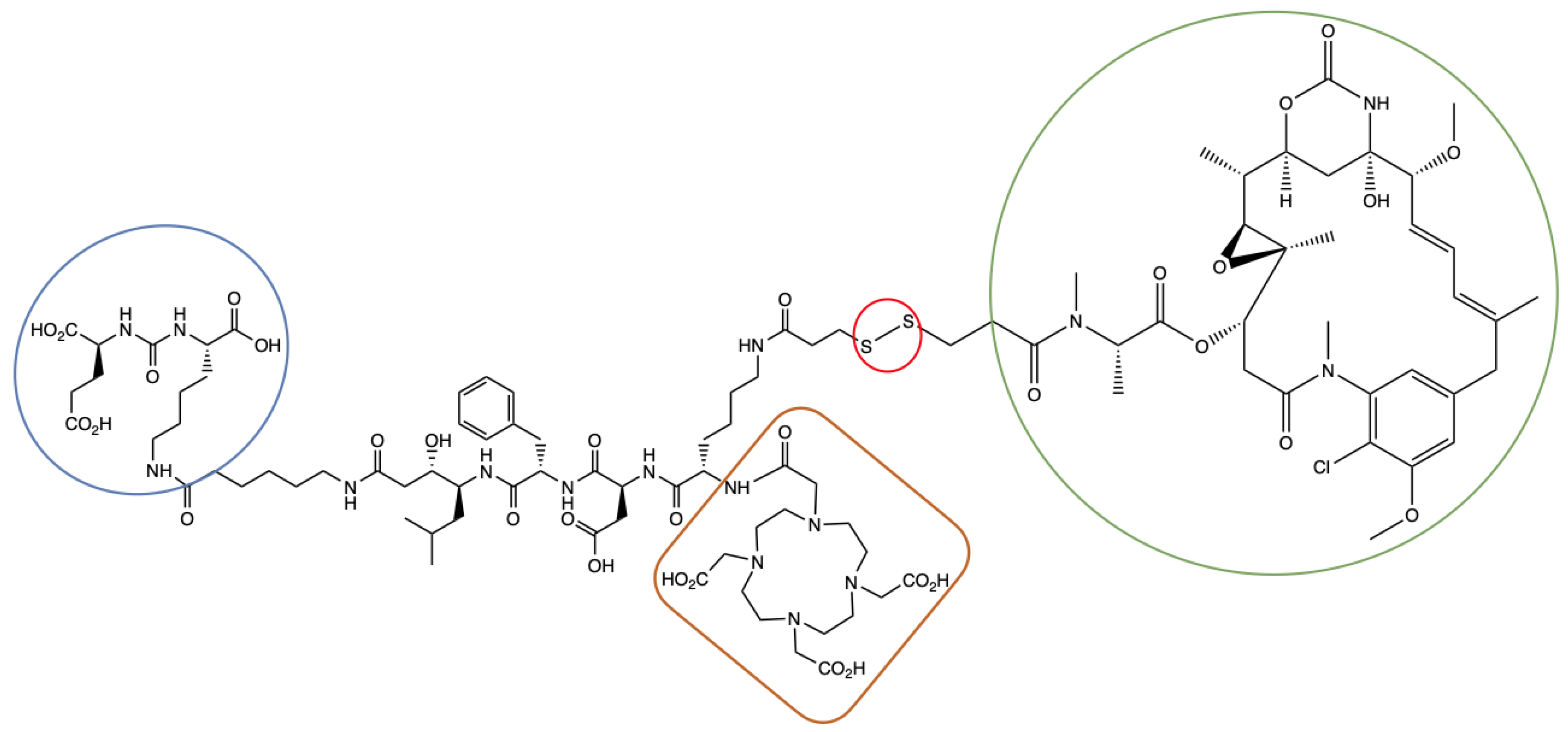
4.2.3. Synthesis of EuK(Ahx-Sta-Phe-Asp-Lys(C(O)-(CH2)2-S-S-DM1)-DOTA) (ePSMA-DM1; (3S,7S,22S,23S,26S,29S)-26-Benzyl-29-((S)-6-(3-((3-(((S)-1-(((14S,16S,32S,33S, 2R,4S,10E,12E,14R)-86-Chloro-14-Hydroxy-85,14-Dimethoxy-33,2,7,10-Tetramethyl-12,6-Dioxo-7-Aza-1(6,4)-Oxazinana-3(2,3)-Oxirana-8(1,3)-Benzenacyclotetradecaphane-10,12-Dien-4-yl)Oxy)-1-Oxopropan-2-yl)(Methyl)Amino)-3-Oxopropyl)Disulfaneyl)Propanamido)-2-(2-(4,7,10-Tris(Carboxymethyl)-1,4,7,10-Tetraazacyclododecan-1-Yl)Acetamido)Hexanamido)-22-Hydroxy-23-Isobutyl-5,13,20,25,28-Pentaoxo-4,6,12,19,24,27-Hexaazatriacontane-1,3,7,30-Tetracarboxylic Acid)
4.3. Radiochemistry
4.3.1. Radiolabeling with [111In]InCl3
4.3.2. Radiolabeling with [177Lu]LuCl3
4.3.3. Stability in Phosphate Buffered Saline (PBS)
4.3.4. Stability in Mouse Serum
4.3.5. Determination of LogD7.4 Value
4.4. Biological Assays
4.4.1. NAALADase Assay
4.4.2. GSH Stability
4.4.3. Cell Culture
4.4.4. Cytotoxicity Assay
4.4.5. Internalization and Cell Uptake Assay
4.5. Animal Studies
4.5.1. Mouse Model
4.5.2. Ex Vivo Biodistribution
4.5.3. SPECT-CT Imaging
4.5.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II—2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer [Formula Presented]. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Debnath, S.; Zhou, N.; McLaughlin, M.; Rice, S.; Pillai, A.K.; Hao, G.; Sun, X. PSMA-Targeting Imaging and Theranostic Agents—Current Status and Future Perspective. Int. J. Mol. Sci. 2022, 23, 1158. [Google Scholar] [CrossRef]
- Wright, G.L.; Haley, C.; Beckett, M.L.; Schellhammer, P.F. Expression of Prostate-Specific Membrane Antigen in Normal, Benign, and Malignant Prostate Tissues. Urol. Oncol. Semin. Orig. Investig. 1995, 1, 18–28. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical Therapy in Cancer: Clinical Advances and Challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Duan, H.; Iagaru, A.; Aparici, C.M. Radiotheranostics—Precision Medicine in Nuclear Medicine and Molecular Imaging. Nanotheranostics 2022, 6, 103–117. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. [177Lu]Lu-PSMA-617 versus Cabazitaxel in Patients with Metastatic Castration-Resistant Prostate Cancer (TheraP): A Randomised, Open-Label, Phase 2 Trial. Lancet 2021, 397, 797–804. [Google Scholar] [CrossRef]
- Pathmanandavel, S.; Crumbaker, M.; Yam, A.O.; Nguyen, A.; Rofe, C.; Hovey, E.; Gedye, C.; Kwan, E.M.; Hauser, C.; Azad, A.A.; et al. 177Lu-PSMA-617 and Idronoxil in Men with End-Stage Metastatic Castration-Resistant Prostate Cancer (LuPIN): Patient Outcomes and Predictors of Treatment Response in a Phase I/II Trial. J. Nucl. Med. 2022, 63, 560–566. [Google Scholar] [CrossRef]
- Suman, S.; Parghane, R.V.; Joshi, A.; Prabhash, K.; Talole, S.; Basu, S. Combined 177Lu-PSMA-617 PRLT and Abiraterone Acetate versus 177Lu-PSMA-617 PRLT Monotherapy in Metastatic Castration-Resistant Prostate Cancer: An Observational Study Comparing the Response and Durability. Prostate 2021, 81, 1225–1234. [Google Scholar] [CrossRef]
- Emmett, L.; Subramaniam, S.; Joshua, A.M.; Crumbaker, M.; Martin, A.; Zhang, A.Y.; Rana, N.; Langford, A.; Mitchell, J.; Yip, S.; et al. ENZA-p Trial Protocol: A Randomized Phase II Trial Using Prostate-Specific Membrane Antigen as a Therapeutic Target and Prognostic Indicator in Men with Metastatic Castration-Resistant Prostate Cancer Treated with Enzalutamide (ANZUP 1901). BJU Int. 2021, 128, 642–651. [Google Scholar] [CrossRef]
- Sandhu, S.; Joshua, A.M.; Emmett, L.; Spain, L.A.; Horvath, L.; Crumbaker, M.; Anton, A.; Wallace, R.; Pasam, A.; Bressel, M.; et al. PRINCE: Phase I Trial of 177Lu-PSMA-617 in Combination with Pembrolizumab in Patients with Metastatic Castration-Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2022, 40, 5017. [Google Scholar] [CrossRef]
- Sandhu, S.; Guo, C.; Hofman, M.S. Radionuclide Therapy in Prostate Cancer: From Standalone to Combination PSMA Theranostics. J. Nucl. Med. 2021, 62, 1660–1668. [Google Scholar] [CrossRef]
- Suman, S.K.; Subramanian, S.; Mukherjee, A. Combination Radionuclide Therapy: A New Paradigm. Nucl. Med. Biol. 2021, 98–99, 40–58. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Galsky, M.D.; Morris, M.J.; Crona, D.J.; George, D.J.; Dreicer, R.; Tse, K.; Petruck, J.; Webb, I.J.; Bander, N.H.; et al. Phase 1/2 Multiple Ascending Dose Trial of the Prostate-Specific Membrane Antigen-Targeted Antibody Drug Conjugate MLN2704 in Metastatic Castration-Resistant Prostate Cancer. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 530.e15–530.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrylak, D.P.; Kantoff, P.; Vogelzang, N.J.; Mega, A.; Fleming, M.T.; Stephenson, J.J.; Frank, R.; Shore, N.D.; Dreicer, R.; McClay, E.F.; et al. Phase 1 Study of PSMA ADC, an Antibody-Drug Conjugate Targeting Prostate-Specific Membrane Antigen, in Chemotherapy-Refractory Prostate Cancer. Prostate 2019, 79, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Guan, X.; Ma, H.; Cong, H.; Zhang, W.; Miao, Z. Small Molecule-Drug Conjugates: A Novel Strategy for Cancer-Targeted Treatment. Eur. J. Med. Chem. 2019, 63, 883–895. [Google Scholar] [CrossRef]
- Olatunji, F.P.; Pun, M.; Herman, J.W.; Romero, O.; Maniatopoulos, M.; Latoche, J.D.; Parise, R.A.; Guo, J.; Beumer, J.H.; Anderson, C.J.; et al. Modular Smart Molecules for PSMA-Targeted Chemotherapy. Mol. Cancer Ther. 2022, 21, 1701–1709. [Google Scholar] [CrossRef]
- Boinapally, S.; Ahn, H.-H.; Cheng, B.; Brummet, M.; Nam, H.; Gabrielson, K.L.; Banerjee, S.R.; Minn, I.; Pomper, M.G. A Prostate-Specific Membrane Antigen (PSMA)-Targeted Prodrug with a Favorable in Vivo Toxicity Profile. Sci. Rep. 2021, 11, 7114. [Google Scholar] [CrossRef]
- Wang, X.; Shirke, A.; Walker, E.; Sun, R.; Ramamurthy, G.; Wang, J.; Shan, L.; Mangadlao, J.; Dong, Z.; Li, J.; et al. Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of, Prostate Cancer. Cancers 2021, 13, 417. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Wang, K.; Santhapuram, H.K.R.; Low, P.S. Prostate-Specific Membrane Antigen Targeted Imaging and Therapy of Prostate Cancer Using a PSMA Inhibitor as a Homing Ligand. Mol. Pharm. 2009, 6, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Leamon, C.P.; Reddy, J.A.; Bloomfield, A.; Dorton, R.; Nelson, M.; Vetzel, M.; Kleindl, P.; Hahn, S.; Wang, K.; Vlahov, I.R. Prostate-Specific Membrane Antigen-Specific Antitumor Activity of a Self-Immolative Tubulysin Conjugate. Bioconjug. Chem. 2019, 30, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Nguyen, T.X.; Kanduluru, A.K.; Venkatesh, C.; Lv, W.; Reddy, P.V.N.; Low, P.S.; Cushman, M. DUPA Conjugation of a Cytotoxic Indenoisoquinoline Topoisomerase i Inhibitor for Selective Prostate Cancer Cell Targeting. J. Med. Chem. 2015, 58, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Machulkin, A.E.; Skvortsov, D.A.; Ivanenkov, Y.A.; Ber, A.P.; Kavalchuk, M.V.; Aladinskaya, A.V.; Uspenskaya, A.A.; Shafikov, R.R.; Plotnikova, E.A.; Yakubovskaya, R.I.; et al. Synthesis and Biological Evaluation of PSMA-Targeting Paclitaxel Conjugates. Bioorg. Med. Chem. Lett. 2019, 29, 2229–2235. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Uspenskaya, A.A.; Zyk, N.Y.; Nimenko, E.A.; Ber, A.P.; Petrov, S.A.; Shafikov, R.R.; Skvortsov, D.A.; Smirnova, G.B.; Borisova, Y.A.; et al. PSMA-Targeted Small-Molecule Docetaxel Conjugate: Synthesis and Preclinical Evaluation. Eur. J. Med. Chem. 2022, 227, 113936. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the Design of Ligand-Targeted Cancer Therapeutics and Imaging Agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.G.; Park, R.; Kim, D.K.; Shin, Y.; Cho, Y.Y.; Lee, H.S. Mertansine Inhibits MRNA Expression and Enzyme Activities of Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyltransferases in Human Hepatocytes and Liver Microsomes. Pharmaceutics 2020, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.T.; Guo, X.; Bařinka, C.; Lupold, S.E.; Pomper, M.G.; Gabrielson, K.; Raman, V.; Artemov, D.; Hapuarachchige, S. Development of 5D3-DM1: A Novel Anti-Prostate-Specific Membrane Antigen Antibody-Drug Conjugate for PSMA-Positive Prostate Cancer Therapy. Mol. Pharm. 2020, 17, 3392–3402. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab Emtansine: Mechanisms of Action and Drug Resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- White, B.H.; Whalen, K.; Kriksciukaite, K.; Alargova, R.; Au Yeung, T.; Bazinet, P.; Brockman, A.; Dupont, M.; Oller, H.; Lemelin, C.A.; et al. Discovery of an SSTR2-Targeting Maytansinoid Conjugate (PEN-221) with Potent Activity In Vitro and In Vivo. J. Med. Chem. 2019, 62, 2708–2719. [Google Scholar] [CrossRef]
- Kumar, A.; Mastren, T.; Wang, B.; Hsieh, J.T.; Hao, G.; Sun, X. Design of a Small-Molecule Drug Conjugate for Prostate Cancer Targeted Theranostics. Bioconjug. Chem. 2016, 27, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J.L.; Supuran, C.T.; Neri, D. A Small-Molecule Drug Conjugate for the Treatment of Carbonic Anhydrase IX Expressing Tumors. Angew. Chem. Int. Ed. 2014, 53, 4231–4235. [Google Scholar] [CrossRef] [PubMed]
- Hillier, S.M.; Maresca, K.P.; Femia, F.J.; Marquis, J.C.; Foss, C.A.; Nguyen, N.; Zimmerman, C.N.; Barrett, J.A.; Eckelman, W.C.; Pomper, M.G.; et al. Preclinical Evaluation of Novel Glutamate-Urea-Lysine Analogues That Target Prostate-Specific Membrane Antigen as Molecular Imaging Pharmaceuticals for Prostate Cancer. Cancer Res. 2009, 69, 6932–6940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-Complex Lipophilicity and the Targeting Property of a Urea-Based PSMA Inhibitor for PET Imaging. Bioconjug. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef]
- Lv, Q.; Yang, J.; Zhang, R.; Yang, Z.; Yang, Z.; Wang, Y.; Xu, Y.; He, Z. Prostate-Specific Membrane Antigen Targeted Therapy of Prostate Cancer Using a DUPA–Paclitaxel Conjugate. Mol. Pharm. 2018, 15, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, W.; Yang, N.; Liu, Y. Learn from Antibody–Drug Conjugates: Consideration in the Future Construction of Peptide-Drug Conjugates for Cancer Therapy. Exp. Hematol. Oncol. 2022, 11, 93. [Google Scholar] [CrossRef]
- Rana, A.; Bhatnagar, S. Advancements in Folate Receptor Targeting for Anti-Cancer Therapy: A Small Molecule-Drug Conjugate Approach. Bioorg. Chem. 2021, 112, 104946. [Google Scholar] [CrossRef]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef]
- Wüstemann, T.; Bauder-Wüst, U.; Schäfer, M.; Eder, M.; Benesova, M.; Leotta, K.; Kratochwil, C.; Haberkorn, U.; Kopka, K.; Mier, W. Design of Internalizing PSMA-Specific Glu-Ureido-Based Radiotherapeuticals. Theranostics 2016, 6, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004; pp. 1–25. [Google Scholar]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor Delivery and in Vivo Processing of Disulfide-Linked and Thioether-Linked Antibody-Maytansinoid Conjugates. Bioconjug. Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, E.A.M.; van Vliet, N.; Dalm, S.U.; de Blois, E.; van Gent, D.C.; Haeck, J.; de Ridder, C.; Stuurman, D.; Konijnenberg, M.W.; van Weerden, W.M.; et al. Extensive Preclinical Evaluation of Lutetium-177-Labeled PSMA-Specific Tracers for Prostate Cancer Radionuclide Therapy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Murce, E.; Beekman, S.; Spaan, E.; Handula, M.; Stuurman, D.; de Ridder, C.; Seimbille, Y. Preclinical Evaluation of a PSMA-Targeting Homodimer with an Optimized Linker for Imaging of Prostate Cancer. Molecules 2023, 28, 4022. [Google Scholar] [CrossRef]
- Felber, V.B.; Valentin, M.A.; Wester, H.J. Design of PSMA Ligands with Modifications at the Inhibitor Part: An Approach to Reduce the Salivary Gland Uptake of Radiolabeled PSMA Inhibitors? EJNMMI Radiopharm. Chem. 2021, 6, 10. [Google Scholar] [CrossRef]
- Debnath, S.; Hao, G.; Guan, B.; Thapa, P.; Hao, J.; Hammers, H.; Sun, X. Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists. Int. J. Mol. Sci. 2022, 23, 7160. [Google Scholar] [CrossRef]
- Derks, Y.H.W.; Rijpkema, M.; Amatdjais-Groenen, H.I.V.; Kip, A.; Franssen, G.M.; Michiel Sedelaar, J.P.; Somford, D.M.; Simons, M.; Laverman, P.; Gotthardt, M.; et al. Photosensitizer-Based Multimodal PSMA-Targeting Ligands for Intraoperative Detection of Prostate Cancer. Theranostics 2021, 11, 1527–1541. [Google Scholar] [CrossRef] [PubMed]
- de Blois, E.; Sze Chan, H.; Konijnenberg, M.; de Zanger, R.; Breeman, A.P.W. Effectiveness of Quenchers to Reduce Radiolysis of 111In- or 177Lu-Labelled Methionine-Containing Regulatory Peptides. Maintaining Radiochemical Purity as Measured by HPLC. Curr. Top. Med. Chem. 2013, 12, 2677–2685. [Google Scholar] [CrossRef]
- de Zanger, R.M.S.; Chan, H.S.; Breeman, W.A.P.; de Blois, E. Maintaining Radiochemical Purity of [177Lu]Lu-DOTA-PSMA-617 for PRRT by Reducing Radiolysis. J. Radioanal. Nucl. Chem. 2019, 321, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Breeman, W.; de Zanger, R.; Chan, H.; Blois, E. Alternative Method to Determine Specific Activity of 177Lu by HPLC. Curr. Radiopharm. 2015, 8, 119–122. [Google Scholar] [CrossRef]
- Chen, K.T.; Nieuwenhuizen, J.; Handula, M.; Seimbille, Y. A Novel Clickable MSAP Agent for Dual Fluorescence/Nuclear Labeling of Biovectors. Org. Biomol. Chem. 2020, 18, 6134–6139. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RCY (iTLC, %) | RCP (HPLC, %) | LogD7.4 | Stability in PBS (%) 1 | Stability in Mouse Serum (%) 1 |
|---|---|---|---|---|---|
| [111In]In-ePSMA-DM1 | 97.2 ± 1.3 2 | 92.6 ± 2.2 2 | −2.47 ± 0.11 2 | 90.9 | 82.9 |
| [177Lu]Lu-ePSMA-DM1 | 96.3 | 94.8 | - | 81.4 | 91.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murce, E.; Spaan, E.; Beekman, S.; van den Brink, L.; Handula, M.; Stuurman, D.; de Ridder, C.; Dalm, S.U.; Seimbille, Y. Synthesis and Evaluation of ePSMA-DM1: A New Theranostic Small-Molecule Drug Conjugate (T-SMDC) for Prostate Cancer. Pharmaceuticals 2023, 16, 1072. https://doi.org/10.3390/ph16081072
Murce E, Spaan E, Beekman S, van den Brink L, Handula M, Stuurman D, de Ridder C, Dalm SU, Seimbille Y. Synthesis and Evaluation of ePSMA-DM1: A New Theranostic Small-Molecule Drug Conjugate (T-SMDC) for Prostate Cancer. Pharmaceuticals. 2023; 16(8):1072. https://doi.org/10.3390/ph16081072
Chicago/Turabian StyleMurce, Erika, Evelien Spaan, Savanne Beekman, Lilian van den Brink, Maryana Handula, Debra Stuurman, Corrina de Ridder, Simone U. Dalm, and Yann Seimbille. 2023. "Synthesis and Evaluation of ePSMA-DM1: A New Theranostic Small-Molecule Drug Conjugate (T-SMDC) for Prostate Cancer" Pharmaceuticals 16, no. 8: 1072. https://doi.org/10.3390/ph16081072