
Immunomodulatory and Antioxidant Drugs in Glaucoma Treatment




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Characteristics of Glaucoma
2.1. Classification, Epidemiology, and Economic Implications
2.2. Symptoms and Diagnostic Features
2.3. Pharmaceutical Approaches to Treatment and Surgical Interventions
- Prostaglandin analogues: examples include bimatoprost, which enhances both trabecular and uveoscleral outflow of AH [22].
- β-blockers: medications like levobunolol and timolol work by reducing the production of AH [22].
- α2-adrenoceptor agonists: drugs such as apraclonidine and brimonidine lower IOP by decreasing aqueous humor production and augmenting trabecular outflow [22].
- Carbonic anhydrase inhibitors: agents like brinzolamide act by reducing the production of aqueous humor [22].
- Miotic agents: pilocarpine, for instance, increases the chamber angle by constricting the pupil and can also provide neuroprotective effects through the activation of muscarinic receptors [48].
- Rho-associated protein kinase (ROCK) inhibitors: Netarsudil is a ROCK inhibitor that targets the ROCK pathway, suppressing fibrotic events in the trabecular meshwork ™ and optimizing aqueous humor flow, thereby reducing IOP [49]. This molecule has been approved for the treatment of glaucoma in the United States (2017) and Europe (2019) in the form of a 0.02% ophthalmic topical formulation for once-daily application [50].
3. Pathophysiology
3.1. Risk Factors
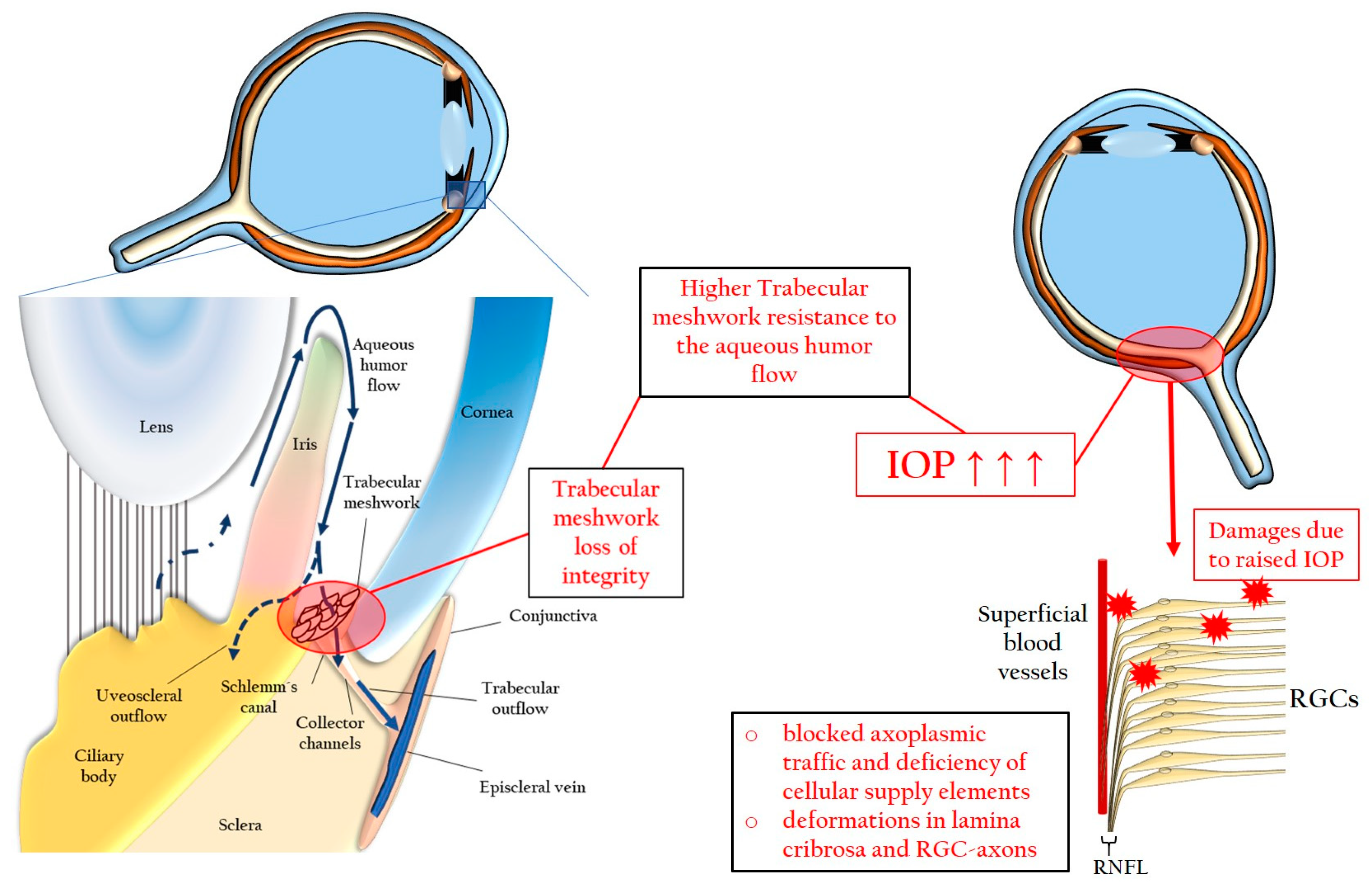
3.1.1. Elevated Intraocular Pressure
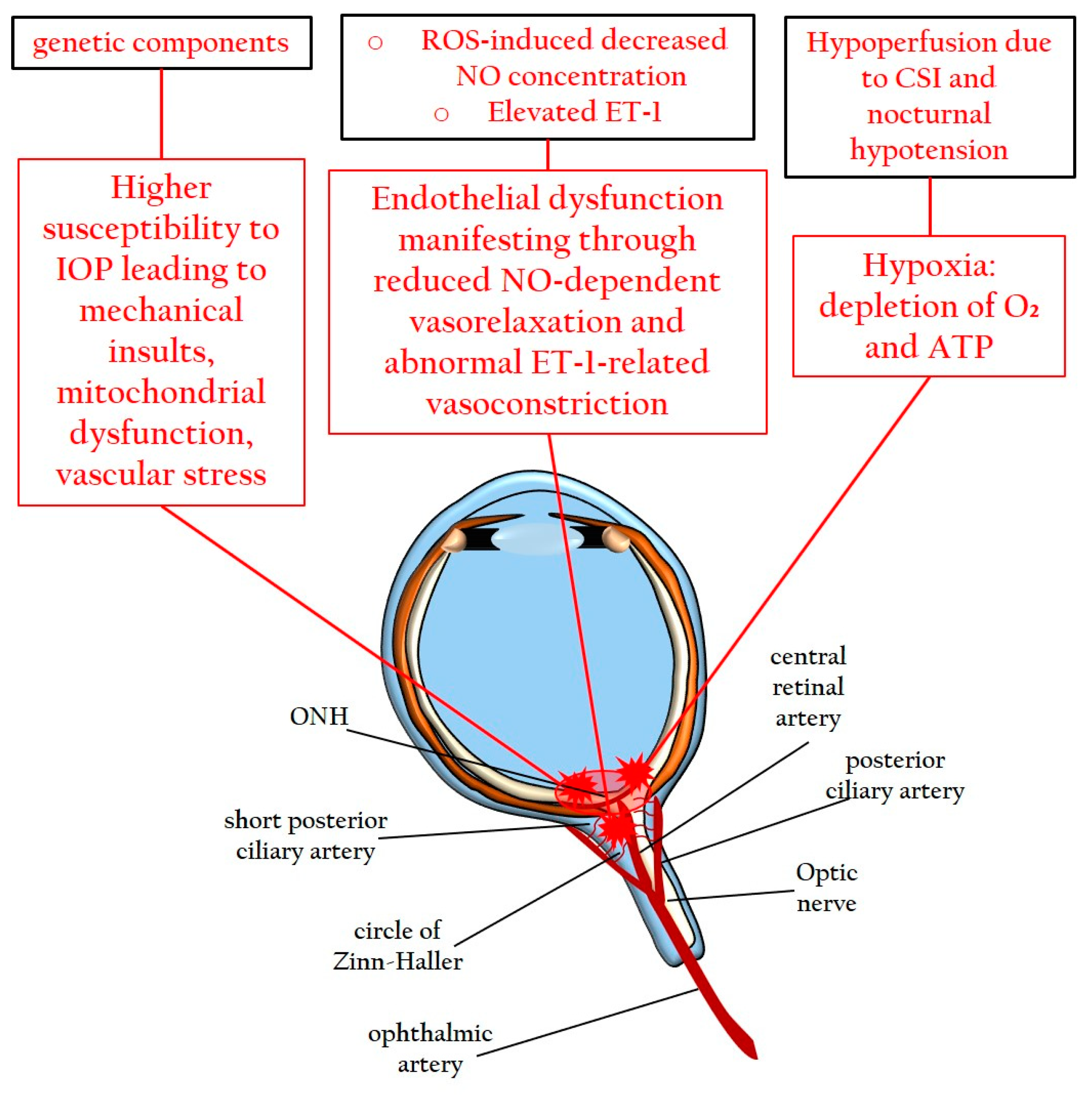
3.1.2. Genetic Factors, Systemic Vascular Dysregulation, and Endothelial Dysfunction
3.2. Pathomechanisms
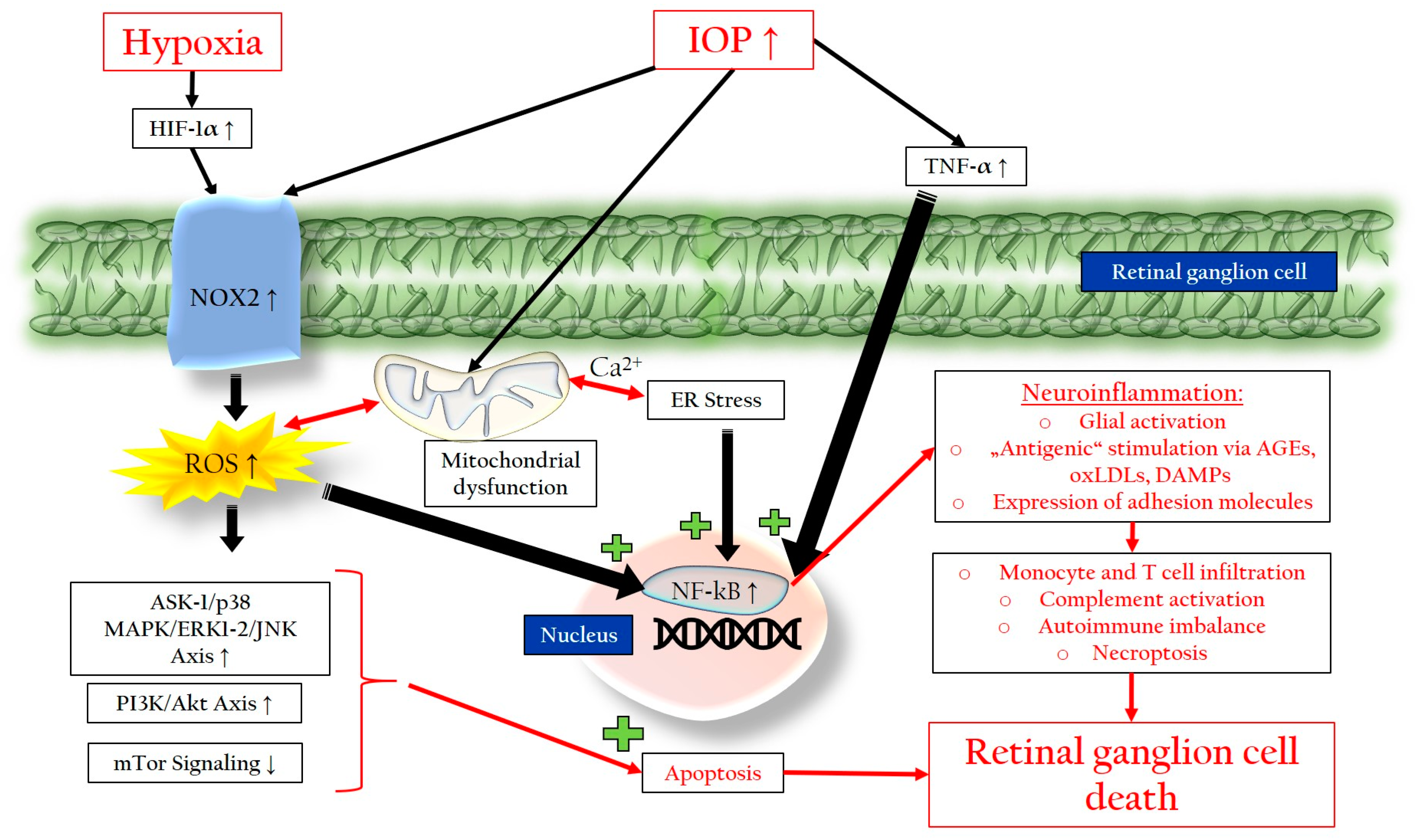
3.2.1. Chronic Oxidative Stress
3.2.2. Mitochondrial Dysfunction
3.2.3. Endoplasmic Reticulum Stress
- The protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2α (eIF2α)/activating transcription factor 4 (ATF4)/CCAAT-enhancer-binding protein homologous protein (CHOP) pathway, which reduces protein translation but can increase ROS production and promote apoptosis [127].
3.2.4. Neuroinflammation and Glial Activation
3.2.5. Autoimmune Imbalance
4. Emerging Curative Strategies: Immunomodulatory and Antioxidants
4.1. Immunomodulatory Candidates for Glaucoma
4.2. Promising Antioxidants for Glaucoma Treatment
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, D.R.; Patella, V.M. Automated Static Perimetry, 2nd ed.; Mosby: Saint Louis, MO, USA, 1999. [Google Scholar]
- Casson, R.J.; Chidlow, G.; Wood, J.P.; Crowston, J.G.; Goldberg, I. Definition of glaucoma: Clinical and experimental concepts. Clin. Exp. Ophthalmol. 2012, 40, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, C. The morphological difference between glaucoma and other optic neuropathies. J. Neuroophthalmol. 2015, 35 (Suppl. S1), S8–S21. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, J.; Li, Y.; Jiang, B. Prevalence of primary open angle glaucoma in the last 20 years: A meta-analysis and systematic review. Sci. Rep. 2021, 11, 13762. [Google Scholar] [CrossRef]
- Acott, T.S.; Keller, K.E.; Kelley, M.J. Role of Proteoglycans in the Trabecular Meshwork. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Cesareo, M.; Giannini, C.; Martucci, A.; Di Marino, M.; Pocobelli, G.; Aiello, F.; Mancino, R.; Nucci, C. Chapter 2-Links between obstructive sleep apnea and glaucoma neurodegeneration. In Progress in Brain Research; Bagetta, G., Nucci, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 257, pp. 19–36. [Google Scholar]
- Leung, D.Y.L.; Tham, C.C. Normal-tension glaucoma: Current concepts and approaches-A review. Clin. Exp. Ophthalmol. 2022, 50, 247–259. [Google Scholar] [CrossRef]
- Killer, H.E.; Pircher, A. Normal tension glaucoma: Review of current understanding and mechanisms of the pathogenesis. Eye 2018, 32, 924–930. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Arch. Ophthalmol. 2004, 122, 1348–1356. [Google Scholar] [CrossRef]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [CrossRef]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog. Retin. Eye Res. 2022, 87, 100998. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Geyer, O.; Levo, Y. Glaucoma is an autoimmune disease. Autoimmun. Rev. 2020, 19, 102535. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Sim, R.H.; Sirasanagandla, S.R.; Das, S.; Teoh, S.L. Treatment of Glaucoma with Natural Products and Their Mechanism of Action: An Update. Nutrients 2022, 14, 534. [Google Scholar] [CrossRef]
- Hsueh, Y.-J.; Chen, Y.-N.; Tsao, Y.-T.; Cheng, C.-M.; Wu, W.-C.; Chen, H.-C. The Pathomechanism, Antioxidant Biomarkers, and Treatment of Oxidative Stress-Related Eye Diseases. Int. J. Mol. Sci. 2022, 23, 1255. [Google Scholar] [CrossRef]
- Foster, P.J.; Buhrmann, R.; Quigley, H.A.; Johnson, G.J. The definition and classification of glaucoma in prevalence surveys. Br. J. Ophthalmol. 2002, 86, 238–242. [Google Scholar] [CrossRef]
- Schuster, A.K.; Erb, C.; Hoffmann, E.M.; Dietlein, T.; Pfeiffer, N. The Diagnosis and Treatment of Glaucoma. Dtsch. Arztebl. Int. 2020, 117, 225–234. [Google Scholar] [CrossRef]
- Flores-Sánchez, B.C.; Tatham, A.J. Acute angle closure glaucoma. Br. J. Hosp. Med. 2019, 80, C174–C179. [Google Scholar] [CrossRef]
- Khazaeni, B.; Khazaeni, L. Acute Closed Angle Glaucoma. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Chen, M.-J. Normal tension glaucoma in Asia: Epidemiology, pathogenesis, diagnosis, and management. Taiwan J. Ophthalmol. 2020, 10, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Belamkar, A.; Harris, A.; Oddone, F.; Verticchio Vercellin, A.; Fabczak-Kubicka, A.; Siesky, B. Asian Race and Primary Open-Angle Glaucoma: Where Do We Stand? J. Clin. Med. 2022, 11, 2486. [Google Scholar] [PubMed]
- Cho, H.-k.; Kee, C. Population-based glaucoma prevalence studies in Asians. Surv. Ophthalmol. 2014, 59, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Patel, S. Angle-closure glaucoma. Dis. Mon. 2014, 60, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Tawfik, M.A.; Waisbourd, M.; Katz, L.J. Primary angle-closure glaucoma: An update. Acta Ophthalmol. 2016, 94, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Number of people with glaucoma worldwide. Br. J. Ophthalmol. 1996, 80, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Wolfram, C.; Breitscheidel, L.; Shlaen, M.; Verboven, Y.; Pfeiffer, N. Direct cost and predictive factors for treatment in patients with ocular hypertension or early, moderate and advanced primary open-angle glaucoma: The CoGIS study in Germany. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2019–2028. [Google Scholar] [CrossRef]
- Thygesen, J.; Aagren, M.; Arnavielle, S.; Bron, A.; Fröhlich, S.J.; Baggesen, K.; Azuara-Blanco, A.; Buchholz, P.; Walt, J.G. Late-stage, primary open-angle glaucoma in Europe: Social and health care maintenance costs and quality of life of patients from 4 countries. Curr. Med. Res. Opin. 2008, 24, 1763–1770. [Google Scholar] [CrossRef]
- McGinley, P.; Ansari, E.; Sandhu, H.; Dixon, T. The cost burden of falls in people with glaucoma in National Health Service Hospital Trusts in the UK. J. Med. Econ. 2020, 23, 106–112. [Google Scholar] [CrossRef]
- Lichter, P.R. Glaucoma clinical trials and what they mean for our patients. Am. J. Ophthalmol. 2003, 136, 136–145. [Google Scholar] [CrossRef]
- Chen, P.P. Blindness in patients with treated open-angle glaucoma. Ophthalmology 2003, 110, 726–733. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Kim, C.S.; Zimmerman, M.B.; Alward, W.L.; Hayreh, S.S. Rate of visual field loss and long-term visual outcome in primary open-angle glaucoma. Am. J. Ophthalmol. 2001, 132, 47–56. [Google Scholar] [CrossRef]
- Rossetti, L.; Digiuni, M.; Montesano, G.; Centofanti, M.; Fea, A.M.; Iester, M.; Frezzotti, P.; Figus, M.; Ferreras, A.; Oddone, F.; et al. Blindness and Glaucoma: A Multicenter Data Review from 7 Academic Eye Clinics. PLoS ONE 2015, 10, e0136632. [Google Scholar] [CrossRef] [PubMed]
- Cronemberger, S.; Lourenço, L.F.; Silva, L.C.; Calixto, N.; Pires, M.C. Prognosis of glaucoma in relation to blindness at a university hospital. Arq. Bras. Oftalmol. 2009, 72, 199–204. [Google Scholar] [CrossRef]
- Ang, G.S.; Eke, T. Lifetime visual prognosis for patients with primary open-angle glaucoma. Eye 2007, 21, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Salmon, J.F. Ten-year outcomes in newly diagnosed glaucoma patients: Mortality and visual function. Br. J. Ophthalmol. 2007, 91, 1282–1284. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.P.; Smith, N.D.; Glen, F.C.; Burton, R.; Garway-Heath, D.F. How does glaucoma look?: Patient perception of visual field loss. Ophthalmology 2013, 120, 1120–1126. [Google Scholar] [CrossRef]
- Aspberg, J.; Heijl, A.; Bengtsson, B. Screening for Open-Angle Glaucoma and Its Effect on Blindness. Am. J. Ophthalmol. 2021, 228, 106–116. [Google Scholar] [CrossRef]
- Jonas, J.B.; Budde, W.M.; Panda-Jonas, S. Ophthalmoscopic evaluation of the optic nerve head. Surv. Ophthalmol. 1999, 43, 293–320. [Google Scholar] [CrossRef]
- Sihota, R.; Angmo, D.; Ramaswamy, D.; Dada, T. Simplifying “target” intraocular pressure for different stages of primary open-angle glaucoma and primary angle-closure glaucoma. Indian J. Ophthalmol. 2018, 66, 495–505. [Google Scholar] [CrossRef]
- Brusini, P. OCT Glaucoma Staging System: A new method for retinal nerve fiber layer damage classification using spectral-domain OCT. Eye 2018, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Vianna, J.R.; Chauhan, B.C. Chapter 7—How to detect progression in glaucoma. In Progress in Brain Research; Bagetta, G., Nucci, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 221, pp. 135–158. [Google Scholar]
- Prum, B.E., Jr.; Rosenberg, L.F.; Gedde, S.J.; Mansberger, S.L.; Stein, J.D.; Moroi, S.E.; Herndon, L.W., Jr.; Lim, M.C.; Williams, R.D. Primary Open-Angle Glaucoma Preferred Practice Pattern(®) Guidelines. Ophthalmology 2016, 123, P41–P111. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.P.; Yuan, H.H.; Zhu, X.; Cui, Y.Y.; Li, H.; Feng, X.M.; Qiu, Y.; Chen, H.Z.; Zhou, W. Activation of muscarinic receptors protects against retinal neurons damage and optic nerve degeneration in vitro and in vivo models. CNS Neurosci. Ther. 2014, 20, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lee, C.; Read, A.T.; Wang, K.; Ha, J.; Kuhn, M.; Navarro, I.; Cui, J.; Young, K.; Gorijavolu, R.; et al. Anti-fibrotic activity of a rho-kinase inhibitor restores outflow function and intraocular pressure homeostasis. eLife 2021, 10, e60831. [Google Scholar] [CrossRef]
- Batra, M.; Gupta, S.; Nair, A.B.; Dhanawat, M.; Sandal, S.; Morsy, M.A. Netarsudil: A new ophthalmic drug in the treatment of chronic primary open angle glaucoma and ocular hypertension. Eur. J. Ophthalmol. 2021, 31, 2237–2244. [Google Scholar] [CrossRef]
- Gulati, V.; Fan, S.; Gardner, B.J.; Havens, S.J.; Schaaf, M.T.; Neely, D.G.; Toris, C.B. Mechanism of Action of Selective Laser Trabeculoplasty and Predictors of Response. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1462–1468. [Google Scholar] [CrossRef]
- Aquino, M.C.; Barton, K.; Tan, A.M.; Sng, C.; Li, X.; Loon, S.C.; Chew, P.T. Micropulse versus continuous wave transscleral diode cyclophotocoagulation in refractory glaucoma: A randomized exploratory study. Clin. Exp. Ophthalmol. 2015, 43, 40–46. [Google Scholar] [CrossRef]
- Collotta, D.; Colletta, S.; Carlucci, V.; Fruttero, C.; Fea, A.M.; Collino, M. Pharmacological Approaches to Modulate the Scarring Process after Glaucoma Surgery. Pharmaceuticals 2023, 16, 898. [Google Scholar] [CrossRef]
- Kroese, M.; Burton, H. Primary open angle glaucoma. The need for a consensus case definition. J. Epidemiol. Community Health 2003, 57, 752–754. [Google Scholar] [CrossRef]
- McMonnies, C. Reactive oxygen species, oxidative stress, glaucoma and hyperbaric oxygen therapy. J. Optom. 2018, 11, 3–9. [Google Scholar] [CrossRef]
- Downs, J.C.; Roberts, M.D.; Burgoyne, C.F. Mechanical environment of the optic nerve head in glaucoma. Optom. Vis. Sci. 2008, 85, 425–435. [Google Scholar] [CrossRef]
- Flammer, J.; Orgül, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.-P.; Stefánsson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Llobet, A.; Gasull, X.; Gual, A. Understanding trabecular meshwork physiology: A key to the control of intraocular pressure? News Physiol. Sci. 2003, 18, 205–209. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A.; Rossi, P.; Traverso, C. Glaucomatous outflow pathway and oxidative stress. Exp. Eye Res. 2007, 84, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Yue, B.Y. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: The trabecular meshwork. J. Cell Physiol. 1999, 180, 182–189. [Google Scholar] [CrossRef]
- Pervan, C.L.; Lautz, J.D.; Blitzer, A.L.; Langert, K.A.; Stubbs, E.B. Rho GTPase signaling promotes constitutive expression and release of TGF-β2 by human trabecular meshwork cells. Exp. Eye Res. 2016, 146, 95–102. [Google Scholar] [CrossRef] [PubMed]
- The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Collaborative Normal-Tension Glaucoma Study Group. Am. J. Ophthalmol. 1998, 126, 498–505. [CrossRef] [PubMed]
- Trivli, A.; Koliarakis, I.; Terzidou, C.; Goulielmos, G.N.; Siganos, C.S.; Spandidos, D.A.; Dalianis, G.; Detorakis, E.T. Normal-tension glaucoma: Pathogenesis and genetics. Exp. Ther. Med. 2019, 17, 563–574. [Google Scholar] [CrossRef]
- Tang, S.; Toda, Y.; Kashiwagi, K.; Mabuchi, F.; Iijima, H.; Tsukahara, S.; Yamagata, Z. The association between Japanese primary open-angle glaucoma and normal tension glaucoma patients and the optineurin gene. Hum. Genet. 2003, 113, 276–279. [Google Scholar] [CrossRef]
- Zhu, G.; Wu, C.J.; Zhao, Y.; Ashwell, J.D. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr. Biol. 2007, 17, 1438–1443. [Google Scholar] [CrossRef]
- Minegishi, Y.; Nakayama, M.; Iejima, D.; Kawase, K.; Iwata, T. Significance of optineurin mutations in glaucoma and other diseases. Prog. Retin. Eye Res. 2016, 55, 149–181. [Google Scholar] [CrossRef]
- Xu, Z.; Jiang, F.; Chen, F. Effects of abnormal optineurin expression on the survival of the rat retinal ganglion cell line RGC-5. Genet. Mol. Res. 2015, 14, 9171–9180. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Ihrke, G.; Piermartiri, T.C.; Shewmaker, F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 2012, 86, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jiang, F.; Chen, F. Distribution and localization of abnormally expressed OPTN proteins in RGC5 retinal ganglion cells and their effects on subcellular morphology. Genet. Mol. Res. 2015, 14, 12093–12101. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, M.L.S.; Kumari, A.; Radha, V.; Swarup, G. E50K-OPTN-induced retinal cell death involves the Rab GTPase-activating protein, TBC1D17 mediated block in autophagy. PLoS ONE 2014, 9, e95758. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Shim, M.S.; Takihara, Y.; Kim, K.Y.; Iwata, T.; Yue, B.Y.; Inatani, M.; Weinreb, R.N.; Perkins, G.A.; Ju, W.K. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci. Rep. 2016, 6, 33830. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, X.; Zhang, H.; Li, N.; Yang, X.; Cheng, W.; Zhao, K. Association of OPA1 polymorphisms with NTG and HTG: A meta-analysis. PLoS ONE 2012, 7, e42387. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, J.Y.; Kim, D.M.; Ko, H.S.; Kim, S.Y.; Yoo, T.; Hwang, S.S.; Park, S.S. Investigations on the association between normal tension glaucoma and single nucleotide polymorphisms of the endothelin-1 and endothelin receptor genes. Mol. Vis. 2006, 12, 1016–1021. [Google Scholar]
- Stroman, G.A.; Stewart, W.C.; Golnik, K.C.; Curé, J.K.; Olinger, R.E. Magnetic resonance imaging in patients with low-tension glaucoma. Arch. Ophthalmol. 1995, 113, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.; Farinelli, A.; Billson, F.; Houang, M.; Stern, M. Comparative study of brain magnetic resonance imaging findings in patients with low-tension glaucoma and control subjects. Ophthalmology 1995, 102, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Tomidokoro, A.; Araie, M.; Tomita, G.; Yamagami, J.; Okubo, T.; Masumoto, T. Visual field damage in normal-tension glaucoma patients with or without ischemic changes in cerebral magnetic resonance imaging. Jpn. J. Ophthalmol. 2004, 48, 340–344. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, M.; Franzone, F.; Lambiase, A.; Bonfiglio, V.; Limoli, P.G.; Artico, M.; Taurone, S.; Vingolo, E.M.; Greco, A.; Polimeni, A. Oxidative Stress Implication in Retinal Diseases-A Review. Antioxidants 2022, 11, 1790. [Google Scholar] [CrossRef]
- Toda, N.; Nakanishi-Toda, M. Nitric oxide: Ocular blood flow, glaucoma, and diabetic retinopathy. Prog. Retin. Eye Res. 2007, 26, 205–238. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Sugiyama, T.; Moriya, S.; Oku, H.; Azuma, I. Association of endothelin-1 with normal tension glaucoma: Clinical and fundamental studies. Surv. Ophthalmol. 1995, 39, S49–S56. [Google Scholar] [CrossRef]
- Cellini, M.; Possati, G.L.; Profazio, V.; Sbrocca, M.; Caramazza, N.; Caramazza, R. Color Doppler imaging and plasma levels of endothelin-1 in low-tension glaucoma. Acta Ophthalmol. Scand. Suppl. 1997, 75, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Sin, B.H.; Song, B.J.; Park, S.P. Aqueous vascular endothelial growth factor and endothelin-1 levels in branch retinal vein occlusion associated with normal tension glaucoma. J. Glaucoma 2013, 22, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.; Harris, A.; Wudunn, D.; Kheradiya, N.; Siesky, B. Dysfunctional regulation of ocular blood flow: A risk factor for glaucoma? Clin. Ophthalmol. 2008, 2, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Galassi, F.; Giambene, B.; Varriale, R. Systemic vascular dysregulation and retrobulbar hemodynamics in normal-tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4467–4471. [Google Scholar] [CrossRef]
- Gericke, A.; Mann, C.; Zadeh, J.K.; Musayeva, A.; Wolff, I.; Wang, M.; Pfeiffer, N.; Daiber, A.; Li, H.; Xia, N.; et al. Elevated Intraocular Pressure Causes Abnormal Reactivity of Mouse Retinal Arterioles. Oxid. Med. Cell Longev. 2019, 2019, 9736047. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Xia, N.; Li, H.; van Beers, T.; Gericke, A.; Prokosch, V. Intraocular Pressure-Induced Endothelial Dysfunction of Retinal Blood Vessels Is Persistent, but Does Not Trigger Retinal Ganglion Cell Loss. Antioxidants 2022, 11, 1864. [Google Scholar] [CrossRef]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into Hypoxia and Oxidative Stress at the Optic Nerve Head in a Rat Model of Glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef]
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and HIFing. Cell 1997, 89, 9–12. [Google Scholar] [CrossRef]
- Rupin, A.; Paysant, J.; Sansilvestri-Morel, P.; Lembrez, N.; Lacoste, J.M.; Cordi, A.; Verbeuren, T.J. Role of NADPH oxidase-mediated superoxide production in the regulation of E-selectin expression by endothelial cells subjected to anoxia/reoxygenation. Cardiovasc. Res. 2004, 63, 323–330. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Yokota, H.; Narayanan, S.P.; Zhang, W.; Liu, H.; Rojas, M.; Xu, Z.; Lemtalsi, T.; Nagaoka, T.; Yoshida, A.; Brooks, S.E.; et al. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8123–8131. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Görlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Kitsati, N.; Mantzaris, M.D.; Galaris, D. Hydroxytyrosol inhibits hydrogen peroxide-induced apoptotic signaling via labile iron chelation. Redox Biol. 2016, 10, 233–242. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Chang, Y.-C.; Chen, P.-H.; Li, C.-Y.; Wu, W.-C.; Kao, Y.-H. MicroRNA-100 Mediates Hydrogen Peroxide-Induced Apoptosis of Human Retinal Pigment Epithelium ARPE-19 Cells. Pharmaceuticals 2021, 14, 314. [Google Scholar] [CrossRef]
- Tezel, G.; Fourth, A.P.O.R.I.C.W.G. The role of glia, mitochondria, and the immune system in glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1001–1012. [Google Scholar] [CrossRef]
- Martin, K.R.; Levkovitch-Verbin, H.; Valenta, D.; Baumrind, L.; Pease, M.E.; Quigley, H.A. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2236–2243. [Google Scholar]
- Zhang, Z.Q.; Xie, Z.; Chen, S.Y.; Zhang, X. Mitochondrial dysfunction in glaucomatous degeneration. Int. J. Ophthalmol. 2023, 16, 811–823. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4903–4911. [Google Scholar] [CrossRef]
- Ju, W.-K.; Kim, K.-Y.; Duong-Polk, K.X.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Mol. Vis. 2010, 16, 1331. [Google Scholar]
- Legros, F.; Lombès, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dai, Y.; Zhang, R.; Shang, K.; Sun, X. Overexpression of Optic Atrophy Type 1 Protects Retinal Ganglion Cells and Upregulates Parkin Expression in Experimental Glaucoma. Front. Mol. Neurosci. 2018, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. Embo J. 2021, 40, e104705. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Vorobjev, I.A.; Popkov, V.A.; Babenko, V.A.; Zorova, L.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Andrianova, N.V.; Plotnikov, E.Y. Lessons from the Discovery of Mitochondrial Fragmentation (Fission): A Review and Update. Cells 2019, 8, 532–541. [Google Scholar] [CrossRef]
- Ju, W.K.; Perkins, G.A.; Kim, K.Y.; Bastola, T.; Choi, W.Y.; Choi, S.H. Glaucomatous optic neuropathy: Mitochondrial dynamics, dysfunction and protection in retinal ganglion cells. Prog. Retin. Eye Res. 2023, 95, 101136. [Google Scholar] [CrossRef]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef]
- Kim, K.; Perkins, G.; Shim, M.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.; Weinreb, R.; Ju, W. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis. 2015, 6, e1839. [Google Scholar] [CrossRef]
- Skeie, J.M.; Nishimura, D.Y.; Wang, C.L.; Schmidt, G.A.; Aldrich, B.T.; Greiner, M.A. Mitophagy: An Emerging Target in Ocular Pathology. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Perkins, G.A.; Kim, K.Y.; Kong, Y.; Lee, Y.; Choi, S.H.; Liu, Y.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Zangwill, L.; et al. Loss of AKAP1 triggers Drp1 dephosphorylation-mediated mitochondrial fission and loss in retinal ganglion cells. Cell Death Dis. 2020, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; You, M.; Fan, C.; Rong, R.; Li, H.; Xia, X. Pathologically high intraocular pressure induces mitochondrial dysfunction through Drp1 and leads to retinal ganglion cell PANoptosis in glaucoma. Redox Biol. 2023, 62, 102687. [Google Scholar] [CrossRef]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Kroeger, H.; Chiang, W.-C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef]
- Hurley, D.J.; Normile, C.; Irnaten, M.; O’Brien, C. The Intertwined Roles of Oxidative Stress and Endoplasmic Reticulum Stress in Glaucoma. Antioxidants 2022, 11, 886. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation Signal of Nuclear Factor-κB in Response to Endoplasmic Reticulum Stress is Transduced via IRE1 and Tumor Necrosis Factor Receptor-Associated Factor 2. Biol. Pharm. Bull. 2003, 26, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.-H.; Burr, L.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in respiratory disease. Clin. Transl. Immunol. 2018, 7, e1019. [Google Scholar] [CrossRef]
- Bariş, M.; Tezel, G. Immunomodulation as a Neuroprotective Strategy for Glaucoma Treatment. Curr. Ophthalmol. Rep. 2019, 7, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5697–5707. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4220–4233. [Google Scholar] [CrossRef]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Investig. 2011, 121, 1429–1444. [Google Scholar] [CrossRef]
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2019, 97, 70–76. [Google Scholar] [CrossRef]
- Mélik Parsadaniantz, S.; Réaux-le Goazigo, A.; Sapienza, A.; Habas, C.; Baudouin, C. Glaucoma: A Degenerative Optic Neuropathy Related to Neuroinflammation? Cells 2020, 9, 535. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 2016, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Soto, I.; Ryan, M.; Graham, L.C.; Smith, R.S.; John, S.W. Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J. Neuroinflamm. 2013, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Anderson, S.R.; Breen, K.T.; Romero, C.O.; Steele, M.R.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Tomlinson, S.; Vetter, M.L. Complement C3-Targeted Gene Therapy Restricts Onset and Progression of Neurodegeneration in Chronic Mouse Glaucoma. Mol. Ther. 2018, 26, 2379–2396. [Google Scholar] [CrossRef]
- Rolle, T.; Ponzetto, A.; Malinverni, L. The Role of Neuroinflammation in Glaucoma: An Update on Molecular Mechanisms and New Therapeutic Options. Front. Neurol. 2021, 11, 612422. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Tummers, B.; Green, D.R. Crashing the computer: Apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ. 2019, 26, 41–52. [Google Scholar] [CrossRef]
- Ko, K.W.; Milbrandt, J.; DiAntonio, A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell. Biol. 2020, 219, e201912047. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Beck, S.; von Thun und Hohenstein-Blaul, N.; Boehm, N.; Ziegler, A.; Vetter, J.M.; Pfeiffer, N.; Grus, F.H. Enhanced Insight into the Autoimmune Component of Glaucoma: IgG Autoantibody Accumulation and Pro-Inflammatory Conditions in Human Glaucomatous Retina. PLoS ONE 2013, 8, e57557. [Google Scholar] [CrossRef]
- Martin, B.W.; Gülgün, T.; Junjie, Y.; Guanghua, P.; Rajkumar, V.P.; Neeraj, A.; Rebecca, M.S.; David, J.C. Induced Autoimmunity to Heat Shock Proteins Elicits Glaucomatous Loss of Retinal Ganglion Cell Neurons via Activated T-Cell-Derived Fas-Ligand. J. Neurosci. 2008, 28, 12085. [Google Scholar] [CrossRef]
- Joachim, S.C.; Grus, F.H.; Pfeiffer, N. Analysis of Autoantibody Repertoires in Sera of Patients with Glaucoma. Eur. J. Ophthalmol. 2003, 13, 752–758. [Google Scholar] [CrossRef]
- Joachim, S.C.; Reichelt, J.; Berneiser, S.; Pfeiffer, N.; Grus, F.H. Sera of glaucoma patients show autoantibodies against myelin basic protein and complex autoantibody profiles against human optic nerve antigens. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 573–580. [Google Scholar] [CrossRef]
- Joachim, S.C.; Pfeiffer, N.; Grus, F.H. Autoantibodies in patients with glaucoma: A comparison of IgG serum antibodies against retinal, optic nerve, and optic nerve head antigens. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 243, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Grus, F.H.; Joachim, S.C.; Hoffmann, E.M.; Pfeiffer, N. Complex autoantibody repertoires in patients with glaucoma. Mol. Vis. 2004, 10, 13–17. [Google Scholar]
- Joachim, S.C.; Wuenschig, D.; Pfeiffer, N.; Grus, F.H. IgG antibody patterns in aqueous humor of patients with primary open angle glaucoma and pseudoexfoliation glaucoma. Mol. Vis. 2007, 13, 1573–1579. [Google Scholar] [PubMed]
- Grus, F.; Joachim, S.; Wax, M.; Pfeiffer, N. Serum autoantibodies in glaucoma patients from Germany and the United States: Further implications for autoimmune mechanisms in the neurodegenerative processes of glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1285. [Google Scholar]
- Wax, M.B.; Tezel, G.; Saito, I.; Gupta, R.S.; Harley, J.B.; Li, Z.; Romano, C. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am. J. Ophthalmol. 1998, 125, 145–157. [Google Scholar] [CrossRef]
- Wax, M.B.; Barrett, D.A.; Pestronk, A. Increased incidence of paraproteinemia and autoantibodies in patients with normal-pressure glaucoma. Am. J. Ophthalmol. 1994, 117, 561–568. [Google Scholar] [CrossRef]
- van Eden, W. Immune tolerance therapies for autoimmune diseases based on heat shock protein T-cell epitopes. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160531. [Google Scholar] [CrossRef]
- Tezel, G.; Seigel, G.M.; Wax, M.B. Autoantibodies to small heat shock proteins in glaucoma. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2277–2287. [Google Scholar]
- Tezel, G.; Wax, M.B. The Mechanisms of hsp27 Antibody-Mediated Apoptosis in Retinal Neuronal Cells. J. Neurosci. 2000, 20, 3552–3562. [Google Scholar] [CrossRef]
- Tezel, G.; Hernandez, M.R.; Wax, M.B. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch. Ophthalmol. 2000, 118, 511–518. [Google Scholar] [CrossRef]
- Wax, M.B.; Tezel, G.; Kawase, K.; Kitazawa, Y. Serum autoantibodies to heat shock proteins in glaucoma patients from Japan and the United States. Ophthalmology 2001, 108, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Antibodies to α B-crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Curr. Eye Res. 2007, 32, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Von Thun Und Hohenstein-Blaul, N.; Bell, K.; Pfeiffer, N.; Grus, F.H. Autoimmune aspects in glaucoma. Eur. J. Pharmacol. 2016, 787, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Wilding, C.; Funke, S.; Pfeiffer, N.; Grus, F.H. Protective effect of 14-3-3 antibodies on stressed neuroretinal cells via the mitochondrial apoptosis pathway. BMC Ophthalmol. 2015, 15, 64. [Google Scholar] [CrossRef]
- Tezel, G. The immune response in glaucoma: A perspective on the roles of oxidative stress. Exp. Eye Res. 2011, 93, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Beutgen, V.M.; Perumal, N.; Pfeiffer, N.; Grus, F.H. Autoantibody Biomarker Discovery in Primary Open Angle Glaucoma Using Serological Proteome Analysis (SERPA). Front. Immunol. 2019, 10, 381. [Google Scholar] [CrossRef]
- Griffith, T.S.; Yu, X.; Herndon, J.M.; Green, D.R.; Ferguson, T.A. CD95-induced apoptosis of lymphocytes in an immune privileged site induces immunological tolerance. Immunity 1996, 5, 7–16. [Google Scholar] [CrossRef]
- Matsumoto, H.; Murakami, Y.; Kataoka, K.; Notomi, S.; Mantopoulos, D.; Trichonas, G.; Miller, J.W.; Gregory, M.S.; Ksander, B.R.; Marshak-Rothstein, A.; et al. Membrane-bound and soluble Fas ligands have opposite functions in photoreceptor cell death following separation from the retinal pigment epithelium. Cell Death Dis. 2015, 6, e1986. [Google Scholar] [CrossRef]
- Krishnan, A.; Fei, F.; Jones, A.; Busto, P.; Marshak-Rothstein, A.; Ksander, B.R.; Gregory-Ksander, M. Overexpression of Soluble Fas Ligand following Adeno-Associated Virus Gene Therapy Prevents Retinal Ganglion Cell Death in Chronic and Acute Murine Models of Glaucoma. J. Immunol. 2016, 197, 4626–4638. [Google Scholar] [CrossRef]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflamm. 2019, 16, 184. [Google Scholar] [CrossRef]
- Brothers, H.M.; Marchalant, Y.; Wenk, G.L. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neurosci. Lett. 2010, 480, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.H.; Boia, R.; Elvas, F.; Martins, T.; Cunha, R.A.; Ambrósio, A.F.; Santiago, A.R. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure–induced transient ischemic injury. Transl. Res. 2016, 169, 112–128. [Google Scholar] [CrossRef]
- Madeira, M.H.; Ortin-Martinez, A.; Nadal-Nícolas, F.; Ambrósio, A.F.; Vidal-Sanz, M.; Agudo-Barriuso, M.; Santiago, A.R. Caffeine administration prevents retinal neuroinflammation and loss of retinal ganglion cells in an animal model of glaucoma. Sci. Rep. 2016, 6, 27532. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jiang, S.; Chen, C.; Lu, G.; Xie, Y.; Sun, X.; Huang, L. Caffeic acid phenethyl ester attenuates nuclear factor-κB-mediated inflammatory responses in Müller cells and protects against retinal ganglion cell death. Mol. Med. Rep. 2019, 19, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Han, C.T.; Sakaguchi, Y.; Lee, J.; Youn, H.Y. Safety evaluation of FM101, an A3 adenosine receptor modulator, in rat, for developing as therapeutics of glaucoma and hepatitis. EXCLI J. 2020, 19, 187–200. [Google Scholar] [CrossRef]
- Ramiro, S.; Radner, H.; van der Heijde, D.; Van Tubergen, A.; Buchbinder, R.; Aletaha, D.; Landewé, R.B. Combination therapy for pain management in inflammatory arthritis (rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, other spondyloarthritis). Cochrane Database Syst. Rev. 2011, CD008886. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef]
- Hashida, N.; Ohguro, N.; Nishida, K. Efficacy and Complications of Intravitreal Rituximab Injection for Treating Primary Vitreoretinal Lymphoma. Transl. Vis. Sci. Technol. 2012, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Q.; Shi, L.; Zhou, X.; Wu, W.; Wang, X.; Wang, L.; Wu, Z. Baicalin prevents fibrosis of human trabecular meshwork cells via inhibiting the MyD88/NF-κB pathway. Eur. J. Pharmacol. 2023, 938, 175425. [Google Scholar] [CrossRef]
- Yang, X.; Zeng, Q.; Barış, M.; Tezel, G. Transgenic inhibition of astroglial NF-κB restrains the neuroinflammatory and neurodegenerative outcomes of experimental mouse glaucoma. J. Neuroinflamm. 2020, 17, 252. [Google Scholar] [CrossRef]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Yanagidaira, T.; Takahashi, Y.; Sano, K.; Murata, T.; Hayashi, T. Development of spontaneous neuropathy in NF-κBp50-deficient mice by calcineurin-signal involving impaired NF-κB activation. Mol. Vis. 2011, 17, 2157–2170. [Google Scholar] [PubMed]
- Becker, S.; Reinehr, S.; Dick, H.B.; Joachim, S.C. Complement activation after induction of ocular hypertension in an animal model. Ophthalmologe 2015, 112, 41–48. [Google Scholar] [CrossRef]
- Kuehn, M.H.; Kim, C.Y.; Ostojic, J.; Bellin, M.; Alward, W.L.; Stone, E.M.; Sakaguchi, D.S.; Grozdanic, S.D.; Kwon, Y.H. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp. Eye Res. 2006, 83, 620–628. [Google Scholar] [CrossRef]
- Howell, G.R.; MacNicoll, K.H.; Braine, C.E.; Soto, I.; Macalinao, D.G.; Sousa, G.L.; John, S.W. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol. Dis. 2014, 71, 44–52. [Google Scholar] [CrossRef]
- Reinehr, S.; Gomes, S.C.; Gassel, C.J.; Asaad, M.A.; Stute, G.; Schargus, M.; Dick, H.B.; Joachim, S.C. Intravitreal Therapy Against the Complement Factor C5 Prevents Retinal Degeneration in an Experimental Autoimmune Glaucoma Model. Front. Pharmacol. 2019, 10, 1381. [Google Scholar] [CrossRef]
- Gibson, L.C.D.; Hastings, S.F.; McPhee, I.; Clayton, R.A.; Darroch, C.E.; Mackenzie, A.; MacKenzie, F.L.; Nagasawa, M.; Stevens, P.A.; MacKenzie, S.J. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur. J. Pharmacol. 2006, 538, 39–42. [Google Scholar] [CrossRef]
- Belforte, N.; Cueva-Vargas, J.L.; Di Polo, A. The phosphodiesterase inhibitor Ibudilast attenuates glial cell reactivity, production of proinflammatory cytokines and neuronal loss in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2665. [Google Scholar]
- Rodgers, K.M.; Deming, Y.K.; Bercum, F.M.; Chumachenko, S.Y.; Wieseler, J.L.; Johnson, K.W.; Watkins, L.R.; Barth, D.S. Reversal of established traumatic brain injury-induced, anxiety-like behavior in rats after delayed, post-injury neuroimmune suppression. J. Neurotrauma 2014, 31, 487–497. [Google Scholar] [CrossRef]
- Ruzafa, N.; Vecino, E. Efecto de las células de Müller en la supervivencia y neuritogénesis de las células ganglionares de la retina. Arch. La Soc. Española Oftalmol. 2015, 90, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Cueva Vargas, J.L.; Belforte, N.; Di Polo, A. The glial cell modulator ibudilast attenuates neuroinflammation and enhances retinal ganglion cell viability in glaucoma through protein kinase A signaling. Neurobiol. Dis. 2016, 93, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.M.; Danesh-Meyer, H.V. Phosphodiesterase inhibitors and the eye. Clin. Exp. Ophthalmol. 2009, 37, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Sela, T.C.; Zahavi, A.; Friedman-Gohas, M.; Weiss, S.; Sternfeld, A.; Ilguisonis, A.; Badash, D.; Geffen, N.; Ofri, R.; BarKana, Y.; et al. Azithromycin and Sildenafil May Have Protective Effects on Retinal Ganglion Cells via Different Pathways: Study in a Rodent Microbead Model. Pharmaceuticals 2023, 16, 486. [Google Scholar] [CrossRef]
- Storgaard, L.; Tran, T.L.; Freiberg, J.C.; Hauser, A.S.; Kolko, M. Glaucoma Clinical Research: Trends in Treatment Strategies and Drug Development. Front. Med. 2021, 8, 733080. [Google Scholar] [CrossRef]
- Abcouwer, S.F.; Lin, C.-m.; Shanmugam, S.; Muthusamy, A.; Barber, A.J.; Antonetti, D.A. Minocycline prevents retinal inflammation and vascular permeability following ischemia-reperfusion injury. J. Neuroinflamm. 2013, 10, 913. [Google Scholar] [CrossRef]
- Levkovitch-Verbin, H.; Kalev-Landoy, M.; Habot-Wilner, Z.; Melamed, S. Minocycline Delays Death of Retinal Ganglion Cells in Experimental Glaucoma and after Optic Nerve Transection. Arch. Ophthalmol. 2006, 124, 520–526. [Google Scholar] [CrossRef]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced Retina Microglial Activation and Improved Optic Nerve Integrity with Minocycline Treatment in the DBA/2J Mouse Model of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1437–1446. [Google Scholar] [CrossRef]
- Bordone, M.P.; González Fleitas, M.F.; Pasquini, L.A.; Bosco, A.; Sande, P.H.; Rosenstein, R.E.; Dorfman, D. Involvement of microglia in early axoglial alterations of the optic nerve induced by experimental glaucoma. J. Neurochem. 2017, 142, 323–337. [Google Scholar] [CrossRef]
- Grotegut, P.; Perumal, N.; Kuehn, S.; Smit, A.; Dick, H.B.; Grus, F.H.; Joachim, S.C. Minocycline reduces inflammatory response and cell death in a S100B retina degeneration model. J. Neuroinflamm. 2020, 17, 375. [Google Scholar] [CrossRef]
- Yu, H.; Zhong, H.; Sun, J.; Li, N.; Chen, J.; Shen, B.; Huang, P.; Shen, X.; Huang, S.; Zhong, Y. Molecular signaling from microglia impacts macroglia autophagy and neurons survival in glaucoma. iScience 2023, 26, 106839. [Google Scholar] [CrossRef] [PubMed]
- Levkovitch-Verbin, H.; Waserzoog, Y.; Vander, S.; Makarovsky, D.; Ilia, P. Minocycline mechanism of neuroprotection involves the Bcl-2 gene family in optic nerve transection. Int. J. Neurosci. 2014, 124, 755–761. [Google Scholar] [CrossRef]
- Kernt, M.; Neubauer, A.S.; Eibl, K.H.; Wolf, A.; Ulbig, M.W.; Kampik, A.; Hirneiss, C. Minocycline is cytoprotective in human trabecular meshwork cells and optic nerve head astrocytes by increasing expression of XIAP, survivin, and Bcl-2. Clin. Ophthalmol. 2010, 4, 591–604. [Google Scholar] [CrossRef]
- Cramer, C.L.; Patterson, A.; Alchakaki, A.; Soubani, A.O. Immunomodulatory indications of azithromycin in respiratory disease: A concise review for the clinician. Postgrad. Med. 2017, 129, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Varano, G.P.; Parisi, V.; Adornetto, A.; Cavaliere, F.; Amantea, D.; Nucci, C.; Corasaniti, M.T.; Morrone, L.A.; Bagetta, G.; Russo, R. Post-ischemic treatment with azithromycin protects ganglion cells against retinal ischemia/reperfusion injury in the rat. Mol. Vis. 2017, 23, 911–921. [Google Scholar]
- Mead, B.; Tomarev, S. Bone Marrow-Derived Mesenchymal Stem Cells-Derived Exosomes Promote Survival of Retinal Ganglion Cells through miRNA-Dependent Mechanisms. Stem. Cells Transl. Med. 2017, 6, 1273–1285. [Google Scholar] [CrossRef]
- Mead, B.; Amaral, J.; Tomarev, S. Mesenchymal Stem Cell-Derived Small Extracellular Vesicles Promote Neuroprotection in Rodent Models of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2018, 59, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Jaganathan, B.G. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biologics 2021, 15, 299–306. [Google Scholar] [CrossRef]
- Vilela, C.A.P.; Messias, A.; Calado, R.T.; Siqueira, R.C.; Silva, M.J.L.; Covas, D.T.; Paula, J.S. Retinal function after intravitreal injection of autologous bone marrow-derived mesenchymal stromal cells in advanced glaucoma. Doc. Ophthalmol. 2021, 143, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Reboussin, É.; Buffault, J.; Brignole-Baudouin, F.; Réaux-Le Goazigo, A.; Riancho, L.; Olmiere, C.; Sahel, J.-A.; Mélik Parsadaniantz, S.; Baudouin, C. Evaluation of neuroprotective and immunomodulatory properties of mesenchymal stem cells in an ex vivo retinal explant model. J. Neuroinflamm. 2022, 19, 63. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef]
- Astafurov, K.; Elhawy, E.; Ren, L.; Dong, C.Q.; Igboin, C.; Hyman, L.; Griffen, A.; Mittag, T.; Danias, J. Oral microbiome link to neurodegeneration in glaucoma. PLoS ONE 2014, 9, e104416. [Google Scholar] [CrossRef] [PubMed]
- Nayyar, A.; Gindina, S.; Barron, A.; Hu, Y.; Danias, J. Do epigenetic changes caused by commensal microbiota contribute to development of ocular disease? A review of evidence. Hum. Genom. 2020, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.S.; Loram, L.C.; Hutchinson, M.R.; Li, C.M.; Zhang, Y.; Maier, S.F.; Huang, Y.; Rice, K.C.; Watkins, L.R. (+)-naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. J. Pain 2012, 13, 498–506. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimazawa, M.; Ojino, K.; Izawa, H.; Takeuchi, H.; Inoue, Y.; Tsuruma, K.; Hara, H. Toll-like receptor 4 inhibitor protects against retinal ganglion cell damage induced by optic nerve crush in mice. J. Pharmacol. Sci. 2017, 133, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zhu, M.N.; Chen, B.J.; Wang, Z.; He, L.Y.; Zhang, R. Inhibitive effect of TAK-242 on Tenon’s capsule fibroblasts proliferation in rat eyes. Int. J. Ophthalmol. 2019, 12, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.P.; Curry, S.; McDowell, C.M. Effects of Toll-like Receptor 4 Inhibition on Transforming Growth Factor-β2 Signaling in the Human Trabecular Meshwork. J. Ocul. Pharmacol. Ther. 2020, 36, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Chen, N.; Wu, J.; Wang, J.; Piri, N.; Chen, F.; Xiao, T.; Zhao, Y.; Sun, D.; Kaplan, H.J.; Shao, H. Short chain fatty acids inhibit endotoxin-induced uveitis and inflammatory responses of retinal astrocytes. Exp. Eye Res. 2021, 206, 108520. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Rep. 1999, 4, 179–184. [Google Scholar] [CrossRef]
- Jung, K.I.; Kim, Y.C.; Park, C.K. Dietary Niacin and Open-Angle Glaucoma: The Korean National Health and Nutrition Examination Survey. Nutrients 2018, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a Metabolic Optic Neuropathy: Making the Case for Nicotinamide Treatment in Glaucoma. J. Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, R.; Kumari, A.; Panwar, A. Astaxanthin: A super antioxidant from microalgae and its therapeutic potential. J. Basic Microbiol. 2022, 62, 1064–1082. [Google Scholar] [CrossRef] [PubMed]
- Cort, A.; Ozturk, N.; Akpinar, D.; Unal, M.; Yucel, G.; Ciftcioglu, A.; Yargicoglu, P.; Aslan, M. Suppressive effect of astaxanthin on retinal injury induced by elevated intraocular pressure. Regul. Toxicol. Pharmacol. 2010, 58, 121–130. [Google Scholar] [CrossRef]
- Kikuchi, K.; Dong, Z.; Shinmei, Y.; Murata, M.; Kanda, A.; Noda, K.; Harada, T.; Ishida, S. Cytoprotective Effect of Astaxanthin in a Model of Normal Intraocular Pressure Glaucoma. J. Ophthalmol. 2020, 2020, 9539681. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Q.; Chu, C.; Liu, S. Astaxanthin protects retinal ganglion cells from acute glaucoma via the Nrf2/HO-1 pathway. J Chem. Neuroanat. 2020, 110, 101876. [Google Scholar] [CrossRef]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int. J. Mol. Sci. 2022, 23, 15105. [Google Scholar] [CrossRef] [PubMed]
- Pirhan, D.; Yüksel, N.; Emre, E.; Cengiz, A.; Kürşat Yıldız, D. Riluzole- and Resveratrol-Induced Delay of Retinal Ganglion Cell Death in an Experimental Model of Glaucoma. Curr. Eye Res. 2016, 41, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.J.; Meng, N. Resveratrol acts via the mitogen-activated protein kinase (MAPK) pathway to protect retinal ganglion cells from apoptosis induced by hydrogen peroxide. Bioengineered 2021, 12, 4878–4886. [Google Scholar] [CrossRef]
- Chronopoulos, P.; Manicam, C.; Zadeh, J.K.; Laspas, P.; Unkrig, J.C.; Göbel, M.L.; Musayeva, A.; Pfeiffer, N.; Oelze, M.; Daiber, A.; et al. Effects of Resveratrol on Vascular Function in Retinal Ischemia-Reperfusion Injury. Antioxidants 2023, 12, 145. [Google Scholar] [CrossRef]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef] [PubMed]
- Sanz-González, S.M.; Raga-Cervera, J.; Aguirre Lipperheide, M.; Zanón-Moreno, V.; Chiner, V.; Ramírez, A.I.; Pinazo-Durán, M.D. Effect of an oral supplementation with a formula containing R-lipoic acid in glaucoma patients. Arch. Soc. Esp. Oftalmol. 2020, 95, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Yue, Y.K.; Mo, B.; Zhao, J.; Yu, Y.J.; Liu, L.; Yue, C.L.; Liu, W. Neuroprotective effect of curcumin against oxidative damage in BV-2 microglia and high intraocular pressure animal model. J. Ocul. Pharmacol. Ther. 2014, 30, 657–664. [Google Scholar] [CrossRef]
- Buccarello, L.; Dragotto, J.; Hassanzadeh, K.; Maccarone, R.; Corbo, M.; Feligioni, M. Retinal ganglion cell loss in an ex vivo mouse model of optic nerve cut is prevented by curcumin treatment. Cell Death Discov. 2021, 7, 394. [Google Scholar] [CrossRef]
- Liu, X.G.; Wu, S.Q.; Li, P.; Yang, H. Advancement in the chemical analysis and quality control of flavonoid in Ginkgo biloba. J Pharm. Biomed. Anal. 2015, 113, 212–225. [Google Scholar] [CrossRef]
- Yu, H.; Dong, L.-H.; Zhang, Y.; Liu, Q. A network pharmacology-based strategy for predicting the protective mechanism of Ginkgo biloba on damaged retinal ganglion cells. Chin. J. Nat. Med. 2022, 20, 54–66. [Google Scholar] [CrossRef]
- Anne-Marie, B.; Beatriz, V.; Ana Isabel, J.; Covadonga, P. Managing Intraocular Pressure: Innovation in Glaucoma Management. In Glaucoma, Parul, I., Ed.; IntechOpen: Rijeka, Croatia, 2016; p. 6. [Google Scholar]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef]
- Quaranta, L.; Riva, I.; Biagioli, E.; Rulli, E.; Rulli, E.; Poli, D.; Legramandi, L. Evaluating the Effects of an Ophthalmic Solution of Coenzyme Q10 and Vitamin E in Open-Angle Glaucoma Patients: A Study Protocol. Adv. Ther. 2019, 36, 2506–2514. [Google Scholar] [CrossRef]
- Laspas, P.; Zhutdieva, M.B.; Brochhausen, C.; Musayeva, A.; Zadeh, J.K.; Pfeiffer, N.; Xia, N.; Li, H.; Wess, J.; Gericke, A. The M(1) muscarinic acetylcholine receptor subtype is important for retinal neuron survival in aging mice. Sci. Rep. 2019, 9, 5222. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.P.; Medina, S.V.; Araujo, E.G. Cholinergic activity modulates the survival of retinal ganglion cells in culture: The role of M1 muscarinic receptors. Int. J. Dev. Neurosci. 2001, 19, 559–567. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, X.; Zhu, L.; Cui, Y.Y.; Wang, H.; Qi, H.; Ren, Q.S.; Chen, H.Z. Neuroprotection of muscarinic receptor agonist pilocarpine against glutamate-induced apoptosis in retinal neurons. Cell Mol. Neurobiol. 2008, 28, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Zhou, Y.; Kelly, M.E.; Casanova, C.; Di Polo, A. Structural and functional neuroprotection in glaucoma: Role of galantamine-mediated activation of muscarinic acetylcholine receptors. Cell Death Dis. 2010, 1, e27. [Google Scholar] [CrossRef]
- Yu, P.; Dong, W.P.; Tang, Y.B.; Chen, H.Z.; Cui, Y.Y.; Bian, X.L. Huperzine A lowers intraocular pressure via the M3 mAChR and provides retinal neuroprotection via the M1 mAChR: A promising agent for the treatment of glaucoma. Ann. Transl. Med. 2021, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Guo, X.; Noro, T.; Harada, C.; Tanaka, K.; Namekata, K.; Harada, T. Valproic acid prevents retinal degeneration in a murine model of normal tension glaucoma. Neurosci. Lett. 2015, 588, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Tribble, J.R.; Kastanaki, E.; Uslular, A.B.; Rutigliani, C.; Enz, T.J.; Williams, P.A. Valproic Acid Reduces Neuroinflammation to Provide Retinal Ganglion Cell Neuroprotection in the Retina Axotomy Model. Front. Cell Dev. Biol. 2022, 10, 903436. [Google Scholar] [CrossRef]
- Mahalingam, K.; Chaurasia, A.K.; Gowtham, L.; Gupta, S.; Somarajan, B.I.; Velpandian, T.; Sihota, R.; Gupta, V. Therapeutic potential of valproic acid in advanced glaucoma: A pilot study. Indian J. Ophthalmol. 2018, 66, 1104–1108. [Google Scholar] [CrossRef]
- Harada, C.; Noro, T.; Kimura, A.; Guo, X.; Namekata, K.; Nakano, T.; Harada, T. Suppression of Oxidative Stress as Potential Therapeutic Approach for Normal Tension Glaucoma. Antioxidants 2020, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, G.; Tolun, F.I.; Gul, M.; Imrek, S. Retinal Oxidative Stress Induced by Intraocular Hypertension in Rats May be Ameliorated by Brimonidine Treatment and N-acetyl Cysteine Supplementation. J. Glaucoma 2009, 18, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Namekata, K.; Kimura, A.; Shitara, H.; Guo, X.; Harada, C.; Mitamura, Y.; Harada, T. Differential effects of N-acetylcysteine on retinal degeneration in two mouse models of normal tension glaucoma. Cell Death Dis. 2019, 10, 75. [Google Scholar] [CrossRef]
- Yang, L.; Tan, P.; Zhou, W.; Zhu, X.; Cui, Y.; Zhu, L.; Feng, X.; Qi, H.; Zheng, J.; Gu, P.; et al. N-acetylcysteine protects against hypoxia mimetic-induced autophagy by targeting the HIF-1α pathway in retinal ganglion cells. Cell Mol. Neurobiol. 2012, 32, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: Is edaravone an effective neuroprotective therapy? Expert Opin. Pharmacother. 2010, 11, 1753–1763. [Google Scholar] [CrossRef]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell. Longev. 2017, 2017, 9208489. [Google Scholar] [CrossRef]
- Akaiwa, K.; Namekata, K.; Azuchi, Y.; Guo, X.; Kimura, A.; Harada, C.; Mitamura, Y.; Harada, T. Edaravone suppresses retinal ganglion cell death in a mouse model of normal tension glaucoma. Cell Death Dis. 2017, 8, e2934. [Google Scholar] [CrossRef]
- Aksar, A.T.; Yuksel, N.; Gok, M.; Cekmen, M.; Caglar, Y. Neuroprotective effect of edaravone in experimental glaucoma model in rats: A immunofluorescence and biochemical analysis. Int. J. Ophthalmol. 2015, 8, 239–244. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin Protects against Neuron Death in In Vitro and In Vivo Models of Parkinson’s Disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem. 2009, 284, 27416–27424. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS ONE 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kwon, Y.W.; Kondo, N.; Bai, J.; Masutani, H.; Nakamura, H.; Fujii, J.; Ohira, A.; Yodoi, J. Cytoprotective effects of geranylgeranylacetone against retinal photooxidative damage. J. Neurosci. 2005, 25, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shinmei, Y.; Dong, Y.; Inafuku, S.; Fukuhara, J.; Ando, R.; Kitaichi, N.; Kanda, A.; Tanaka, K.; Noda, K.; et al. Effect of geranylgeranylacetone on the protection of retinal ganglion cells in a mouse model of normal tension glaucoma. Heliyon 2016, 2, e00191. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Leng, Y.; Jiang, Q.; Wang, Z.; Luo, P.; Zhang, C.; Chen, L.; Wang, Y.; Wang, H.; Yue, X.; et al. Eye Drops of Metformin Prevents Fibrosis After Glaucoma Filtration Surgery in Rats via Activating AMPK/Nrf2 Signaling Pathway. Front. Pharmacol. 2020, 11, 1038. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, M.; Yao, F.; Xia, X.; Duan, T.; Meng, J.; Huang, Y.; He, Y.; Saro, A.; Huang, J. Valdecoxib Protects against Cell Apoptosis Induced by Endoplasmic Reticulum Stress via the Inhibition of PERK-ATF4-CHOP Pathway in Experimental Glaucoma. Int. J. Mol. Sci. 2022, 23, 12983. [Google Scholar] [CrossRef]
- Feillet, F.; Leonard, J.V. Alternative pathway therapy for urea cycle disorders. J. Inherit. Metab. Dis. 1998, 21 (Suppl. S1), 101–111. [Google Scholar] [CrossRef]
- Brown, C.R.; Hong-Brown, L.Q.; Biwersi, J.; Verkman, A.S.; Welch, W.J. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996, 1, 117–125. [Google Scholar] [CrossRef]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson’s disease. PLoS ONE 2012, 7, e38113. [Google Scholar] [CrossRef]
- Dong, Y.; Li, L.; Xia, T.; Wang, L.; Xiao, L.; Ding, N.; Wu, Y.; Lu, K. Oxidative Stress Can Be Attenuated by 4-PBA Caused by High-Fat or Ammonia Nitrogen in Cultured Spotted Seabass: The Mechanism Is Related to Endoplasmic Reticulum Stress. Antioxidants 2022, 11, 1276. [Google Scholar] [CrossRef]
- Zode, G.S.; Kuehn, M.H.; Nishimura, D.Y.; Searby, C.C.; Mohan, K.; Grozdanic, S.D.; Bugge, K.; Anderson, M.G.; Clark, A.F.; Stone, E.M.; et al. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J. Clin. Investig. 2011, 121, 3542–3553. [Google Scholar] [CrossRef] [PubMed]
- Maddineni, P.; Kasetti, R.B.; Kodati, B.; Yacoub, S.; Zode, G.S. Sodium 4-Phenylbutyrate Reduces Ocular Hypertension by Degrading Extracellular Matrix Deposition via Activation of MMP9. Int. J. Mol. Sci. 2021, 22, 10095. [Google Scholar] [CrossRef]
- Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants 2021, 10, 238. [Google Scholar] [CrossRef]
- Deliyanti, D.; Wilkinson-Berka, J.L. Inhibition of NOX1/4 with GKT137831: A potential novel treatment to attenuate neuroglial cell inflammation in the retina. J. Neuroinflamm. 2015, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Wikström, P.; Walum, E.; Thermos, K. Effect of NADPH oxidase inhibitors in an experimental retinal model of excitotoxicity. Exp. Eye Res. 2020, 200, 108232. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Inoue, T.; Ohira, S.; Awai-Kasaoka, N.; Kameda, T.; Inoue-Mochita, M.; Tanihara, H. Inhibition of Rho Kinase Induces Antioxidative Molecules and Suppresses Reactive Oxidative Species in Trabecular Meshwork Cells. J. Ophthalmol. 2017, 2017, 7598140. [Google Scholar] [CrossRef]
- Chen, W.; Yang, X.; Fang, J.; Zhang, Y.; Zhu, W.; Yang, X. Rho-Associated Protein Kinase Inhibitor Treatment Promotes Proliferation and Phagocytosis in Trabecular Meshwork Cells. Front. Pharmacol. 2020, 11, 302. [Google Scholar] [CrossRef]
- Kamiya, T.; Omae, T.; Nakabayashi, S.; Takahashi, K.; Tanner, A.; Yoshida, A. Effect of Rho Kinase Inhibitor Ripasudil (K-115) on Isolated Porcine Retinal Arterioles. J. Ocul. Pharmacol. Ther. 2021, 37, 104–111. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buonfiglio, F.; Pfeiffer, N.; Gericke, A. Immunomodulatory and Antioxidant Drugs in Glaucoma Treatment. Pharmaceuticals 2023, 16, 1193. https://doi.org/10.3390/ph16091193
Buonfiglio F, Pfeiffer N, Gericke A. Immunomodulatory and Antioxidant Drugs in Glaucoma Treatment. Pharmaceuticals. 2023; 16(9):1193. https://doi.org/10.3390/ph16091193
Chicago/Turabian StyleBuonfiglio, Francesco, Norbert Pfeiffer, and Adrian Gericke. 2023. "Immunomodulatory and Antioxidant Drugs in Glaucoma Treatment" Pharmaceuticals 16, no. 9: 1193. https://doi.org/10.3390/ph16091193
APA StyleBuonfiglio, F., Pfeiffer, N., & Gericke, A. (2023). Immunomodulatory and Antioxidant Drugs in Glaucoma Treatment. Pharmaceuticals, 16(9), 1193. https://doi.org/10.3390/ph16091193