

s-Triazine Derivatives Functionalized with Alkylating 2-Chloroethylamine Fragments as Promising Antimicrobial Agents: Inhibition of Bacterial DNA Gyrases, Molecular Docking Studies, and Antibacterial and Antifungal Activity

 , , and
, , and
Abstract
:1. Introduction
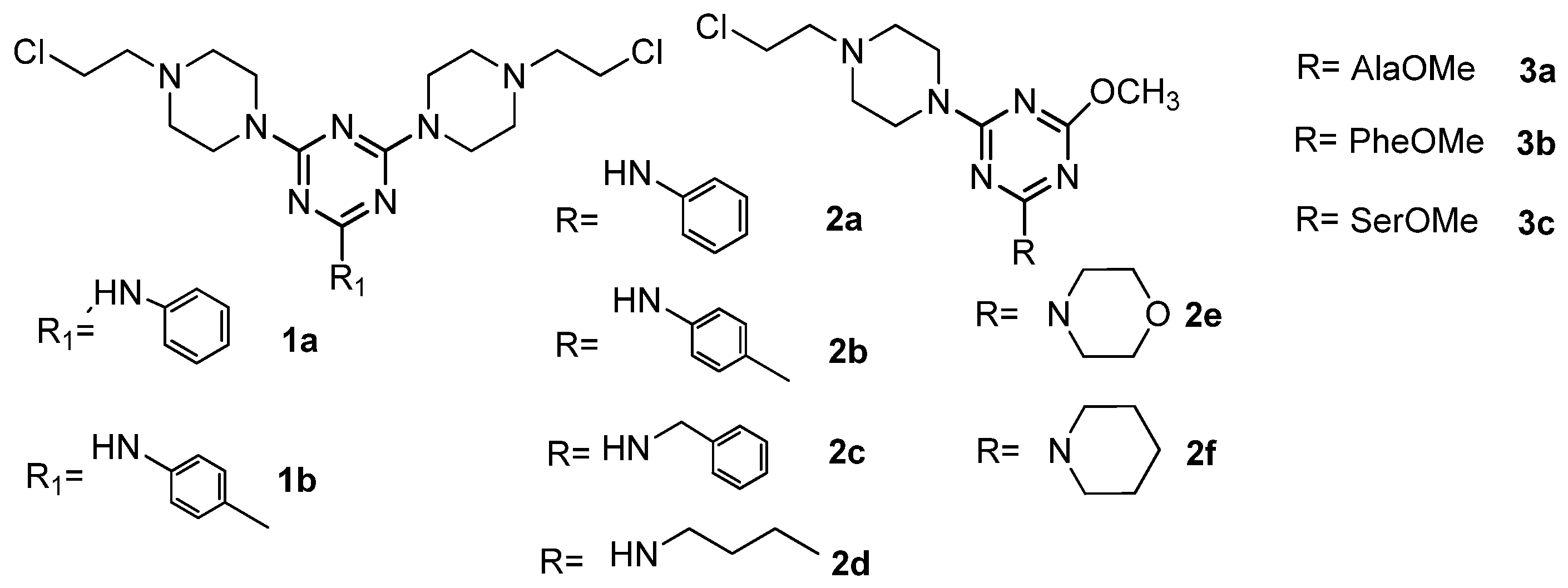
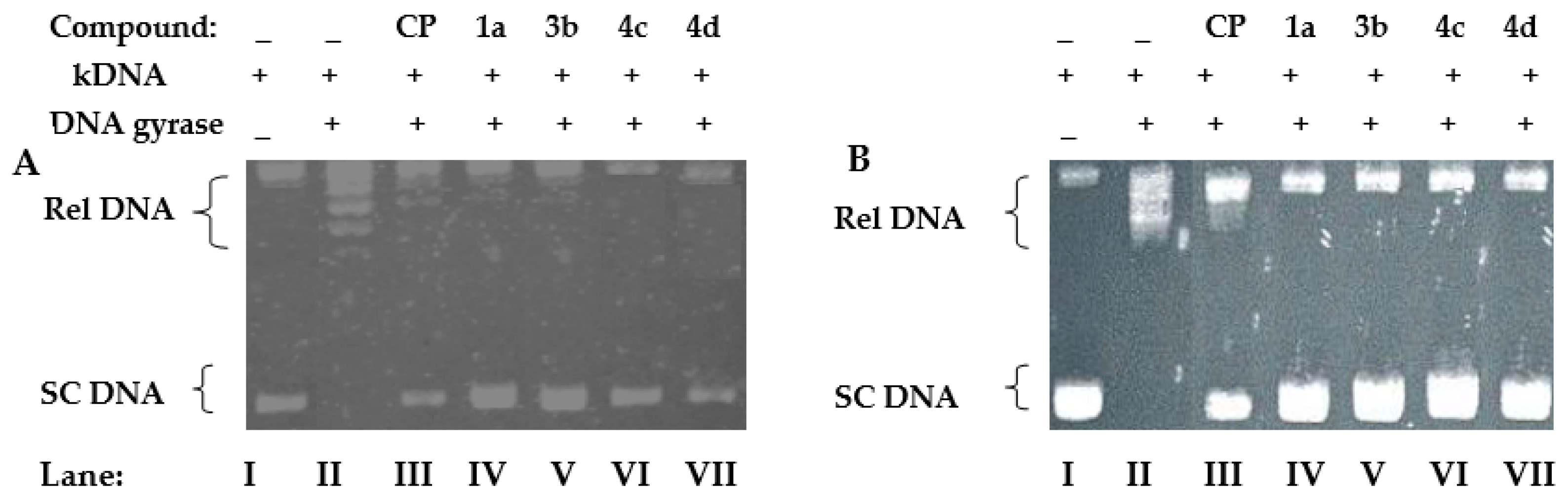
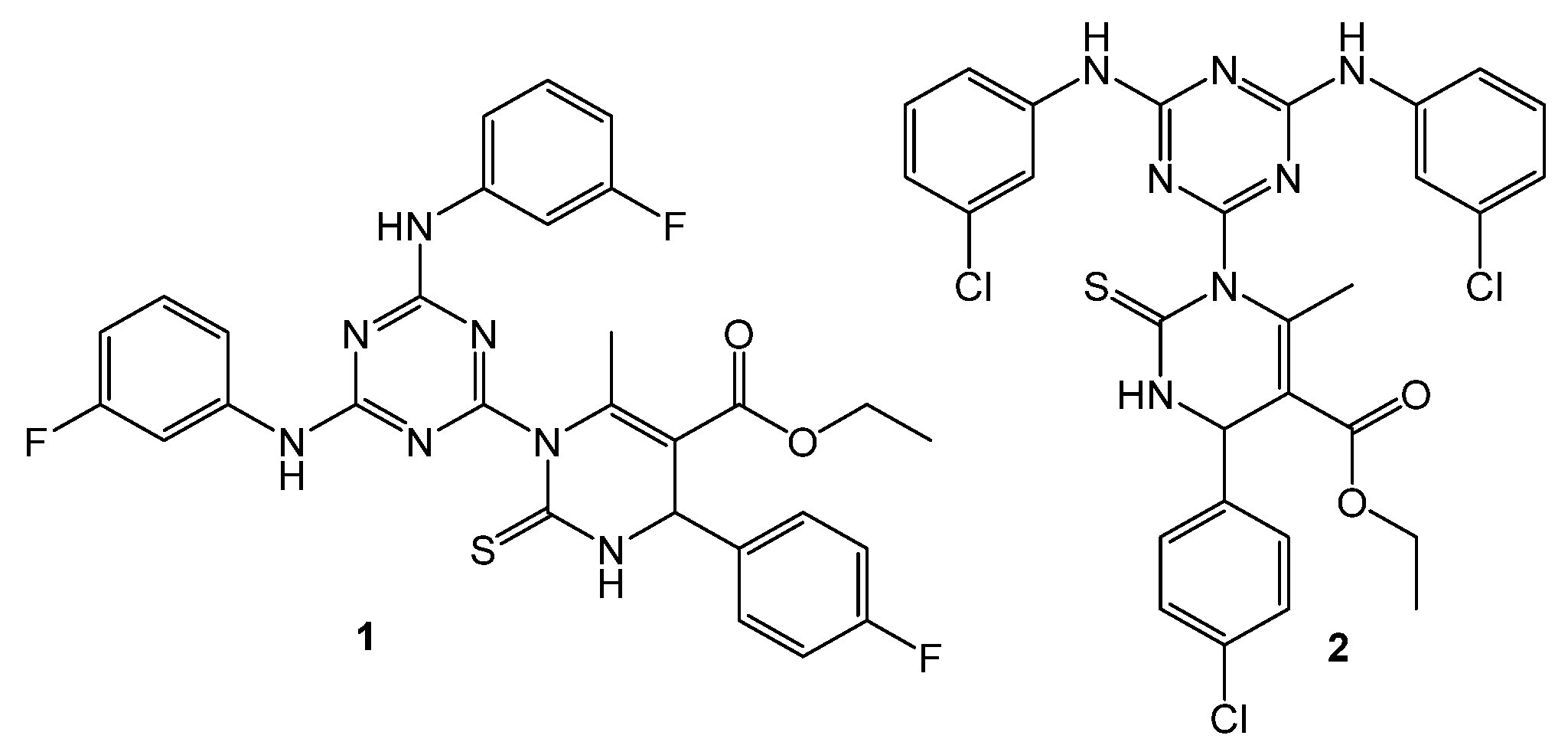
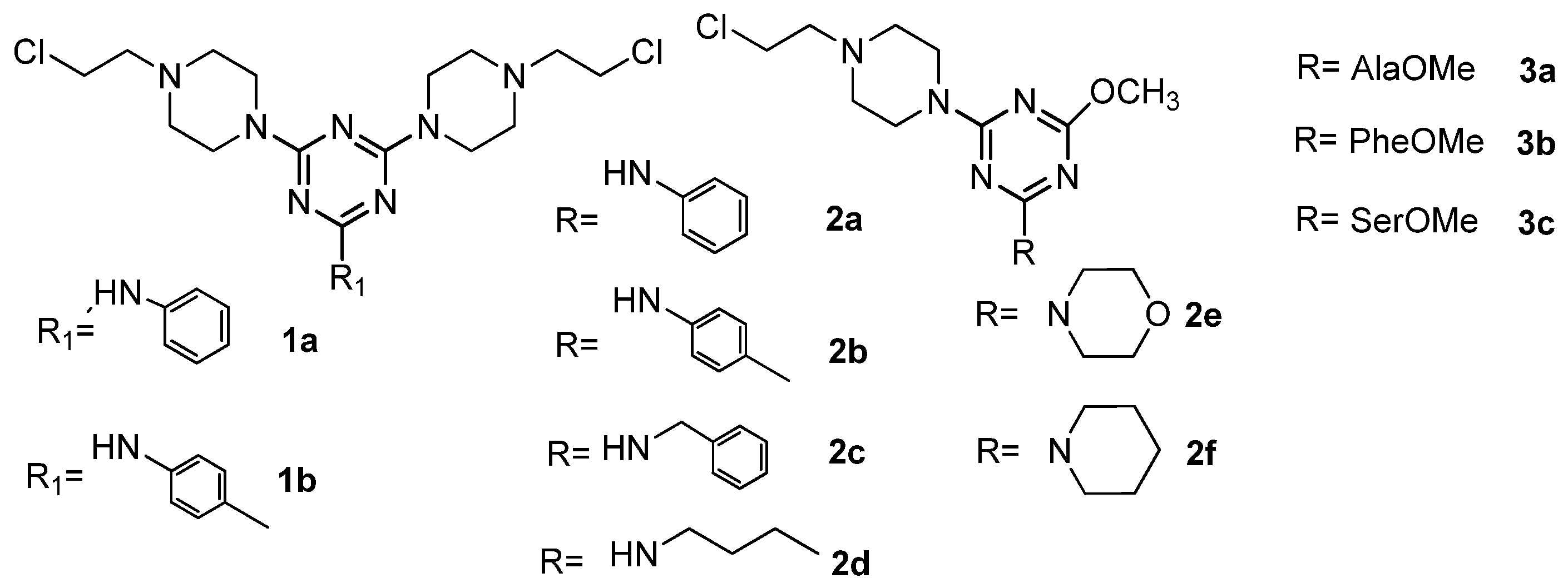
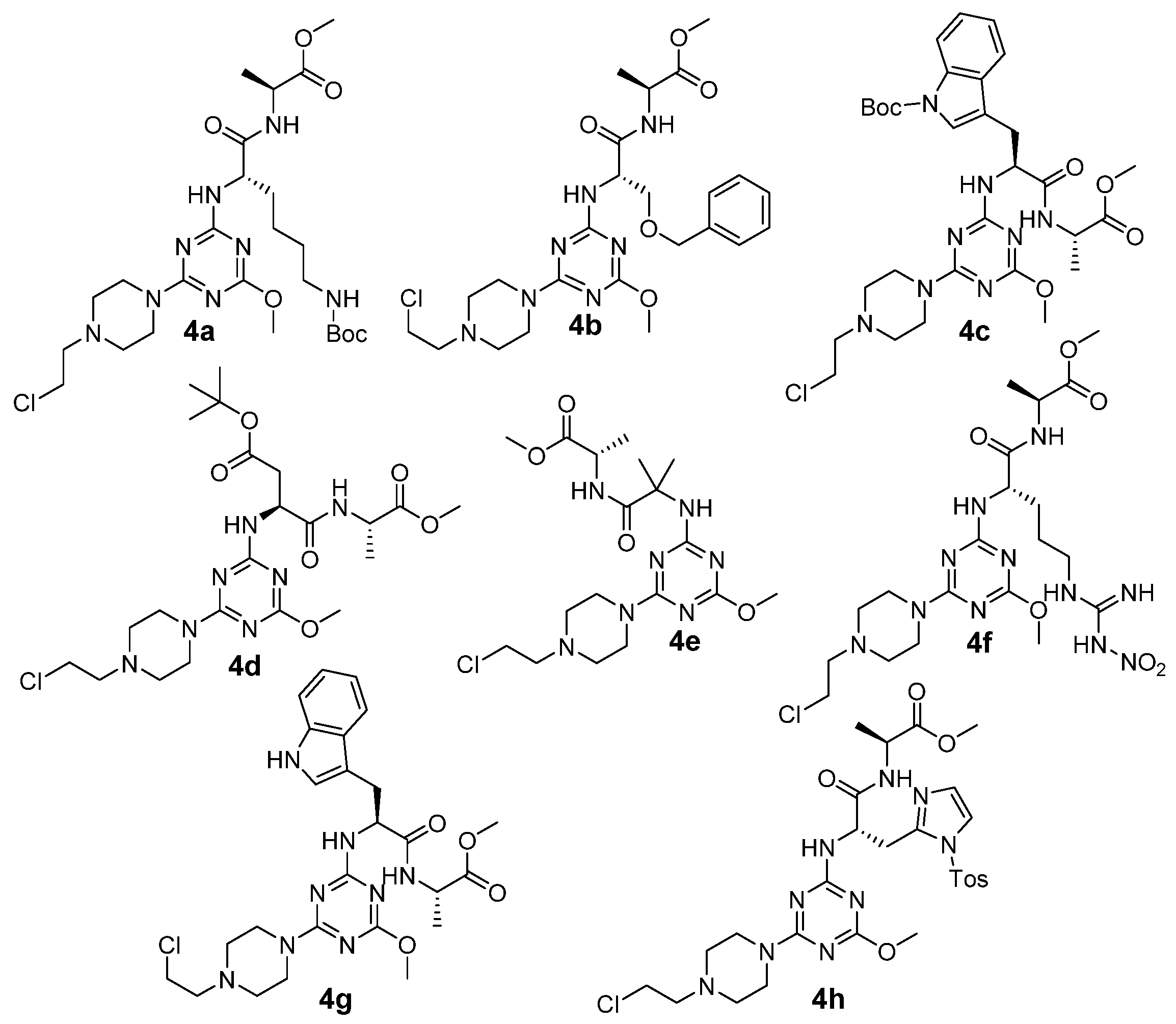
2. Results
3. Discussion
4. Materials and Methods
4.1. Study on Selected Cell Strains of Fungi and Bacteria
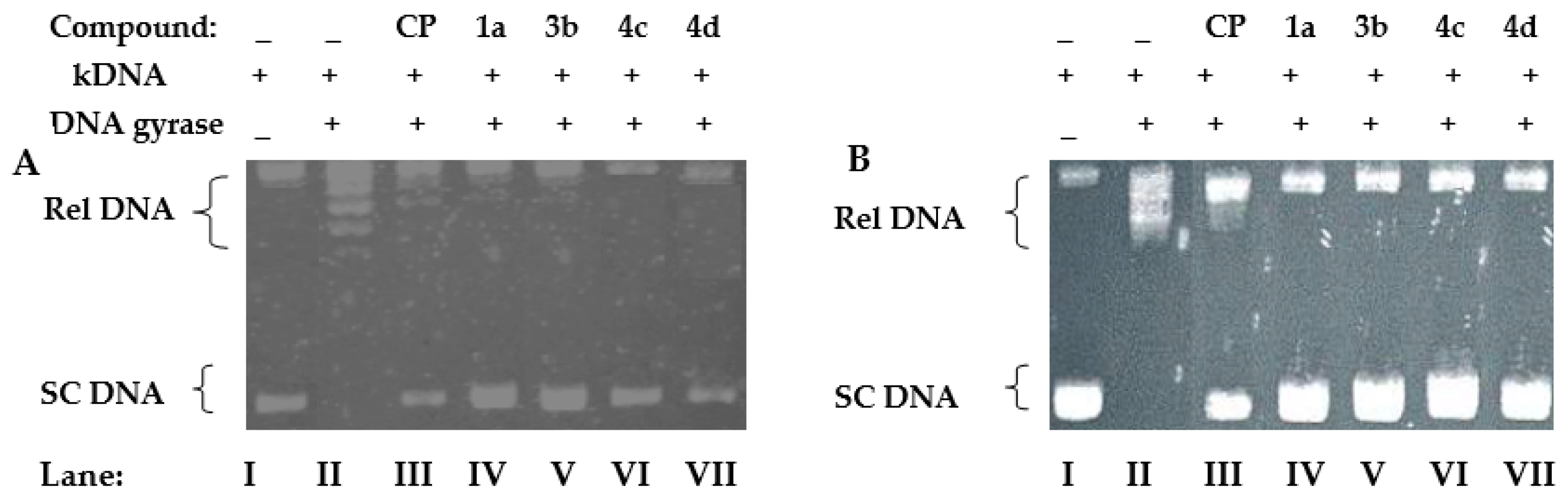
4.2. Relaxation Assay of Escherichia coli and Staphylococcus aureus Gyrases
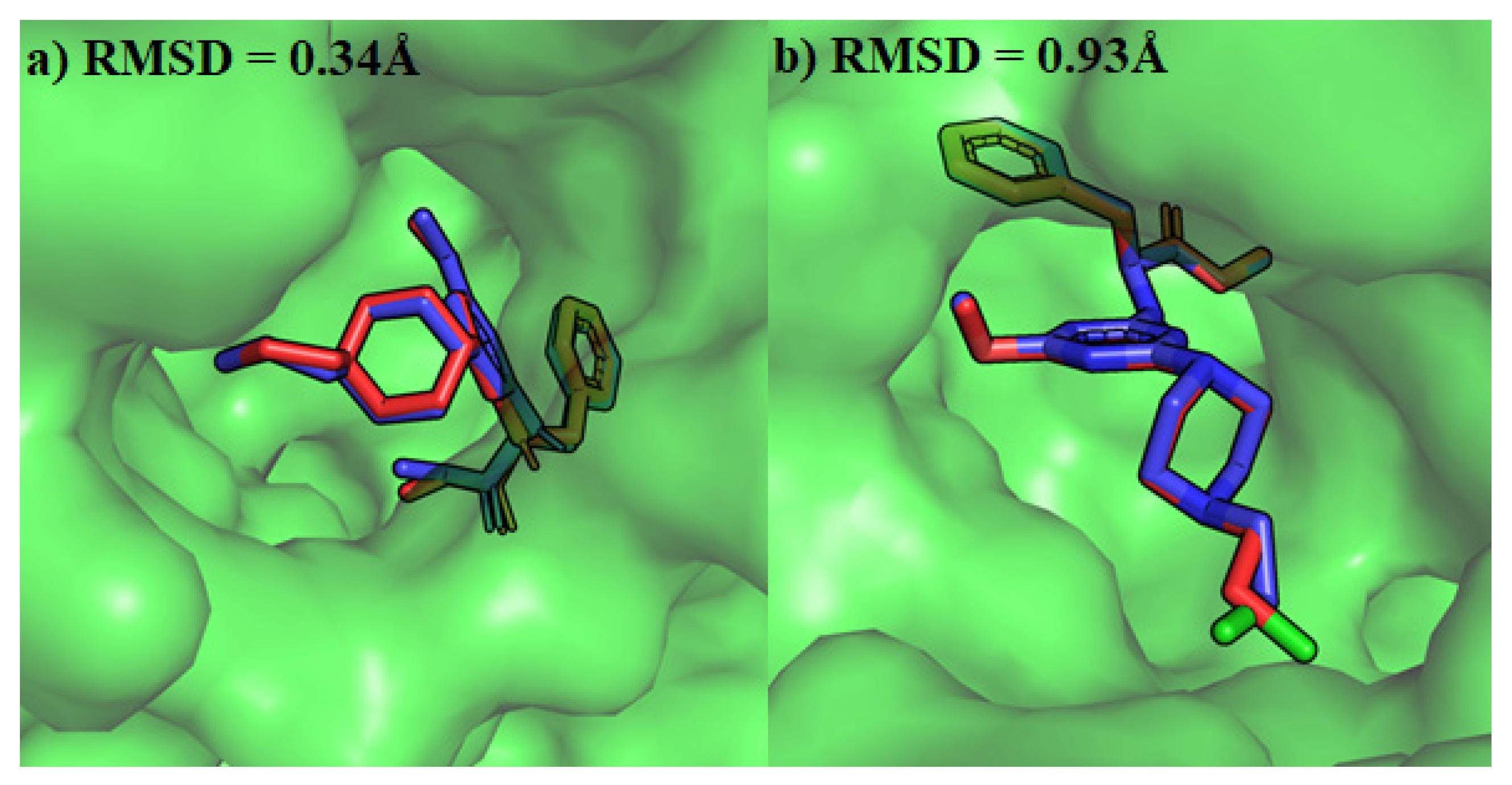
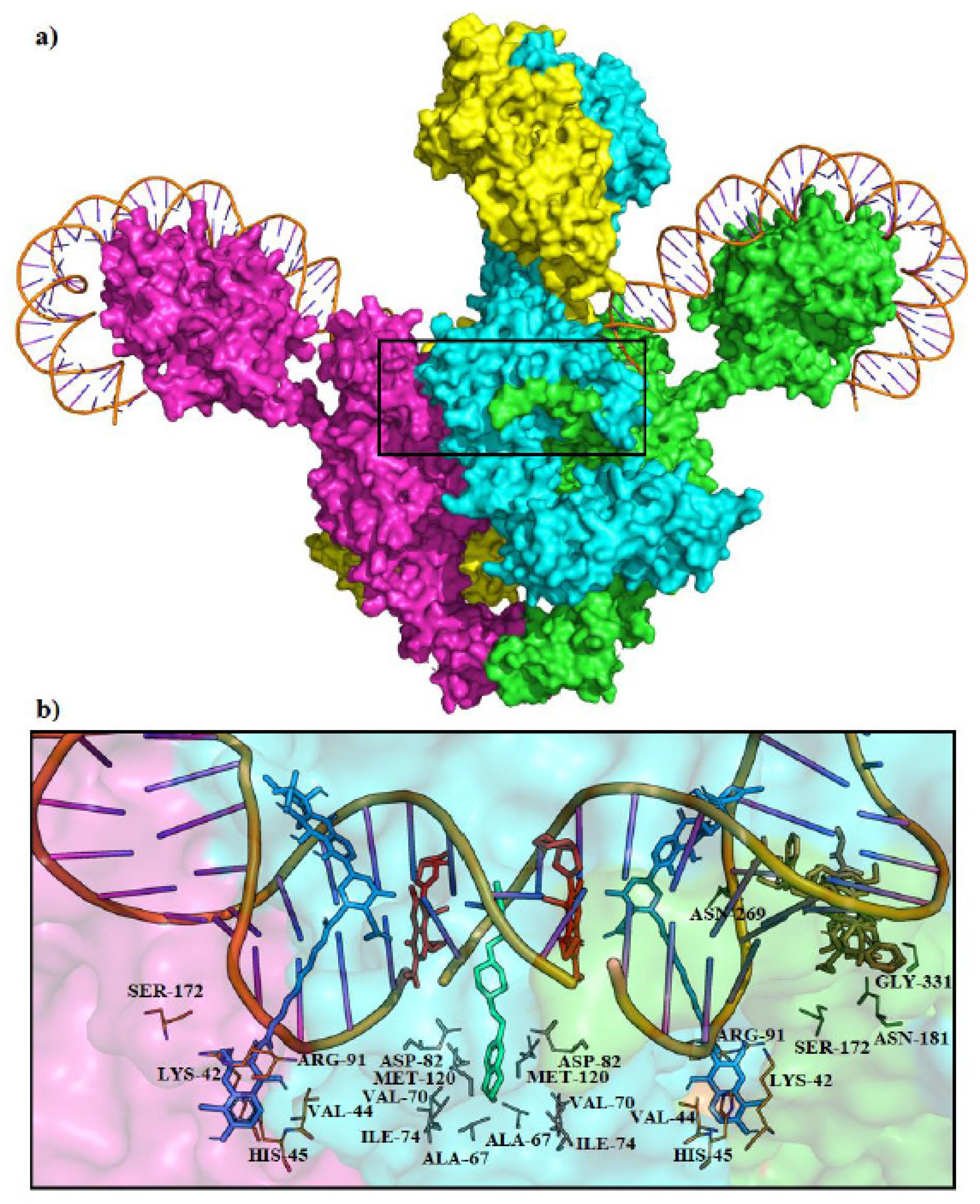
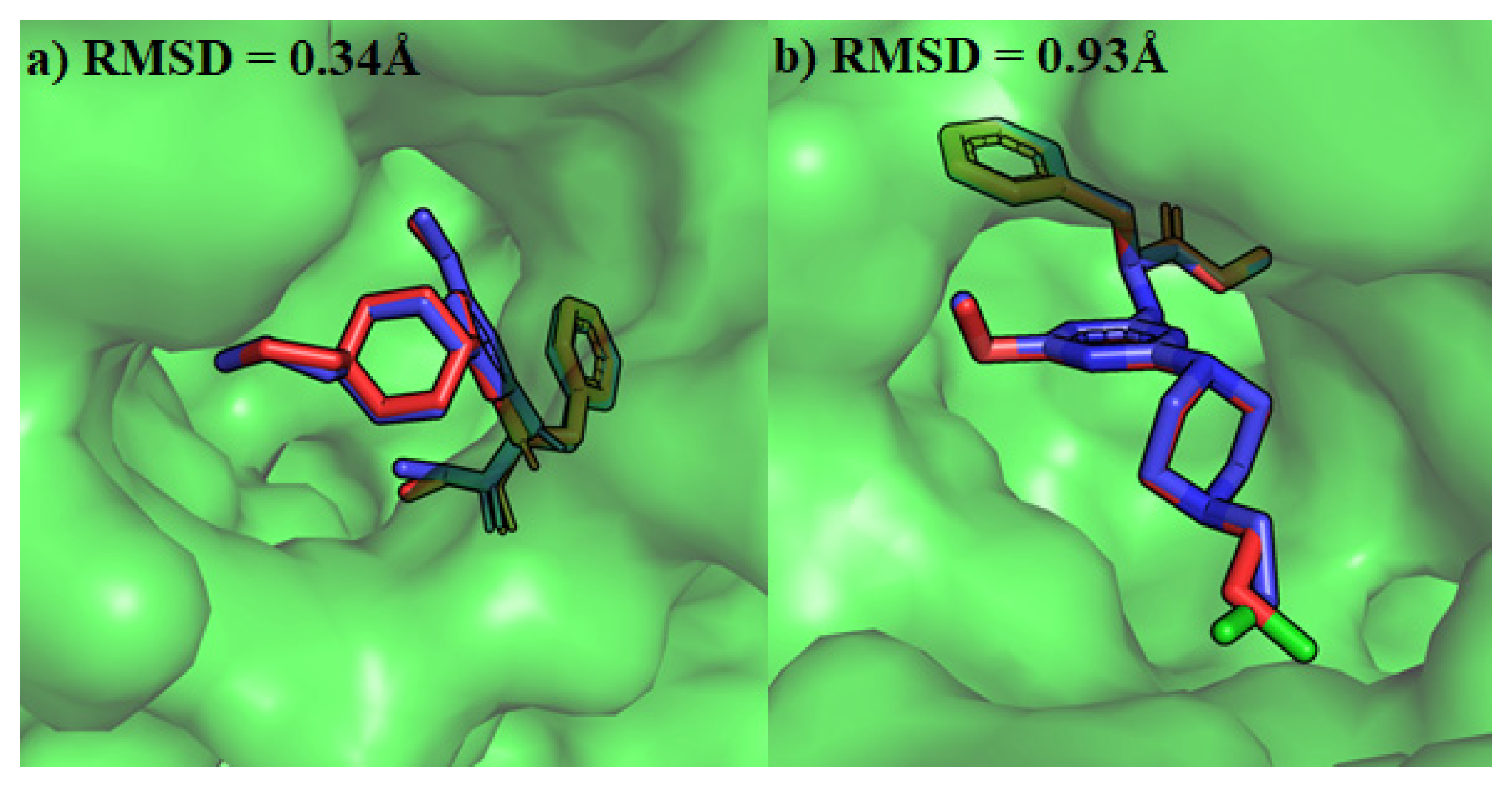
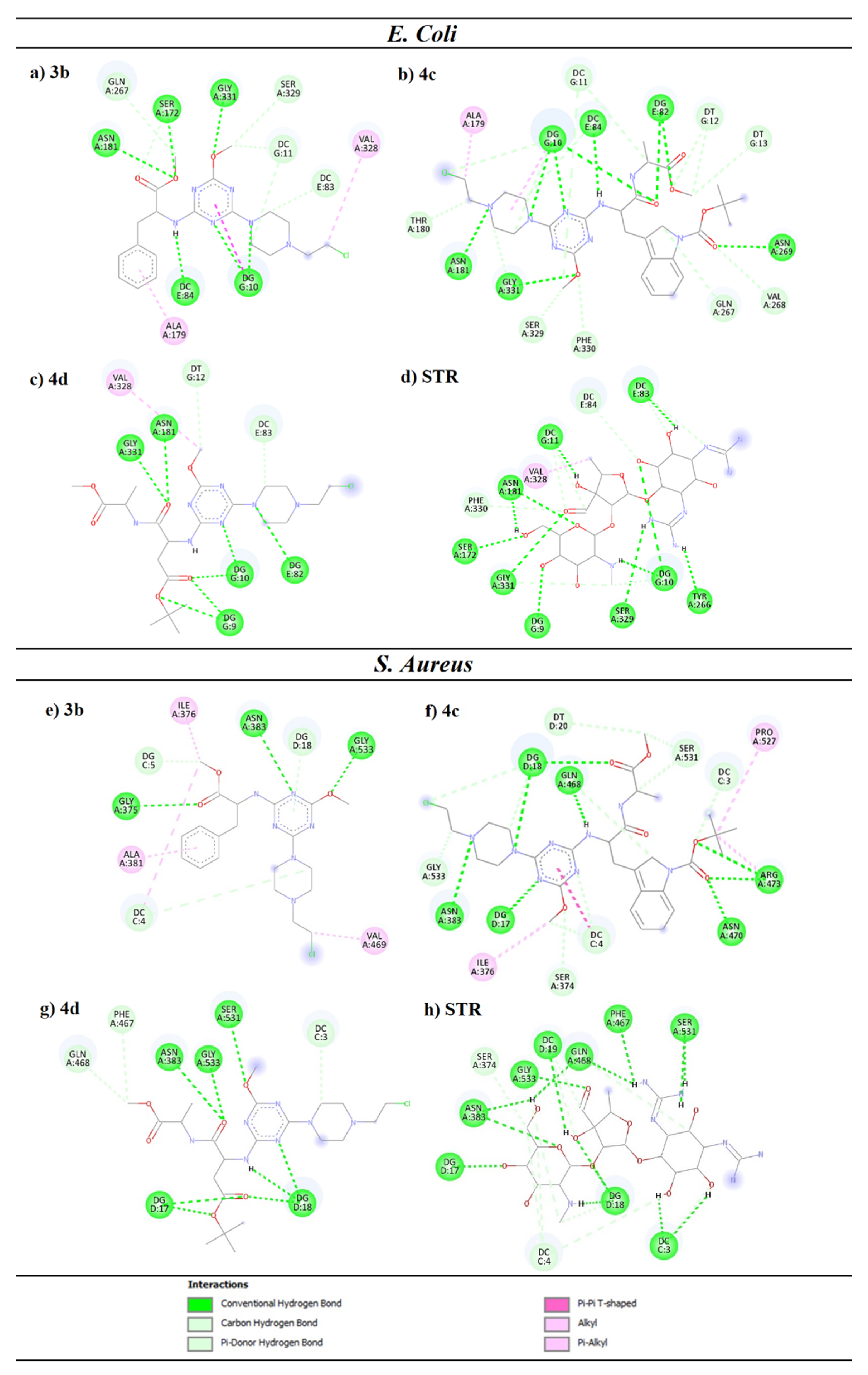
4.3. Molecular Docking of Escherichia coli and Staphylococcus aureus Gyrases
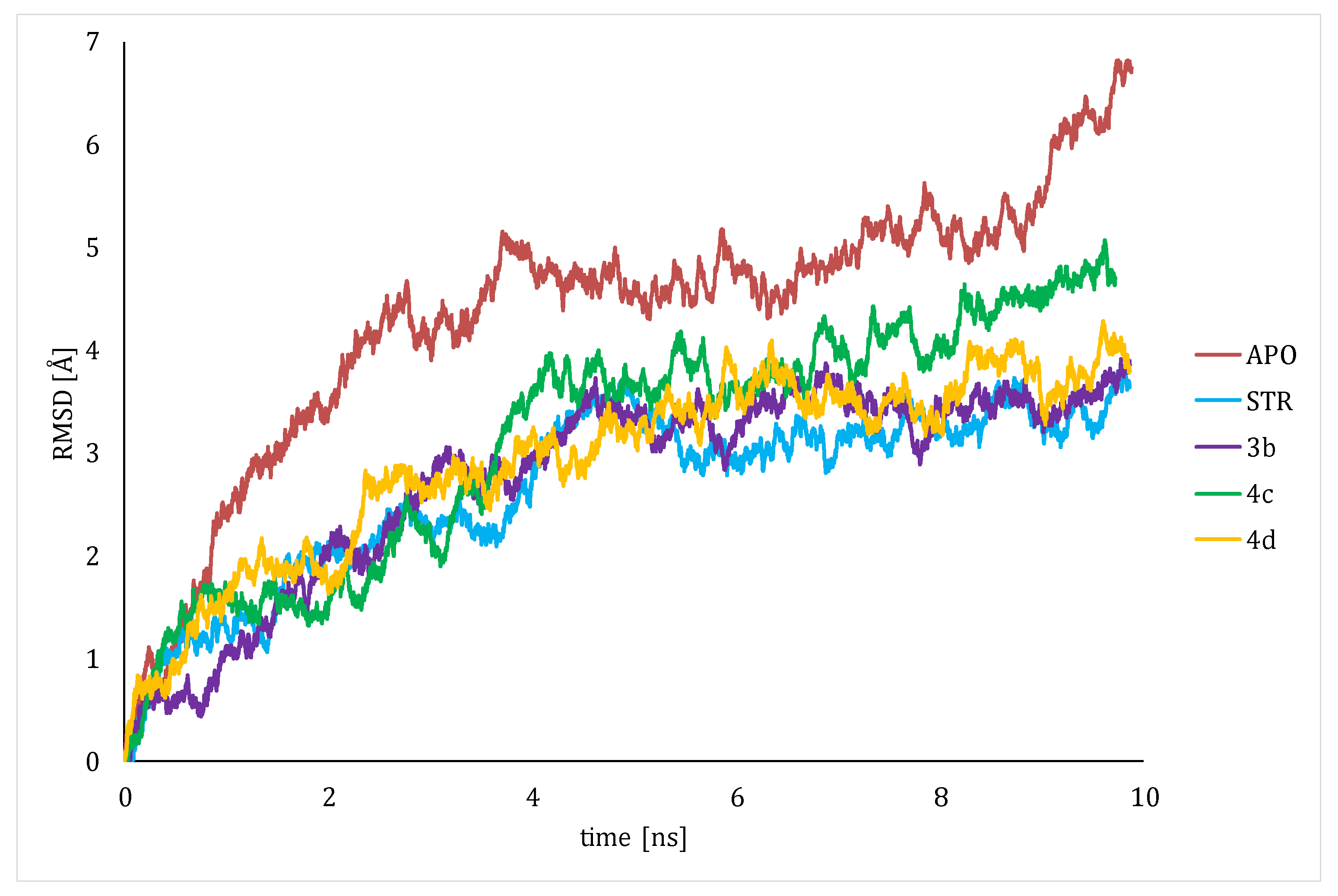
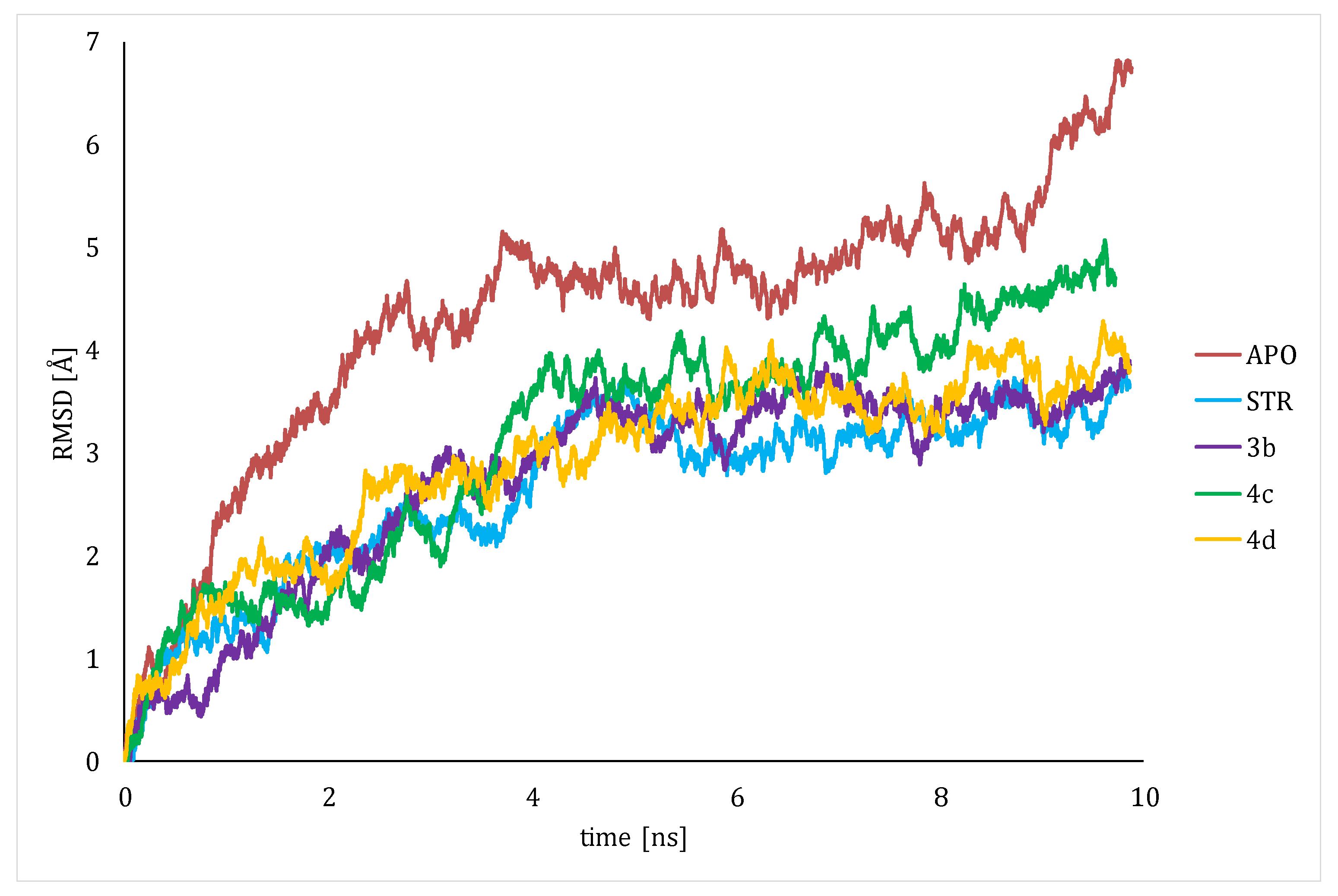
4.4. Molecular Dynamics
4.5. Pharmacokinetic Properties
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report: 2021; World Health Organization: Geneva, Switzerland, 2021; Available online: https://apps.who.int/iris/handle/10665/341666 (accessed on 12 April 2023).
- Sharma, A.; Sheyi, R.; de la Torre, B.G.; El-Faham, A.; Albericio, F. s-Triazine: A Privileged Structure for Drug Discovery and Bioconjugation. Molecules 2021, 26, 864. [Google Scholar] [CrossRef] [PubMed]
- Maliszewski, D.; Drozdowska, D. Recent Advances in the Biological Activity of s-Triazine Core Compounds. Pharmaceuticals 2022, 15, 221. [Google Scholar] [CrossRef] [PubMed]
- Drug Bank Stat. Available online: https://go.drugbank.com/stats (accessed on 30 May 2023).
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef]
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017; Available online: www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 30 May 2023).
- Edwards, M.J.; Flatman, R.H.; Mitchenall, L.A.; Stevenson, C.E.M.; Le, T.B.K.; Clarke, T.A.; McKay, A.; Fiedler, H.-P.; Buttner, M.; Lawson, D.; et al. A Crystal Structure of the Bifunctional Antibiotic Simocyclinone D8, Bound to DNA Gyrase. Science 2009, 326, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Singh, O.M.; Smith, C.V.; Skarzynski, T.; Maxwell, A.; Wonacott, A.J.; Wigley, D.B. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996, 15, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Flatman, R.H.; Howells, A.J.; Heide, L.; Fiedler, H.P.; Maxwell, A. Simocyclinone D8, an Inhibitor of DNA Gyrase with a Novel Mode of Action. Antimicrob. Agents Chemother. 2005, 49, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Sohi, M.K.; Veselkov, D.A.; Pan, X.S.; Sawhney, R.; Thompson, A.W.; McAuley, K.; Fisher, M.; Sanderson, M. Structural insight into the quinolone–DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009, 16, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. [Google Scholar] [CrossRef]
- Kolarič, A.; Germe, T.; Hrast, M.; Stevenson, C.E.M.; Lawson, D.M.; Burton, N.P.; Vörös, J.; Maxwell, A.; Minovski, N.; Anderluh, M. Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds. Nat. Commun. 2021, 12, 150. [Google Scholar] [CrossRef]
- Masih, A.; Shrivastava, J.K.; Bhat, H.R.; Singh, U.P. Potent antibacterial activity of dihydydropyrimidine-1,3,5-triazines via inhibition of DNA gyrase and antifungal activity with favourable metabolic profile. Chem. Biol. Drug Des. 2020, 96, 861–869. [Google Scholar] [CrossRef]
- Younis, M.H.; Mohammed, E.R.; Mohamed, A.R.; Abdel-Aziz, M.M.; Georgey, H.H.; Abdel Gawad, N.M. Design, synthesis and anti-Mycobacterium tuberculosis evaluation of new thiazolidin-4-one and thiazolo[3,2-a][1,3,5]triazine derivatives. Bioorg. Chem. 2022, 124, 105807. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Noonikara-Poyil, A.; Joshi, S.D.; Patil, S.A.; Patil, S.A.; Lewis, A.M.; Bugarin, A. Synthesis, molecular docking studies, and in vitro evaluation of 1,3,5-triazine derivatives as promising antimicrobial agents. J. Mol. Struct. 2020, 1220, 128687. [Google Scholar] [CrossRef]
- Mekheimer, R.A.; Abuo-Rahma, G.E.D.A.; Abd-Elmonem, M.; Yahia, R.; Hisham, M.; Hayallah, A.M.; Mostafa, S.M.; Abo-Elsoud, F.A.; Sadek, K.U. New s-Triazine/Tetrazole conjugates as potent antifungal and antibacterial agents: Design, molecular docking and mechanistic study. J. Mol. Struct. 2022, 1267, 133615. [Google Scholar] [CrossRef]
- Patel, R.V.; Kumari, P.; Rajani, D.P.; Chikhalia, K.H. Synthesis and studies of novel 2-(4-cyano-3-trifluoromethylphenyl amino)-4-(quinoline-4-yloxy)-6-(piperazinyl/piperidinyl)-s-triazines as potential antimicrobial, antimycobacterial and anticancer agents. Eur. J. Med. Chem. 2011, 46, 4354–4365. [Google Scholar] [CrossRef] [PubMed]
- Gahtori, P.; Das, A.; Mishra, R. Design, synthesis and antibacterial activity of substituted phenylthiazolyl s-triazines. Pharm. Chem. J. 2011, 1. [Google Scholar] [CrossRef]
- Desai, N.C.; Makwana, A.H.; Senta, R.D. Synthesis, characterization and antimicrobial activity of some novel 4-(4-(arylamino)-6-(piperidin-1-yl)-1,3,5-triazine-2-ylamino)-N-(pyrimidin-2-yl)benzenesulfonamides. J. Saudi Chem. Soc. 2016, 20, 686–694. [Google Scholar] [CrossRef]
- Liu, H.; Long, S.; Rakesh, K.P.; Zha, G.F. Structure-activity relationships (SAR) of triazine derivatives: Promising antimicrobial agents. Eur. J. Med. Chem. 2020, 185, 111804. [Google Scholar] [CrossRef] [PubMed]
- Cuartas, V.; Robledo, S.M.; Vélez, I.D.; Crespo, M.D.P.; Sortino, M.; Zacchino, S.; Nogueras, M.; Cobo, J.; Upegui, Y.; Pineda, T.; et al. New thiazolyl-pyrazoline derivatives bearing nitrogen mustard as potential antimicrobial and antiprotozoal agents. Arch. Pharm. 2020, 353, e1900351. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kolesinska, B.; Swiontek, M.; Lipinski, W.; Drozdowska, D.; Kaminski, Z.J. Synthesis of Arylamino-1,3,5-triazines Functionalized with Alkylatin 2-chloroethylamine Fragments and Studies of their Cytotoxicity on the Breast Cancer MCF-7 Cell Line. Anticancer Agents Med. Chem. 2016, 16, 1435–1444. [Google Scholar] [CrossRef]
- Maliszewski, D.; Wróbel, A.; Kolesińska, B.; Frączyk, J.; Drozdowska, D. 1,3,5-Triazine Nitrogen Mustards with Different Peptide Group as Innovative Candidates for AChE and BACE1 Inhibitors. Molecules 2021, 26, 3942. [Google Scholar] [CrossRef]
- Mounnissamy, V.M.; Priya, B. Triazine Derivatives and its Pharmacological Potential—A Review. Int. J. Pharm. Sci. Rev. Res. 2020, 62, 143–147. [Google Scholar]
- Majeed Ganai, A.; Khan Pathan, T.; Hampannavar, G.A.; Pawar, C.; Obakachi, V.A.; Kushwaha, B.; Kushwaha, N.D.; Karpoormath, R. Recent Advances on the s-Triazine Scaffold with Emphasis on Synthesis, Structure-Activity and Pharmacological Aspects: A Concise Review. ChemistrySelect 2021, 6, 1616–1660. [Google Scholar] [CrossRef]
- Chen, S.C.A.; Sorrell, T.C. Antifungal agents. Med. J. Aust. 2007, 187, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.E. Current Concepts in Antifungal Pharmacology. Mayo Clin. Proc. 2011, 86, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Li, J.; Yang, Z.; Zhang, D.; Tian, J.; Gu, B. Discovery of novel 1,3,5-triazines as potent antibacterial agent against urinary tract infection-causing clinical isolates of Escherichia coli via inhibition of DNA Gyrase. Chem. Biol. Drug Des. 2023, 101, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Castro, W.; Navarro, M.; Biot, C. Medicinal potential of ciprofloxacin and its derivatives. Future Med. Chem. 2013, 5, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Vibhute, S.; Li, L.; Okumu, A.; Ratigan, S.C.; Nolan, S.; Papa, J.L.; Mann, C.A.; English, A.; Chen, A.; et al. Optimization of TopoIV Potency, ADMET Properties, and hERG Inhibition of 5-Amino-1,3-dioxane-Linked Novel Bacterial Topoisomerase Inhibitors: Identification of a Lead with In Vivo Efficacy against MRSA. J. Med. Chem. 2021, 64, 15214–15249. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.G.; Bax, B.; Chan, P.F.; Osheroff, N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. ACS Infect. Dis. 2019, 5, 570–581. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- M07-A10; Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. 10th ed. Approved Standard. Clinical & Laboratory Standards Institute: Wayne, PA, USA, 2015; Volume 35.
- Pfaller, M.A. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard—Second Edition; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2002. [Google Scholar]
- Sánchez-Linares, I.; Pérez-Sánchez, H.; Cecilia, J.M.; García, J.M. High-Throughput parallel blind Virtual Screening using BINDSURF. BMC Bioinform. 2012, 13, S13. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D., Jr.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.; Bouguet-Bonnet, S.; Pastor, R.W.; MacKerell, A.D. Importance of the CMAP correction to the CHARMM22 protein force field: Dynamics of hen lysozyme. Biophys. J. 2006, 90, L36–L38. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jo, S.; Brooks, I.I.I.C.L.; Lee, H.S.; Im, W. CHARMM-GUI Ligand Reader and Modeler for CHARMM Force Field Generation of Small Molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Mod. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibacterial Activities | Antifungal Activities | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | A | B | C | D | E | F | G | H | I |
| 1a | 250 | 250 | 250 | 125 | 250 | 250 | 500 | 250 | 250 |
| 1b | 250 | 250 | 125 | 62.50 | 250 | 250 | 500 | 250 | 125 |
| 2a | 250 | 250 | 250 | 250 | 250 | 250 | 500 | 250 | 250 |
| 2b | 250 | 250 | 250 | 125 | 250 | 250 | 250 | 250 | 125 |
| 2c | 250 | 250 | 250 | 250 | 250 | 250 | 250 | 250 | 250 |
| 2d | 250 | 125 | 125 | 125 | 250 | 250 | 500 | 250 | 125 |
| 2e | 500 | 500 | 500 | 500 | 250 | 250 | 250 | 250 | 500 |
| 2f | 250 | 250 | 250 | 125 | 250 | 250 | 500 | 250 | 125 |
| 3a | 500 | 500 | 250 | 250 | 250 | 250 | 500 | 250 | 250 |
| 3b | 500 | 62.50 | 31.25 | 7.81 | 250 | 250 | 500 | 250 | 7.81 |
| 3c | 250 | 250 | 250 | 250 | 250 | 250 | 500 | 250 | 250 |
| 4a | 250 | 500 | 250 | 250 | 250 | 250 | 500 | 250 | 250 |
| 4b | 250 | 250 | 250 | 250 | 250 | 250 | 500 | 250 | 125 |
| 4c | 125 | 125 | 62.50 | 62.50 | 250 | 250 | 500 | 250 | 62.50 |
| 4d | 125 | 62.50 | 31.25 | 15.62 | 250 | 250 | 500 | 250 | 15.62 |
| 4e | 250 | 250 | 250 | 250 | 250 | 250 | 500 | 250 | 250 |
| 4f | 250 | 500 | 500 | 250 | 250 | 500 | 250 | 250 | 125 |
| 4g | 250 | 500 | 250 | 250 | 250 | 250 | 500 | 250 | 250 |
| 4h | 250 | 500 | 500 | 250 | 500 | 250 | 250 | 250 | 250 |
| Streptomycin | 15.62 | 31.25 | 7.81 | 31.25 | - | - | - | - | - |
| Ketoconazole | - | - | - | - | 62.50 | 15.62 | 62.52 | 31.25 | 250 |
| Nystatin | 3.90 | 3.90 | 3.90 | 1.95 | 250 | ||||
| Gyrase | Compound | Binding Energy (kcal/mol) | Inhibition Constant (μM) | Ligand Efficiency |
|---|---|---|---|---|
| E. coli | 3b | −8.8 | 0.354 | −0.29 |
| 4d | −8.8 | 0.354 | −0.24 | |
| 4c | −10.0 | 0.047 | −0.22 | |
| S. aureus | 3b | −9.2 | 0.180 | −0.31 |
| 4d | −8.6 | 0.496 | −0.24 | |
| 4c | −10.2 | 0.033 | −0.23 |
| E. coli | S. aureus | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Residue | H-Bond Length [Å] | No. | Residue | H-Bond Length [Å] | No. | Residue | H-Bond Length [Å] | No. | Residue | H-Bond Length [Å] |
| 3b | DG10 | 2.33 | 4c | DG82 | 2.62 | 3b | Gly-375 | 2.88 | 4c | Asn-470 | 2.32 |
| DG10 | 2.73 | DG82 | 2.97 | Asn-383 | 2.73 | Arg-473 | 2.8 | ||||
| DC84 | 2.25 | DC84 | 2.28 | Gly-533 | 2.21 | Arg-473 | 2.9 | ||||
| Ser-172 | 2.68 | Asn-181 | 2.47 | 4d | DG17 | 2.33 | STR | DC3 | 1.77 | ||
| Asn-181 | 2.77 | Asn-269 | 2.88 | DG17 | 3.07 | DC3 | 2.46 | ||||
| Gly-331 | 2.41 | Gly-331 | 2.57 | DG18 | 2.52 | DG17 | 2.97 | ||||
| 4d | DG9 | 2.38 | STR | DG9 | 2.35 | DG18 | 2.7 | DG18 | 2.42 | ||
| DG9 | 2.34 | DG10 | 2.34 | DG18 | 2.72 | DG18 | 2.11 | ||||
| DG10 | 3 | DG10 | 3.1 | Asn-383 | 2.75 | DC19 | 2.63 | ||||
| DG10 | 2.42 | DC11 | 2.08 | Gly-533 | 2.12 | Asn-383 | 2.84 | ||||
| DG82 | 2.54 | DC83 | 3.06 | Ser-531 | 2.51 | Asn-383 | 2.94 | ||||
| Asn-181 | 2.46 | Ser-172 | 2.54 | 4c | DG17 | 3.01 | Phe-467 | 2.42 | |||
| Gly-331 | 2.04 | Asn-181 | 2.83 | DG18 | 2.23 | Gln-468 | 2.57 | ||||
| 4c | DG10 | 2.63 | Tyr-266 | 2.41 | DG18 | 2.89 | Gln-468 | 2.84 | |||
| DG10 | 2.76 | Ser-329 | 2.84 | Asn-383 | 2.78 | Ser-531 | 2.33 | ||||
| DG10 | 2.84 | Gly-331 | 2.93 | Gln-468 | 1.91 | Gly-533 | 2.25 | ||||
| Ligand | Donor | Acceptor | Occupancy | Ligand | Donor | Acceptor | Occupancy |
|---|---|---|---|---|---|---|---|
| 3b | LYS-276 | 3b | 77.20% | 4d | ARG-272 | 4d | 87.10% |
| ASN-269 | 3b | 19.71% | GLY-331 | 4d | 49.20% | ||
| GLY-331 | 3b | 17.49% | DG-10 | 4d | 7.60% | ||
| DG-82 | 3b | 15.94% | DG-82 | 4d | 5.20% | ||
| 3b | SER-329 | 11.21% | STR | STR | DC-84 | 51.68% | |
| DG-10 | 3b | 6.86% | STR | DG-85 | 47.72% | ||
| DT-13 | 3b | 5.31% | GLY-331 | STR | 22.78% | ||
| 4c | GLY-331 | 4c | 34.90% | STR | DC-83 | 19.66% | |
| SER-172 | 4c | 32.20% | DG-82 | STR | 16.79% | ||
| DG-82 | 4c | 19.30% | ASN-181 | STR | 9.35% | ||
| 4c | DC-83 | 14.50% | STR | SER-329 | 9.35% | ||
| ASN-181 | 4c | 13.20% | DG-10 | STR | 8.27% | ||
| GLY-173 | 4c | 12.10% | STR | DC-11 | 7.31% | ||
| DG-10 | 4c | 9.80% | ARG-237 | STR | 6.95% | ||
| ARG-272 | 4c | 5.60% | STR | GLN-325 | 6.83% | ||
| ASN-269 | STR | 6.24% | |||||
| DG-9 | STR | 5.52% | |||||
| STR | DT-12 | 5.04% |
| Ligand | Hbond D/A | AlogP | Acute Oral Toxicity Log (1/(mol/kg)) | Carcinogenicity/Hepa/Skin/Respiratory/Mitochondrial/Nephro-Toxicity | Plasma Protein Binding (100%) | Blood-Brain Barrier | Human Intestinal Absorption | Ames Mutagenesis | P-Glycoprotein Inhibition/Substrate |
|---|---|---|---|---|---|---|---|---|---|
| 3b | 1/9 | 1.61 | 2.304 | n/n/n/y/y/n | 0.775 | y | y | n | y/y |
| 4c | 2/11 | 2.85 | 2.528 | n/n/n/y/y/n | 0.761 | y | y | n | y/y |
| 4d | 2/12 | 0.6 | 2.146 | n/y/n/y/y/y | 0.405 | y | y | y | y/y |
| STR | 12/15 | −8.16 | 2.123 | n/y/y/y/y/y | 0.177 | n | n | y | n/n |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maliszewski, D.; Demirel, R.; Wróbel, A.; Baradyn, M.; Ratkiewicz, A.; Drozdowska, D. s-Triazine Derivatives Functionalized with Alkylating 2-Chloroethylamine Fragments as Promising Antimicrobial Agents: Inhibition of Bacterial DNA Gyrases, Molecular Docking Studies, and Antibacterial and Antifungal Activity. Pharmaceuticals 2023, 16, 1248. https://doi.org/10.3390/ph16091248
Maliszewski D, Demirel R, Wróbel A, Baradyn M, Ratkiewicz A, Drozdowska D. s-Triazine Derivatives Functionalized with Alkylating 2-Chloroethylamine Fragments as Promising Antimicrobial Agents: Inhibition of Bacterial DNA Gyrases, Molecular Docking Studies, and Antibacterial and Antifungal Activity. Pharmaceuticals. 2023; 16(9):1248. https://doi.org/10.3390/ph16091248
Chicago/Turabian StyleMaliszewski, Dawid, Rasime Demirel, Agnieszka Wróbel, Maciej Baradyn, Artur Ratkiewicz, and Danuta Drozdowska. 2023. "s-Triazine Derivatives Functionalized with Alkylating 2-Chloroethylamine Fragments as Promising Antimicrobial Agents: Inhibition of Bacterial DNA Gyrases, Molecular Docking Studies, and Antibacterial and Antifungal Activity" Pharmaceuticals 16, no. 9: 1248. https://doi.org/10.3390/ph16091248
APA StyleMaliszewski, D., Demirel, R., Wróbel, A., Baradyn, M., Ratkiewicz, A., & Drozdowska, D. (2023). s-Triazine Derivatives Functionalized with Alkylating 2-Chloroethylamine Fragments as Promising Antimicrobial Agents: Inhibition of Bacterial DNA Gyrases, Molecular Docking Studies, and Antibacterial and Antifungal Activity. Pharmaceuticals, 16(9), 1248. https://doi.org/10.3390/ph16091248