
Novel Aminopyrimidine-2,4-diones, 2-Thiopyrimidine-4-ones, and 6-Arylpteridines as Dual-Target Inhibitors of BRD4/PLK1: Design, Synthesis, Cytotoxicity, and Computational Studies
 ,
,  ,
, 
Abstract
:
1. Introduction
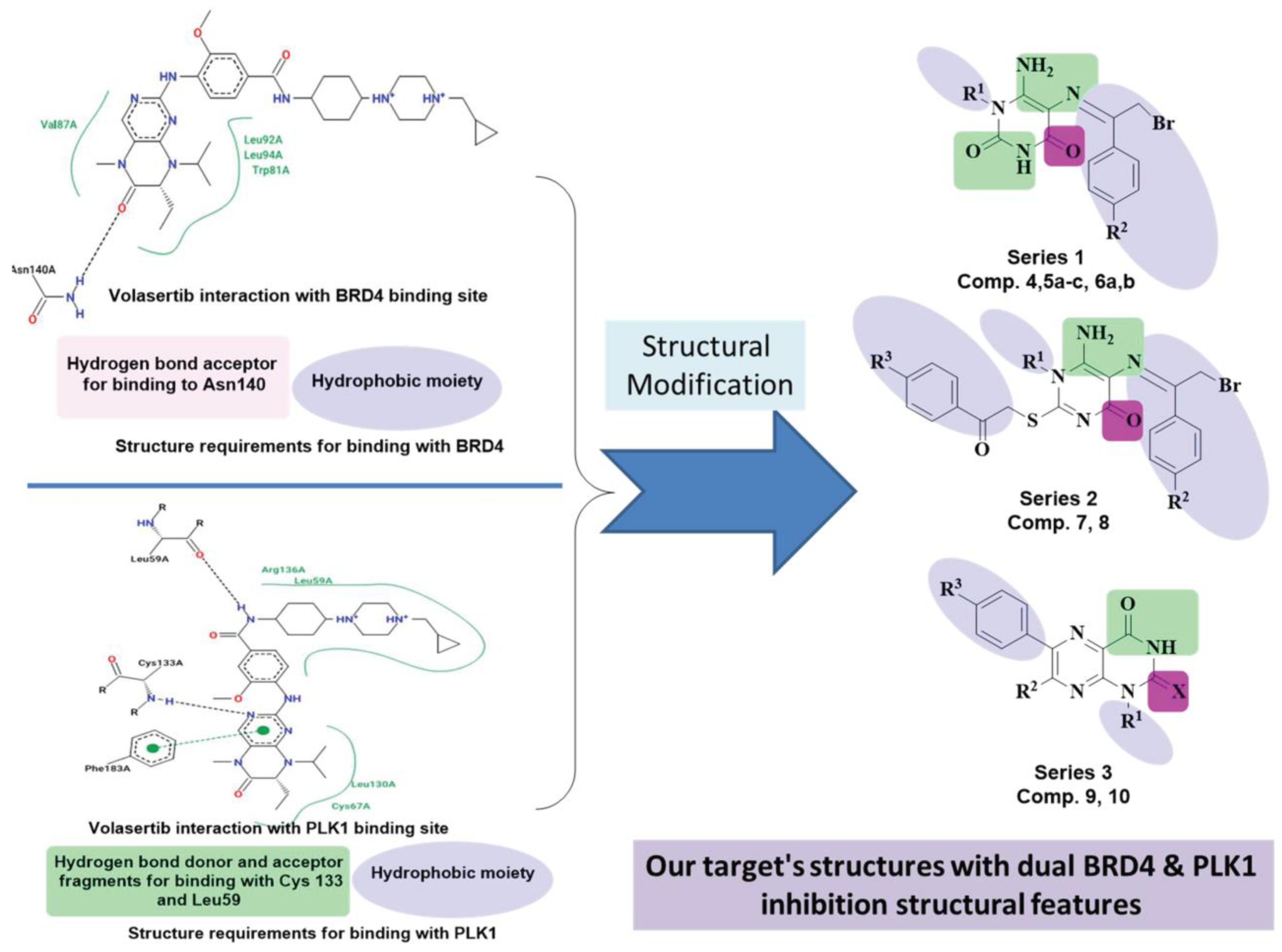
Rationale Strategy for Designing the New Targets
2. Results and Discussion
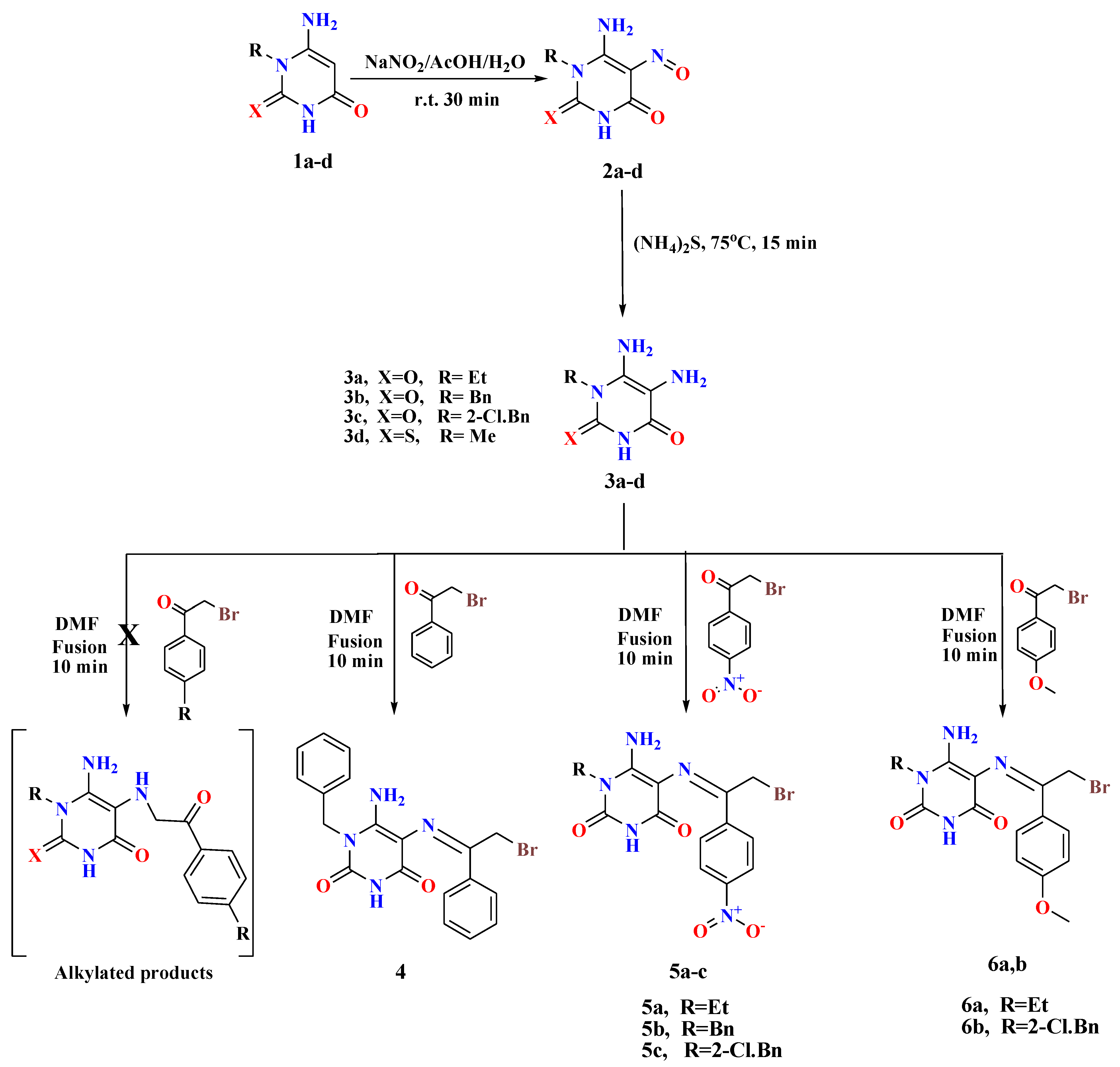
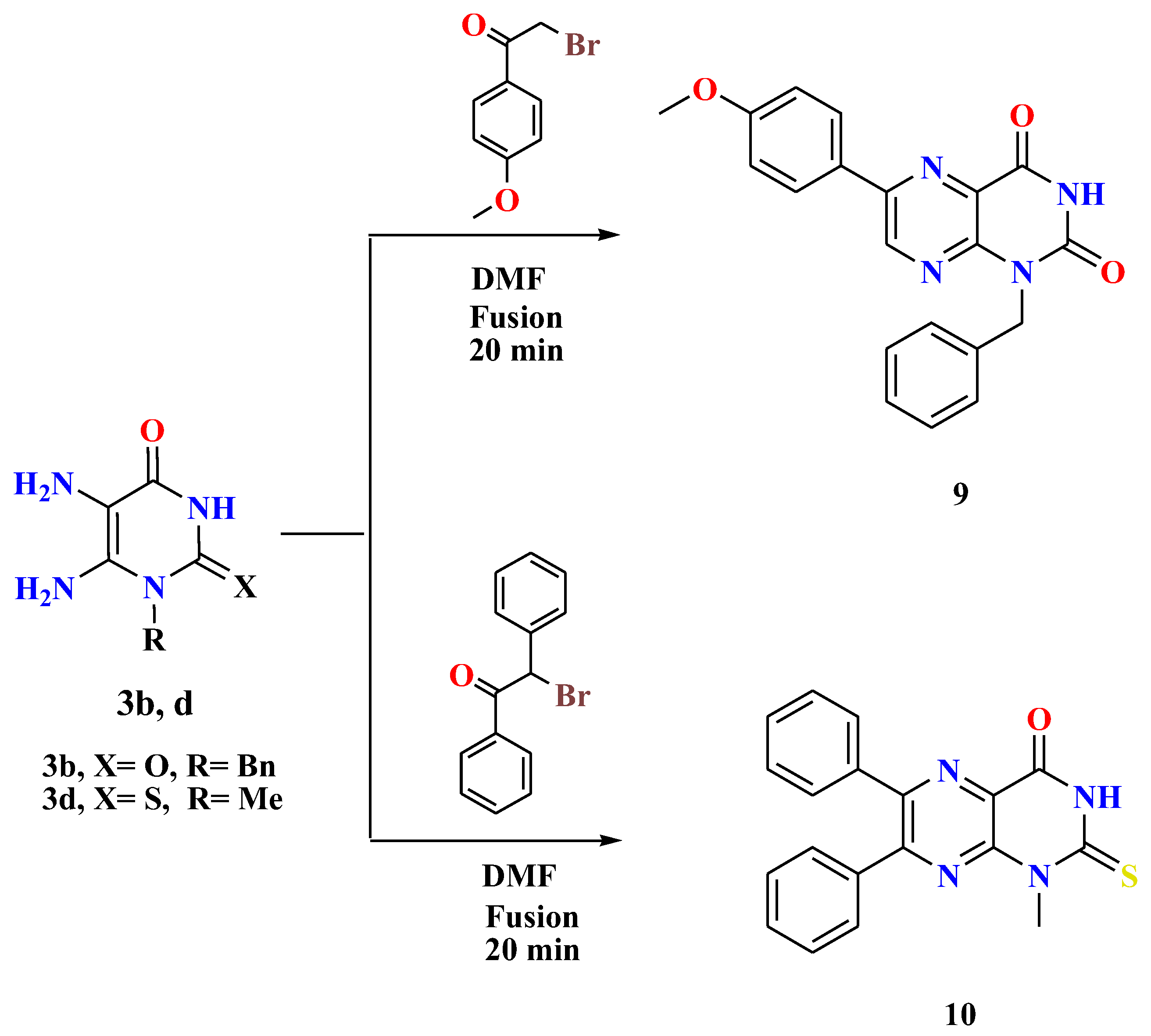
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In vitro Cytotoxicity Screening
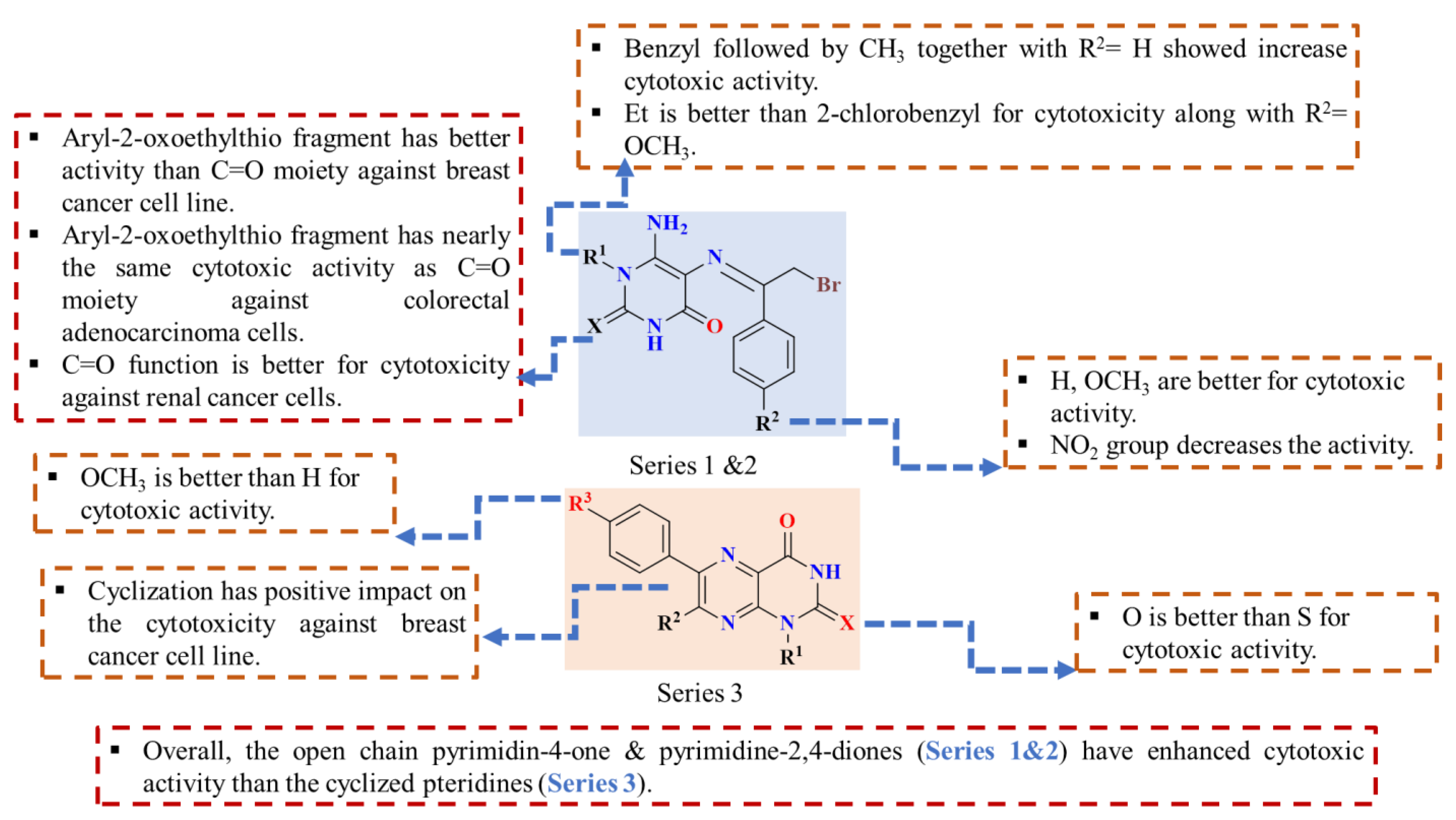
2.2.2. Structure–Activity Relationship
2.2.3. Selectivity Indices
2.2.4. BRD4 Inhibitory Activity Assay
2.2.5. PLK1 Inhibitory Activity Assay
2.2.6. Cell Cycle and Apoptosis Screening
2.2.7. Estimation of Apoptotic and Anti-Apoptotic Gene Markers
2.2.8. Computational Studies
Molecular Docking Simulation
The Predicted Drug-Likeness and ADME Properties
3. Materials and Methods
3.1. Chemistry
- (E)-6-Amino-1-benzyl-5-((2-bromo-1-phenylethylidene)amino)pyrimidine-2,4(1H,3H)-dione (4)
- Buff solid, Yield: 56%; m.p. 221–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H, NH uracil), 8.13 (s, 2H, NH2), 8.05–8.03 (m, 2H, ArH), 7.48–7.46 (m, 3H, ArH), 7.36 (t, J = 7.3 Hz, 2H, ArH), 7.30–7.24 (m, 3H, ArH), 5.50 (s, 2H, CH2-ph), 5.23 (s, 2H, CH2-Br). 13C NMR (100 MHz, DMSO-d6) δ 160.03, 158.99, 154.16, 148.17, 135.84, 134.39, 130.35, 129.27, 128.47, 128.41, 128.36, 127.66, 127.32, 127.16, 126.47, 126.20, 96.87, 65.42, 45.16. MS: m/z (rel. int. %) = 415 (M+.+2, 29), 413 (M+., 37), 493 (63), 492 (85), 339 (74), 310 (72), 249 (73), 204 (75), 176 (100), 91 (32), 71 (39). Anal. Calcd (%) for C19H17BrN4O2 (413.28): C, 55.22; H, 4.15; N, 13.56. Found: C, 55.43; H, 4.26; N, 13.49.
- (E)-6-Amino-5-((2-bromo-1-(4-nitrophenyl)ethylidene)amino)-1-ethylpyrimidine-2,4(1H,3H)-dione (5a)
- Orange solid, Yield: 78%; m.p. 216–218 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H, NH uracil), 8.57 (s, 2H, NH2), 8.33–8.27 (m, 4H, ArH), 5.58 (s, 2H, CH2-Br), 3.99 (q, J = 7.0 Hz, 2H, CH2), 1.17 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 162.32, 154.47, 149.45, 148.24, 147.03, 139.73, 127.60, 123.58, 97.57, 66.10, 35.79, 12.17. MS: m/z (rel. int. %) = 398 (M+.+2, 45), 396 (M+., 51), 378 (27), 376 (25), 360 (83), 358 (77), 355 (72), 351 (100), 302 (98), 290 (50), 288 (68). Anal. Calcd (%) for C14H14BrN5O4 (396.20): C, 42.44; H, 3.56; N, 17.68. Found: C, 42.63; H, 3.70; N, 17.92.
- (E)-6-Amino-1-benzyl-5-((2-bromo-1-(4-nitrophenyl)ethylidene)amino)pyrimidine-2,4(1H,3H)-dione (5b)
- Canary yellow solid, Yield: 74%; m.p. 266–268 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.11 (s, 1H, NH uracil), 8.46 (d, J = 9.0 Hz, 1H, ArH), 8.39 (d, J = 9.0 Hz, 1H, ArH), 8.29–8.27 (m, 2H, ArH), 7.93 (s, 2H, NH2), 7.45 (d, J = 7.3 Hz, 1H, ArH), 7.35–7.32 (m, 2H, ArH), 7.27–7.22 (m, 2H, ArH), 5.47 (s, 2H, CH2-ph), 5.21 (s, 2H, CH2-Br). 13C NMR (100 MHz, DMSO-d6) δ 165.08, 157.17, 156.53, 153.30, 148.63, 140.71, 137.69, 129.26, 129.23, 128.91, 128.78, 128.73, 127.66, 127.50, 126.68, 124.54, 85.49, 62.95, 42.85. MS: m/z (rel. int. %) = 460 (M+.+2, 11), 458 (M+.+14), 429 (39), 397 (36), 254 (30), 232 (35), 230 (37), 98 (65), 96 (100). Anal. Calcd (%) for C19H16BrN5O4 (458.27): C, 49.80; H, 3.52; N, 15.28. Found: C, 50.07; H, 3.68; N, 15.43.
- (E)-6-Amino-5-((2-bromo-1-(4-nitrophenyl)ethylidene)amino)-1-(2-chlorobenzyl) pyrimidine-2,4(1H,3H)-dione (5c)
- Orange solid, Yield: 71%; m.p. 235–236 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H, NH uracil, exchangeable), 8.31-8.25 (m, 4H, ArH), 8.02 (s, 2H, NH2, exchangeable), 7.53 (dd, J = 7.5, 1.6 Hz, 1H, ArH), 7.33–7.29 (m, 2H, ArH), 6.85–6.81 (m, 1H, ArH), 5.50 (s, 2H, CH2-Ar), 5.17 (s, 2H, CH2-Br). 13C NMR (100 MHz, DMSO-d6) δ 163.07, 157.02, 151.76, 148.54, 147.53, 143.83, 133.95, 132.33, 131.79, 130.30, 130.17, 129.68, 128.22, 127.81, 125.95, 124.45, 76.85, 61.58, 46.68. MS: m/z (rel. int. %) = 496 (M+.+4, 7), 494 (M+.+2, 23), 492 (M+., 15), 365 (36), 364 (100), 362 (45), 57 (56). Anal. Calcd (%) for C19H15BrClN5O4 (492.71): C, 46.32; H, 3.07; N, 14.21. Found: C, 46.56; H, 3.29; N, 14.42.
- (E)-6-Amino-5-((2-bromo-1-(4-methoxyphenyl)ethylidene)amino)-1-ethylpyrimidine-2,4(1H,3H)-dione (6a)
- Yellow solid, Yield: 55%; m.p. 244–246 °C;1H NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H, NH uracil), 8.94 (s, 2H, NH2), 8.06 (d, J = 8.9 Hz, 2H, ArH), 7.05 (d, J = 8.9 Hz, 2H, ArH), 5.61 (s, 2H, CH2-Br), 4.01 (q, J = 7.0 Hz, 2H, CH2), 3.84 (s, 3H, O-CH3), 1.19 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 161.85, 161.54, 154.80, 150.28, 147.38, 128.62, 126.21, 114.01, 97.28, 66.64, 55.49, 38.45, 12.01. MS: m/z (rel. int. %) = 383 (M+.+2, 14), 381 (M+., 20), 359 (56), 346 (94), 315 (71), 314 (100), 277 (60), 76 (86). Anal. Calcd (%) for C15H17BrN4O3 (381.23): C, 47.26; H, 4.49; N, 14.70. Found: C, 47.43; H, 4.57; N, 14.86.
- (E)-6-Amino-5-((2-bromo-1-(4-methoxyphenyl)ethylidene)amino)-1-(2-chlorobenzyl) pyrimidine-2,4(1H,3H)-dione (6b)
- Buff solid, Yield: 68%; m.p. 232–234 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H, NH uracil), 8.00 (d, J = 8.9 Hz, 2H, ArH), 7.76 (s, 2H, NH2), 7.54–7.51 (m, 1H, ArH), 7.35-7.28 (m, 2H, ArH), 7.00 (d, J = 9.0 Hz, 2H, ArH), 6.87–6.84 (m, 1H, ArH), 5.39 (s, 2H, CH2-Ar), 5.17 (s, 2H, CH2-Br), 3.82 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 162.06, 160.97, 155.91, 153.90, 150.28, 147.35, 136.33, 131.36, 129.28, 128.58, 127.83, 127.33, 126.61, 125.31, 114.69, 113.71, 97.08, 64.94, 55.27, 44.21. MS: m/z (rel. int. %) = 481 (M+.+4, 34), 479 (M+.+2, 65), 477 (M+., 52), 475 (33), 463 (52), 420 (83), 412 (35), 410 (29), 373 (48), 364 (48), 362 (32), 360 (32), 358 (48), 347 (51), 308 (35), 306 (30), 304 (29), 250 (45), 235 (50), 222 (66), 203 (71), 193 (53), 184 (50), 166 (52), 160 (86), 159 (100), 158 (83). Anal. Calcd (%) for C20H18BrClN4O3 (477.74): C, 50.28; H, 3.80; N, 11.73. Found: C, 50.44; H, 3.75; N, 11.94.
- General procedures for the synthesis of 6-amino-5-((2-bromo-1-arylethylidene)amino)-1-methyl-2-((2-oxo-2-arylethyl)thio)pyrimidin-4(1H)-ones (7 and 8)
- 5,6-Diamino-1-methylthiouracil 3d (1.2 mmol) was fused with double the amount of appropriate phenacyl bromides (2.4 mmol) in the presence of drops of DMF (1 mL) for 10 min. After cooling the reaction mixture, the formed residue was washed with methanol, collected by filtration, and recrystallized from DMF to afford the desired compounds 7 and 8 in interesting yields.
- (E)-6-Amino-5-((2-bromo-1-phenylethylidene)amino)-1-methyl-2-((2-oxo-2-phenylethyl) thio)pyrimidin-4(1H)-one (7)
- Yellow solid, Yield: 56%; m.p. 229–230 °C;1H NMR (400 MHz, DMSO-d6) δ 8.99 (s, 2H, NH2), 8.13 (d, J = 8.6 Hz, 2H, ArH), 8.08 (d, J = 8.6 Hz, 2H, ArH), 7.73 (t, J = 7.4 Hz, 1H, ArH), 7.63–7.57 (m, 3H, ArH), 7.52 (t, J = 7.4 Hz, 2H, ArH), 5.63 (s, 2H, S-CH2), 5.04 (s, 2H, CH2-Br), 3.80 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 191.76, 176.83, 161.84, 156.16, 154.66, 137.85, 135.51, 133.94, 133.19, 132.12, 131.70, 129.32, 128.93, 128.60, 128.41, 127.84, 127.34, 105.40, 66.44, 36.46, 34.48. MS: m/z (rel. int. %) = 473 (M+.+2, 27), 471 (M+., 30), 345 (40), 342 (92), 291 (39), 273 (38), 235 (100), 145 (39). Anal. Calcd (%) for C21H19BrN4O2S (471.37): C, 53.51; H, 4.06; N, 11.89. Found: C, 53.75; H, 4.28; N, 12.11.
- (E)-6-Amino-5-((2-bromo-1-(4-nitrophenyl)ethylidene)amino)-1-methyl-2-((2-(4-nitro phenyl)-2-oxoethyl)thio)pyrimidin-4(1H)-one (8)
- Brown solid, Yield: 69%; m.p. 214-216 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.42–8.37 (m, 5H, ArH), 8.34 (s, 2H, NH2), 8.32–8.30 (m, 3H, ArH), 5.68 (s, 2H, S-CH2), 5.10 (s, 2H, CH2-Br), 3.80 (s, 3H, CH3). MS: m/z (rel. int. %) = 563 (M+.+2, 23), 561 (M+., 28), 501 (46), 593 (54), 463 (79), 457 (47), 455 (45), 407 (93), 400 (98), 397 (85), 395 (86), 390 (97), 377 (85), 212 (52), 211 (100). Anal. Calcd (%) for C21H17BrN6O6 S(561.37): C, 44.93; H, 3.05; N, 14.97. Found: C, 45.17; H, 3.24; N, 15.13.
- General procedures for the synthesis of 1-alkyl-6-arylpteridines 9 and 10
- A mixture of 5,6-diaminouracil/thiouracil 3b,d (1.2 mmol) and the appropriate phenacyl bromides (1.2 mmol) was heated under fusion in the presence of drops of DMF (0.7 mL) for 20 min. By cooling the reaction mixture, washing the formed residue with methanol, collecting it by filtration, and recrystallizing it from DMF, the desired compounds 9 and 10 were produced in high yields.
- 1-Benzyl-6-(4-methoxyphenyl)pteridine-2,4(1H,3H)-dione (9)
- Orange solid, Yield: 65%; m.p. > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H, NH uracil), 9.13 (s, 1H, ArH), 8.18 (d, J = 8.9 Hz, 2H, ArH), 7.41 (d, J = 7.6 Hz, 2H, ArH), 7.30 (t, J = 7.6 Hz, 2H, ArH), 7.26–7.21 (m, 1H, ArH), 7.11 (d, J = 8.9 Hz, 2H, ArH), 5.41 (s, 2H, CH2), 3.84 (s, 3H, O-CH3). 13C NMR (100 MHz, DMSO-d6) δ 162.08, 159.82, 152.96, 150.35, 148.24, 136.26, 129.47, 128.37, 127.29, 127.09, 126.80, 114.76, 55.53, 44.07. MS: m/z (rel. int. %) = 360 (M+., 68), 348 (56), 347 (57), 341 (80), 309 (53), 308 (57), 301 (100), 298 (46), 272 (82), 256 (88), 77 (58), 46 (79). Anal. Calcd (%) for C20H16N4O3 (360.37): C, 66.66; H, 4.48; N, 15.55. Found: C, 66.85; H, 4.60; N, 15.79.
- 1-Methyl-6,7-diphenyl-2-thioxo-2,3-dihydropteridin-4(1H)-one (10)
- Yellow solid, Yield: 69%; m.p. 286–287 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.49 (s, 1H, NH uracil), 8.15 (d, J = 7.9 Hz, 2H, ArH), 7.56–7.49 (m, 4H, ArH), 7.45–7.37 (m, 2H, ArH), 7.22–6.91 (m, 2H, ArH), 3.85 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 173.79, 156.63, 152.40, 150.33, 150.18, 130.57, 129.03, 128.32, 126.53, 111.96, 35.06. MS: m/z (rel. int. %) = 346 (M+., 63), 339 (50), 307 (100), 247 (73), 117 (41), 55 (54). Anal. Calcd (%) for C19H14N4OS (346.41): C, 65.88; H, 4.07; N, 16.17. Found: C, 65.71; H, 4.29; N, 16.40.
3.2. Biological Evaluation
3.2.1. In Vitro Cytotoxicity Screening
3.2.2. In Vitro BRD4 and PLK1 Enzymes Inhibition Assays
3.2.3. Cell Cycle and Apoptosis Screening
3.2.4. Estimation of Apoptotic and Anti-Apoptotic Gene Markers
3.3. Computational Studies
3.3.1. Molecular Docking Simulation
3.3.2. The Predicted Drug-Likeness and ADME Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Elkady, H.; El-Adl, K.; Sakr, H.; Abdelraheem, A.S.; Eissa, S.I.; El-Zahabi, M.A. Novel promising benzoxazole/benzothiazole-derived immunomodulatory agents: Design, synthesis, anticancer evaluation, and in silico ADMET analysis. Arch. Der Pharm. 2023, 356, e2300097. [Google Scholar] [CrossRef] [PubMed]
- Penna, L.S.; Henriques, J.A.P.; Bonatto, D. Anti-mitotic agents: Are they emerging molecules for cancer treatment? Pharmacol. Ther. 2017, 173, 67–82. [Google Scholar] [CrossRef]
- Mabrouk, R.R.; Abdallaha, A.E.; Mahdy, H.A.; El-Kalyoubi, S.A.; Kamal, O.J.; Abdelghany, T.M.; Zayed, M.F.; Alshaeri, H.K.; Alasmari, M.M.; El-Zahabi, M.A. Design, Synthesis, and Biological Evaluation of New Potential Unusual Modified Anticancer Immunomodulators for Possible Non-Teratogenic Quinazoline-Based Thalidomide Analogs. Int. J. Mol. Sci. 2023, 24, 12416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Saleh, A.M.; Mahdy, H.A.; El-Zahabi, M.A.; Mehany, A.B.M.; Khalifa, M.M.; Eissa, I.H. Design, synthesis, in silico studies, and biological evaluation of novel pyrimidine-5-carbonitrile derivatives as potential anti-proliferative agents, VEGFR-2 inhibitors and apoptotic inducers. RSC Adv. 2023, 13, 22122–22147. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022. [Google Scholar] [CrossRef]
- Watts, E.; Heidenreich, D.; Tucker, E.; Raab, M.; Strebhardt, K.; Chesler, L.; Knapp, S.; Bellenie, B.; Hoelder, S. Designing dual inhibitors of anaplastic lymphoma kinase (ALK) and bromodomain-4 (BRD4) by tuning kinase selectivity. J. Med. Chem. 2019, 62, 2618–2637. [Google Scholar] [CrossRef]
- Mao, F.; Li, J.; Luo, Q.; Wang, R.; Kong, Y.; Carlock, C.; Liu, Z.; Elzey, B.D.; Liu, X. Plk1 Inhibition Enhances the Efficacy of BET Epigenetic Reader Blockade in Castration-Resistant Prostate CancerJQ1 in CRPC Treatment. Mol. Cancer Ther. 2018, 17, 1554–1565. [Google Scholar] [CrossRef]
- Wyce, A.; Ganji, G.; Smitheman, K.N.; Chung, C.-w.; Korenchuk, S.; Bai, Y.; Barbash, O.; Le, B.; Craggs, P.D.; McCabe, M.T. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE 2013, 8, e72967. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, C.; Wang, C.; Zhou, X.; Li, W.; Zhang, J.-Y.; Wang, M.; Xu, Y.; Sun, L.-P. Design, synthesis and anticancer evaluation of 3-methyl-1H-indazole derivatives as novel selective bromodomain-containing protein 4 inhibitors. Bioorg. Med. Chem. 2022, 55, 116592. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-P.; Picaud, S.; Fujisawa, T.; Hou, H.; Savitsky, P.; Uusküla-Reimand, L.; Gupta, G.D.; Abdouni, H.; Lin, Z.-Y.; Tucholska, M. Interactome rewiring following pharmacological targeting of BET bromodomains. Mol. Cell 2019, 73, 621–638.e17. [Google Scholar] [CrossRef] [PubMed]
- Vann, K.R.; Pal, D.; Morales, G.A.; Burgoyne, A.M.; Durden, D.L.; Kutateladze, T.G. Design of thienopyranone-based BET inhibitors that bind multiple synthetic lethality targets. Sci. Rep. 2020, 10, 12027. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Borrett, M.J.; Innes, B.T.; Voronova, A.; Ketela, T.; Kaplan, D.R.; Bader, G.D.; Miller, F.D. Developmental emergence of adult neural stem cells as revealed by single-cell transcriptional profiling. Cell Rep. 2017, 21, 3970–3986. [Google Scholar] [CrossRef]
- Li, Z.; Mei, S.; Liu, J.; Huang, J.; Yue, H.; Ge, T.; Wang, K.; He, X.; Gu, Y.-C.; Hu, C. Design, synthesis, and biological evaluation of novel dihydropteridone derivatives possessing oxadiazoles moiety as potent inhibitors of PLK1. Eur. J. Med. Chem. 2023, 251, 115242. [Google Scholar] [CrossRef]
- Lan, R.; Lin, G.; Yin, F.; Xu, J.; Zhang, X.; Wang, J.; Wang, Y.; Gong, J.; Ding, Y.-H.; Yang, Z. Dissecting the phenotypes of Plk1 inhibition in cancer cells using novel kinase inhibitory chemical CBB2001. Lab. Investig. 2012, 92, 1503–1514. [Google Scholar] [CrossRef]
- Hu, R.; Wang, W.-L.; Yang, Y.-Y.; Hu, X.-T.; Wang, Q.-W.; Zuo, W.-Q.; Xu, Y.; Feng, Q.; Wang, N.-Y. Identification of a selective BRD4 PROTAC with potent antiproliferative effects in AR-positive prostate cancer based on a dual BET/PLK1 inhibitor. Eur. J. Med. Chem. 2022, 227, 113922. [Google Scholar] [CrossRef]
- Bhujbal, S.P.; Kim, H.; Bae, H.; Hah, J.-M. Design and Synthesis of Aminopyrimidinyl Pyrazole Analogs as PLK1 Inhibitors Using Hybrid 3D-QSAR and Molecular Docking. Pharmaceuticals 2022, 15, 1170. [Google Scholar] [CrossRef]
- Yim, H.; Erikson, R.L. Polo-like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Mol. Cell. Biol. 2009, 29, 2609–2621. [Google Scholar] [CrossRef]
- Yu, Z.; Deng, P.; Chen, Y.; Liu, S.; Chen, J.; Yang, Z.; Chen, J.; Fan, X.; Wang, P.; Cai, Z. Inhibition of the PLK1-Coupled Cell Cycle Machinery Overcomes Resistance to Oxaliplatin in Colorectal Cancer. Adv. Sci. 2021, 8, 2100759. [Google Scholar] [CrossRef]
- Takashima, Y.; Kikuchi, E.; Kikuchi, J.; Suzuki, M.; Kikuchi, H.; Maeda, M.; Shoji, T.; Furuta, M.; Kinoshita, I.; Dosaka-Akita, H. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int. J. Cancer 2020, 146, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, Y.; Yang, D.; Gong, Y.; Rao, F.; Liu, R.; Danna, Y.; Li, J.; Fan, J.; Chen, J. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine 2019, 41, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yosief, H.O.; Dai, L.; Huang, H.; Dhawan, G.; Zhang, X.; Muthengi, A.M.; Roberts, J.; Buckley, D.L.; Perry, J.A. Structure-guided design and development of potent and selective dual bromodomain 4 (BRD4)/polo-like kinase 1 (PLK1) inhibitors. J. Med. Chem. 2018, 61, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lindner, S.; Bei, Y.; Garcia, H.D.; Timme, N.; Althoff, K.; Odersky, A.; Schramm, A.; Lissat, A.; Künkele, A. Synergistic activity of BET inhibitor MK-8628 and PLK inhibitor Volasertib in preclinical models of medulloblastoma. Cancer Lett. 2019, 445, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Yu, Y.; Ma, Z. Novel strategies targeting bromodomain-containing protein 4 (BRD4) for cancer drug discovery. Eur. J. Med. Chem. 2020, 200, 112426. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Seal, J.; Lamotte, Y.; Donche, F.; Bouillot, A.; Mirguet, O.; Gellibert, F.; Nicodeme, E.; Krysa, G.; Kirilovsky, J.; Beinke, S. Identification of a novel series of BET family bromodomain inhibitors: Binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968–2972. [Google Scholar] [CrossRef]
- Peters, U.; Cherian, J.; Kim, J.H.; Kwok, B.H.; Kapoor, T.M. Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nat. Chem. Biol. 2006, 2, 618–626. [Google Scholar] [CrossRef]
- Hyoda, T.; Tsujioka, T.; Nakahara, T.; Suemori, S.i.; Okamoto, S.; Kataoka, M.; Tohyama, K. Rigosertib induces cell death of a myelodysplastic syndrome-derived cell line by DNA damage-induced G2/M arrest. Cancer Sci. 2015, 106, 287–293. [Google Scholar] [CrossRef]
- Yuan, J.; Sanhaji, M.; Krämer, A.; Reindl, W.; Hofmann, M.; Kreis, N.-N.; Zimmer, B.; Berg, T.; Strebhardt, K. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am. J. Pathol. 2011, 179, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Pettit, K.; Odenike, O. Defining and treating older adults with acute myeloid leukemia who are ineligible for intensive therapies. Front. Oncol. 2015, 5, 280. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Müller, S.; O’mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yap, J.L.; Yoshioka, M.; Lanning, M.E.; Fountain, R.N.; Raje, M.; Scheenstra, J.A.; Strovel, J.W.; Fletcher, S. BRD4 structure–activity relationships of dual PLK1 kinase/BRD4 bromodomain inhibitor BI-2536. ACS Med. Chem. Lett. 2015, 6, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Masson, F.; Simeoni, L.; Homem-de-Mello, M. Biological activity of dihydropyrimidinone (DHPM) derivatives. A Syst. Rev. 2018, 143, 1779. [Google Scholar]
- Kaur, R.; Kaur, P.; Sharma, S.; Singh, G.; Mehndiratta, S.; MS Bedi, P.; Nepali, K. Anti-cancer pyrimidines in diverse scaffolds: A review of patent literature. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 23–71. [Google Scholar] [CrossRef]
- Farooqi, S.I.; Arshad, N.; Channar, P.A.; Perveen, F.; Saeed, A.; Larik, F.A.; Javed, A.; Yamin, M. New aryl Schiff bases of thiadiazole derivative of ibuprofen as DNA binders and potential anticancer drug candidates. J. Biomol. Struct. Dyn. 2021, 39, 3548–3564. [Google Scholar] [CrossRef]
- Chazin, E.d.L.; Sanches, P.d.S.; Lindgren, E.B.; Vellasco Junior, W.T.; Pinto, L.C.; Burbano, R.M.R.; Yoneda, J.D.; Leal, K.Z.; Gomes, C.R.B.; Wardell, J.L. Synthesis and biological evaluation of novel 6-hydroxy-benzo [d][1, 3] oxathiol-2-one Schiff bases as potential anticancer agents. Molecules 2015, 20, 1968–1983. [Google Scholar] [CrossRef]
- Butler-Fernández, K.M.; Ramos, Z.; Francis-Malavé, A.M.; Bloom, J.; Dharmawardhane, S.; Hernández, E. Synthesis, anti-cancer and anti-migratory evaluation of 3, 6-dibromocarbazole and 5-bromoindole derivatives. Molecules 2019, 24, 2686. [Google Scholar] [CrossRef]
- Duque, A.; Rakic, P. Different effects of bromodeoxyuridine and [3H] thymidine incorporation into DNA on cell proliferation, position, and fate. J. Neurosci. 2011, 31, 15205–15217. [Google Scholar] [CrossRef]
- Cal, M.; Matyjaszczyk, I.; Litwin, I.; Augustyniak, D.; Ogórek, R.; Ko, Y.; Ułaszewski, S. The anticancer drug 3-bromopyruvate induces DNA damage potentially through reactive oxygen species in yeast and in human cancer cells. Cells 2020, 9, 1161. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.M.; Bikowitz, M.J.; Chan, A.; Zhu, J.-Y.; Grassie, D.; Becker, A.; Berndt, N.; Gunawan, S.; Lawrence, N.J.; Schonbrunn, E. Differential BET bromodomain inhibition by dihydropteridinone and pyrimidodiazepinone kinase inhibitors. J. Med. Chem. 2021, 64, 15772–15786. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.n.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; El-Sebaey, S.A.; Rashad, A.; Al-Ghulikah, H.A.; Ghorab, M.M.; Elfeky, S.M. Synthesis, DFT calculations, and anti-proliferative evaluation of pyrimidine and selenadiazolopyrimidine derivatives as dual Topoisomerase II and HSP90 inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 2198163. [Google Scholar] [CrossRef] [PubMed]
- El-Kalyoubi, S.; Elbaramawi, S.S.; Zordok, W.A.; Malebari, A.M.; Safo, M.K.; Ibrahim, T.S.; Taher, E.S. Design and synthesis of uracil/thiouracil based quinoline scaffolds as topoisomerases I&II inhibitors for chemotherapy: A new hybrid navigator with DFT calculation. Bioorg. Chem. 2023, 136, 106560. [Google Scholar] [PubMed]
- El-Kalyoubi, S.; Agili, F.; Adel, I.; Tantawy, M.A. Novel uracil derivatives depicted potential anticancer agents: In vitro, molecular docking, and ADME study. Arab. J. Chem. 2022, 15, 103669. [Google Scholar] [CrossRef]
- Abdel-Latif, E.; Mustafa, H.; Etman, H.; Fadda, A. Synthesis of new purine, pteridine, and other pyrimidine derivatives. Russ. J. Org. Chem. 2007, 43, 443–448. [Google Scholar] [CrossRef]
- Kolos, N.; Chebanov, V.; Orlov, V.; Surov, Y.N. Synthesis and study of aromatic derivatives of 5, 6-dihydro-4-pteridinol. Chem. Heterocycl. Compd. 2001, 37, 755–762. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.A.; Fayed, E.A.; Abdel-Razek, A.S. One pot synthesis, antimicrobial and antioxidant activities of fused uracils: Pyrimidodiazepines, lumazines, triazolouracil and xanthines. Chem. Cent. J. 2017, 11, 66. [Google Scholar] [CrossRef]
- Qandil, A.M.; Fakhouri, L.I. α-Anilinoketones, Esters and Amides: A Chemical Study. Pharmaceuticals 2012, 5, 591–612. [Google Scholar] [CrossRef]
- Rasmussen, L.; Foulks, Z.; Burton, C.; Shi, H. Establishing pteridine metabolism in a progressive isogenic breast cancer cell model. Metabolomics 2022, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Pfleiderer, W. Pteridine, LXI. Synthese und Eigenschaften von Thiolumazinen. Chem. Berichte 1974, 107, 3377–3394. [Google Scholar] [CrossRef]
- Kong, B.; Zhu, Z.; Li, H.; Hong, Q.; Wang, C.; Ma, Y.; Zheng, W.; Jiang, F.; Zhang, Z.; Ran, T. Discovery of 1-(5-(1H-benzo [d] imidazole-2-yl)-2, 4-dimethyl-1H-pyrrol-3-yl) ethan-1-one derivatives as novel and potent bromodomain and extra-terminal (BET) inhibitors with anticancer efficacy. Eur. J. Med. Chem. 2022, 227, 113953. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-Y.; Xu, Y.; Xiao, K.-J.; Zuo, W.-Q.; Zhu, Y.-X.; Hu, R.; Wang, W.-L.; Shi, Y.-J.; Yu, L.-T.; Liu, Z.-H. Design, synthesis, and biological evaluation of 4, 5-dihydro-[1, 2, 4] triazolo [4, 3-f] pteridine derivatives as novel dual-PLK1/BRD4 inhibitors. Eur. J. Med. Chem. 2020, 191, 112152. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, X.; Guan, P.; Song, S.; You, G.; Xia, C.; Liu, T. Recent progress in agents targeting polo-like kinases: Promising therapeutic strategies. Eur. J. Med. Chem. 2021, 217, 113314. [Google Scholar] [CrossRef]
- Li, Q.; Li, J.; Cai, Y.; Zou, Y.; Chen, B.; Zou, F.; Mo, J.; Han, T.; Guo, W.; Huang, W. Design, synthesis and biological evaluation of novel 6-phenyl-1, 3a, 4, 10b-tetrahydro-2H-benzo [c] thiazolo [4, 5-e] azepin-2-one derivatives as potential BRD4 inhibitors. Bioorg. Med. Chem. 2020, 28, 115601. [Google Scholar] [CrossRef]
- Jung, M.; Philpott, M.; Müller, S.; Schulze, J.; Badock, V.; Eberspächer, U.; Moosmayer, D.; Bader, B.; Schmees, N.; Fernández-Montalván, A. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. In silico prediction of aqueous solubility using simple QSPR models: The importance of phenol and phenol-like moieties. J. Chem. Inf. Model. 2012, 52, 2950–2957. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117. [Google Scholar] [CrossRef] [PubMed]
- El-Kalyoubi, S.; Agili, F. Synthesis, In Silico Prediction and in vitro Evaluation of Antitumor Activities of Novel Pyrido [2, 3-d] pyrimidine, Xanthine and Lumazine Derivatives. Molecules 2020, 25, 5205. [Google Scholar] [CrossRef] [PubMed]
- El-Kalyoubi, S.; Agili, F.; Zordok, W.A.; El-Sayed, A.S. Synthesis, in silico prediction and in vitro evaluation of antimicrobial activity, dft calculation and theoretical investigation of novel xanthines and uracil containing imidazolone derivatives. Int. J. Mol. Sci. 2021, 22, 10979. [Google Scholar] [CrossRef] [PubMed]
- Van de Loosdrecht, A.; Beelen, R.; Ossenkoppele, G.; Broekhoven, M.; Langenhuijsen, M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]
- McBride, A.A.; McPhillips, M.G.; Oliveira, J.G. Brd4: Tethering, segregation and beyond. Trends Microbiol. 2004, 12, 527–529. [Google Scholar] [CrossRef]
- Fabbro, M.; Zhou, B.-B.; Takahashi, M.; Sarcevic, B.; Lal, P.; Graham, M.E.; Gabrielli, B.G.; Robinson, P.J.; Nigg, E.A.; Ono, Y. Cdk1/Erk2-and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev. Cell 2005, 9, 477–488. [Google Scholar] [CrossRef]
- El-Zoghbi, M.S.; El-Sebaey, S.A.; Al-Ghulikah, H.A.; Sobh, E.A. Design, synthesis, docking, and anticancer evaluations of new thiazolo [3, 2-a] pyrimidines as topoisomerase II inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 2175209. [Google Scholar] [CrossRef]
- Al-Ghulikah, H.A.; El-Sebaey, S.A.; Bass, A.K.; El-Zoghbi, M.S. New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molecular Docking, and In Silico ADME Profile Studies. Molecules 2022, 27, 7485. [Google Scholar] [CrossRef]
- Lowe, B.; Avila, H.A.; Bloom, F.R.; Gleeson, M.; Kusser, W. Quantitation of gene expression in neural precursors by reverse-transcription polymerase chain reaction using self-quenched, fluorogenic primers. Anal. Biochem. 2003, 315, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Elfeky, S.M.; Almehmadi, S.J.; Tawfik, S.S. Synthesis, in-silico, and in-vitro study of novel chloro methylquinazolinones as PI3K-δ inhibitors, cytotoxic agents. Arab. J. Chem. 2022, 15, 103614. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds No. | IC50 Value (µM) a | ||||||
|---|---|---|---|---|---|---|---|
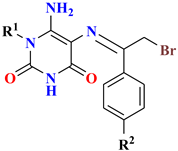 | |||||||
| R1 | R2 | R3 | X | MDA-MB-231 | HT-29 | U-937 | |
| 4 | Benzyl | H | - | - | 0.78 ± 0.01 | 0.76 ± 0 | 0.95 ± 0 |
| 5a | Et- | -NO2 | - | - | 3.24 ± 0.03 | 3.09 ± 0.01 | 6.08 ± 0.11 |
| 5b | Benzyl | -NO2 | - | - | 2.37 ± 0.06 | 3.66 ± 0.05 | 6.29 ± 0.26 |
| 5c | 2-Cl-benzyl | -NO2 | - | - | 3.28 ± 0.04 | 2.24 ± 0.04 | 6.17 ± 0.15 |
| 6a | Et- | -OCH3 | - | - | 0.63 ± 0.01 | 0.75 ± 0.01 | 0.97 ± 0.01 |
| 6b | 2-Cl-benzyl | -OCH3 | - | - | 1 ± 0.02 | 1.69 ± 0.01 | 1.12 ± 0.02 |
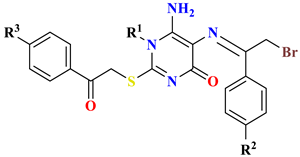 | |||||||
| 7 | -CH3 | H | H | - | 0.4 ± 0.01 | 0.79 ± 0.01 | 1.85 ± 0.04 |
| 8 | -CH3 | -NO2 | -NO2 | - | 2.31 ± 0.06 | 6.48 ± 0.08 | 7.77 ± 0.04 |
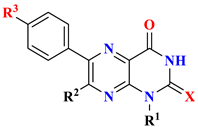 | |||||||
| 9 | Benzyl | H | -OCH3 | O | 2.07 ± 0.03 | 4.11 ± 0.05 | 4.42 ± 0.18 |
| 10 | -CH3 | phenyl | H | S | 3.4 ± 0.02 | 5.63 ± 0.13 | 8.37 ± 0.23 |
| Methotrexate | 2.79 ± 0.06 | 0.99 ± 0.02 | 1.22 ± 0.02 | ||||
| Compounds No. | In Vitro Cytotoxicity against Normal Cells IC50 Value (µM) * | Selectivity Indices (SI) ** | ||
|---|---|---|---|---|
| Vero Cells | MDA-MB-231 | HT-29 | U-937 | |
| 4 | 1.73 ± 0.03 | 2.22 | 2.28 | 1.82 |
| 6a | 2.06 ± 0.03 | 3.27 | 2.75 | 2.12 |
| 6b | 1.95 ± 0.05 | 1.95 | 1.15 | 1.74 |
| 7 | 3.92 ± 0.08 | 9.8 | 5.00 | 2.12 |
| Methotrexate | 10.16 ± 0.3 | 3.64 | 10.26 | 8.33 |
| Compounds No. | BRD4 * IC50 (µM) ± SD | PLK1 * IC50 (µM) ± SD |
|---|---|---|
| 4 | 0.029 ± 0.001 | 0.094 ± 0.004 |
| 6a | 0.141 ± 0.006 | 0.218 ± 0.01 |
| 6b | 0.077 ± 0.003 | 0.204 ± 0.009 |
| 7 | 0.042 ± 0.002 | 0.02 ± 0.001 |
| 9 | 0.179 ± 0.008 | 0.041 ± 0.001 |
| Volasertib | 0.017 ± 0.001 | 0.025 ± 0.001 |
| Compound No./Tested Cancer Cell | Apoptotic Genes | Anti-Apoptotic Genes | |
|---|---|---|---|
| BAX | Caspase-3 | Bcl-2 | |
| 7/MDA-MB-231 | 7.325 | 3.904 | 0.301 |
| Volasertib/MDA-MB-231 | 5.332 | 5.531 | 0.351 |
| Control/MDA-MB-231 | 1 | 1 | 1 |
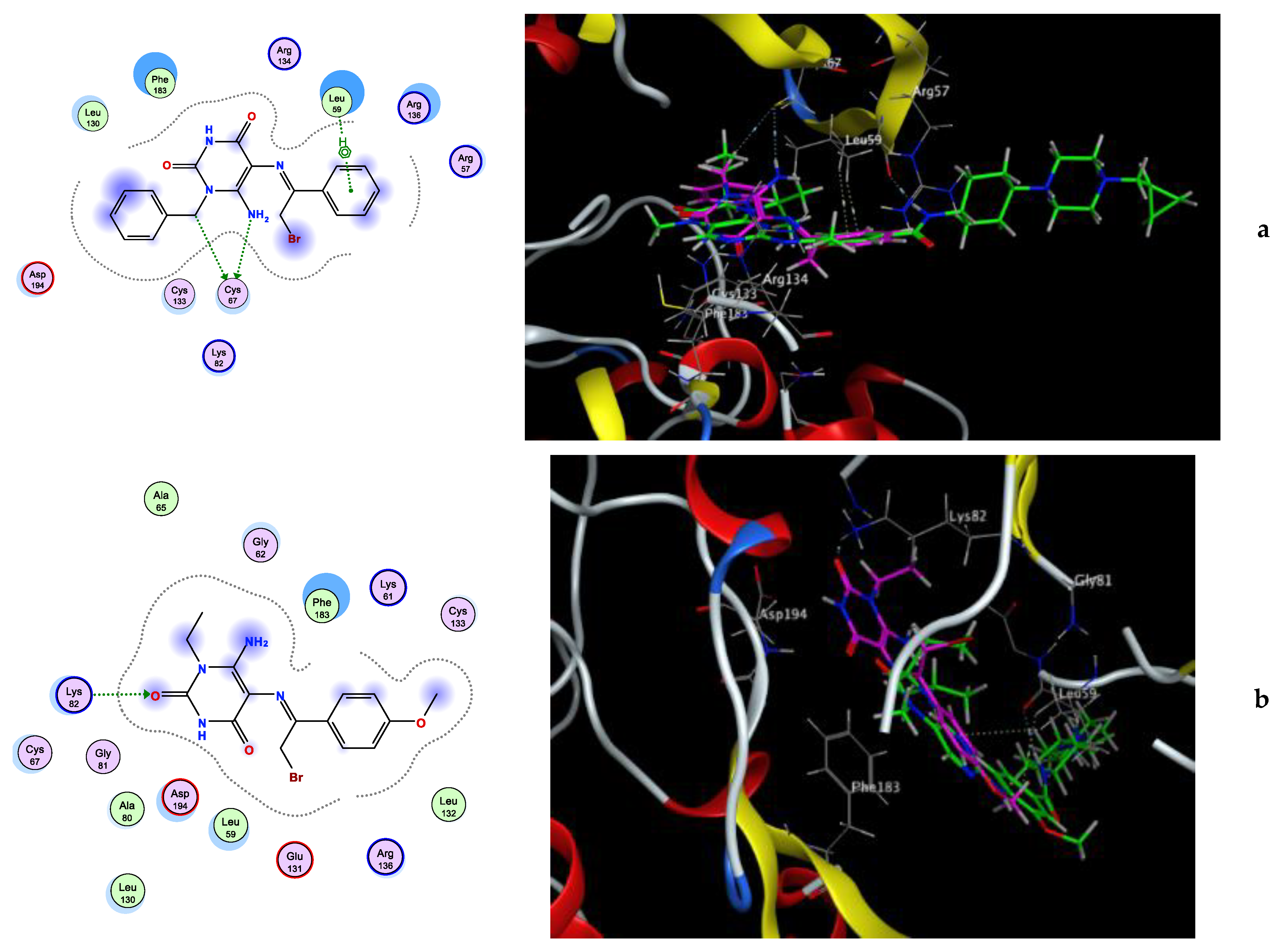
| Compounds | Docking Score (kcal/mol) | Amino Acids H-Bond (Bond Length Å) | Amino Acids Hydrophobic Interactions |
|---|---|---|---|
| 4 | −7.45 | Asn140 (2.12) | Tyr97, Val87, Pro82, Leu92, Ile146, Trp81 |
| 6a | −6.35 | Asn140 (2.57) | Tyr97, Val87, Ile146, Trp81, Leu92, Leu94 |
| 6b | −6.99 | Asn140 (2.10, 2.12) | Trp81, Leu94, Ile146, Leu92, Tyr97 |
| 7 | −7.89 | Cys136 (3.00) | Asn140, Trp81, Pro82, Ile146, Leu92, Leu94, Try97, Val87 |
| 9 | −5.48 | Asn140 (2.08), Pro82 (2.04) | Val87, Leu92, Tyr97, Cys136 |
| Volasertib | −7.93 | Asn140 (2.95) | Val87, Pro82, Gln85, Tyr97, Leu92, Trp81, Ile146, Leu94 |
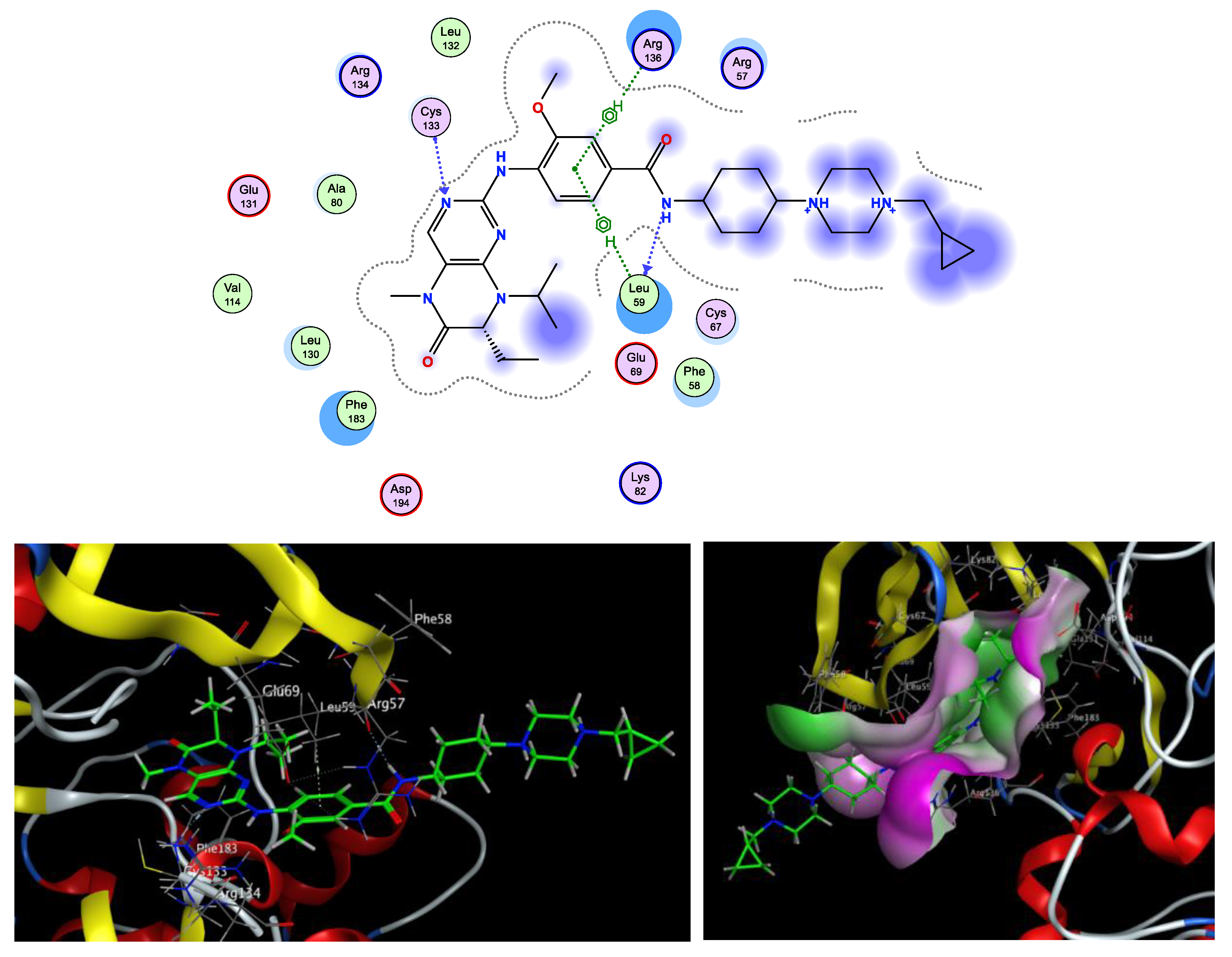
| Compounds | Docking Score (kcal/mol) | Amino Acids H-Bond (Bond Length Å) | Amino Acids Hydrophobic Interactions |
|---|---|---|---|
| 4 | −6.88 | Cys67 (3.11,3.12) | Cys133, Leu59, Phe183, Cys67 |
| 6a | −5.94 | Lys82 (2.20) | Phe183, Leu59, Cys67, Lys82, Leu130, Asp194 |
| 6b | −6.09 | - | Leu59, Arg57, Phe183, Asp194, Lys82 |
| 7 | −7.93 | Asn181 (2.26) | Lys82, Phe183, Leu59, Asp194, Glu140, Glu180 |
| 9 | −7.45 | Arg134 (2.07), Cys133 (2.14), Leu132 (2.28) | Arg134, Leu59, Arg57, Phe183, Arg136 |
| Volasertib | −8.79 | Cys133 (1.86), Leu59 (2.08) | Phe183, Phe59, Leu59, Cys67, Arg136, Arg57 |
| Cpds No. | a M.W. (g/mol) | b iLog Po/w | c Log S | d TPSA (Å) | e HBA | f HBD | g NRB | Lipinski Violations |
|---|---|---|---|---|---|---|---|---|
| 4 | 413.27 | 2.40 | −4.36 ** | 93.24 | 3 | 2 | 5 | 0 |
| 6a | 381.22 | 2.39 | −3.36 * | 102.47 | 4 | 2 | 5 | 0 |
| 6b | 477.74 | 3.23 | −5.18 ** | 102.47 | 4 | 2 | 6 | 0 |
| 7 | 471.37 | 3.09 | −5.99 ** | 115.64 | 4 | 1 | 7 | 0 |
| 9 | 360.37 | 2.52 | −3.80 * | 89.87 | 5 | 1 | 4 | 0 |
| Volasertib | 618.81 | 5.84 | −6.55 *** | 106.17 | 7 | 2 | 11 | 2 |
| Cpds No. | BBB Permeant | GI Absorption | Cytochrome P450 (CYP2D6 Inhibitor) |
|---|---|---|---|
| 4 | No | High | No |
| 6a | No | High | No |
| 6b | No | High | No |
| 7 | No | High | No |
| 9 | No | High | No |
| Volasertib | No | High | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Kalyoubi, S.; El-Sebaey, S.A.; Elfeky, S.M.; AL-Ghulikah, H.A.; El-Zoghbi, M.S. Novel Aminopyrimidine-2,4-diones, 2-Thiopyrimidine-4-ones, and 6-Arylpteridines as Dual-Target Inhibitors of BRD4/PLK1: Design, Synthesis, Cytotoxicity, and Computational Studies. Pharmaceuticals 2023, 16, 1303. https://doi.org/10.3390/ph16091303
El-Kalyoubi S, El-Sebaey SA, Elfeky SM, AL-Ghulikah HA, El-Zoghbi MS. Novel Aminopyrimidine-2,4-diones, 2-Thiopyrimidine-4-ones, and 6-Arylpteridines as Dual-Target Inhibitors of BRD4/PLK1: Design, Synthesis, Cytotoxicity, and Computational Studies. Pharmaceuticals. 2023; 16(9):1303. https://doi.org/10.3390/ph16091303
Chicago/Turabian StyleEl-Kalyoubi, Samar, Samiha A. El-Sebaey, Sherin M. Elfeky, Hanan A. AL-Ghulikah, and Mona S. El-Zoghbi. 2023. "Novel Aminopyrimidine-2,4-diones, 2-Thiopyrimidine-4-ones, and 6-Arylpteridines as Dual-Target Inhibitors of BRD4/PLK1: Design, Synthesis, Cytotoxicity, and Computational Studies" Pharmaceuticals 16, no. 9: 1303. https://doi.org/10.3390/ph16091303