
The Discovery of Novel Agents against Staphylococcus aureus by Targeting Sortase A: A Combination of Virtual Screening and Experimental Validation

 ,
,
Abstract
:
1. Introduction
2. Results
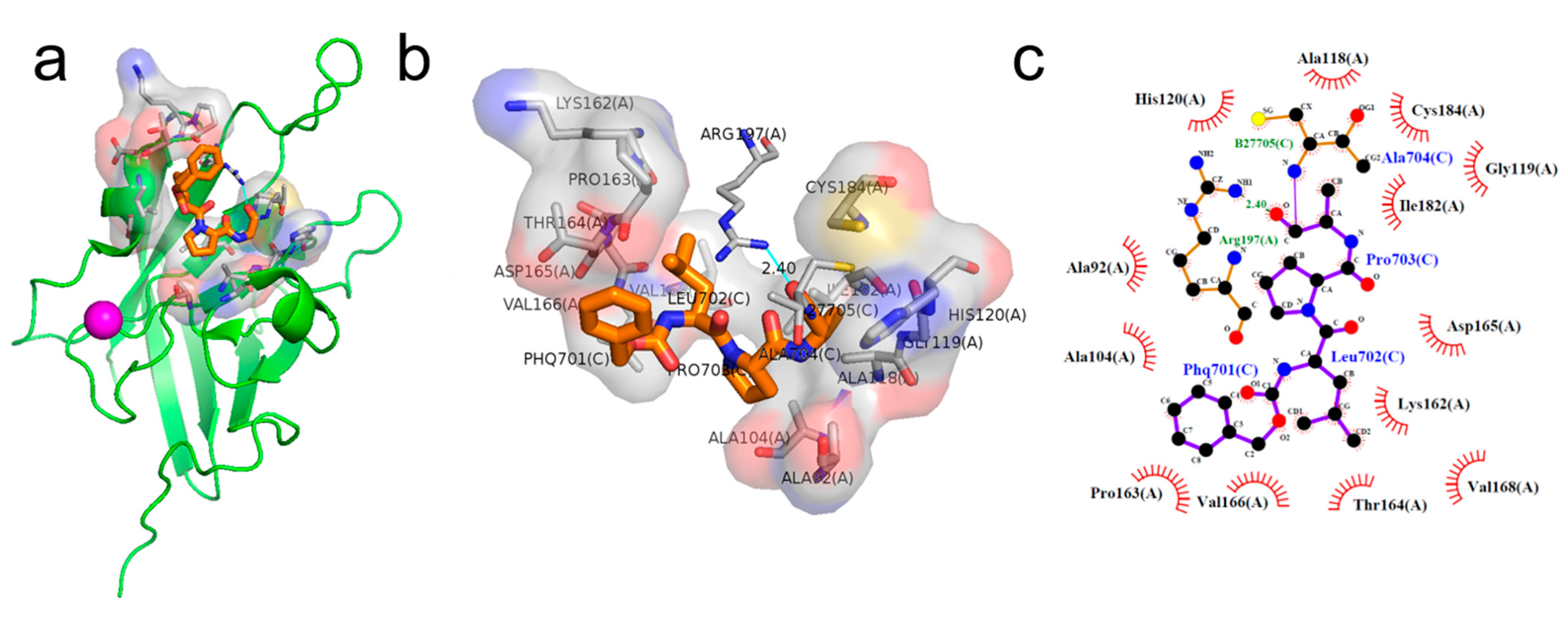
2.1. Binding Pose of the LPXTG Sequence with SrtA
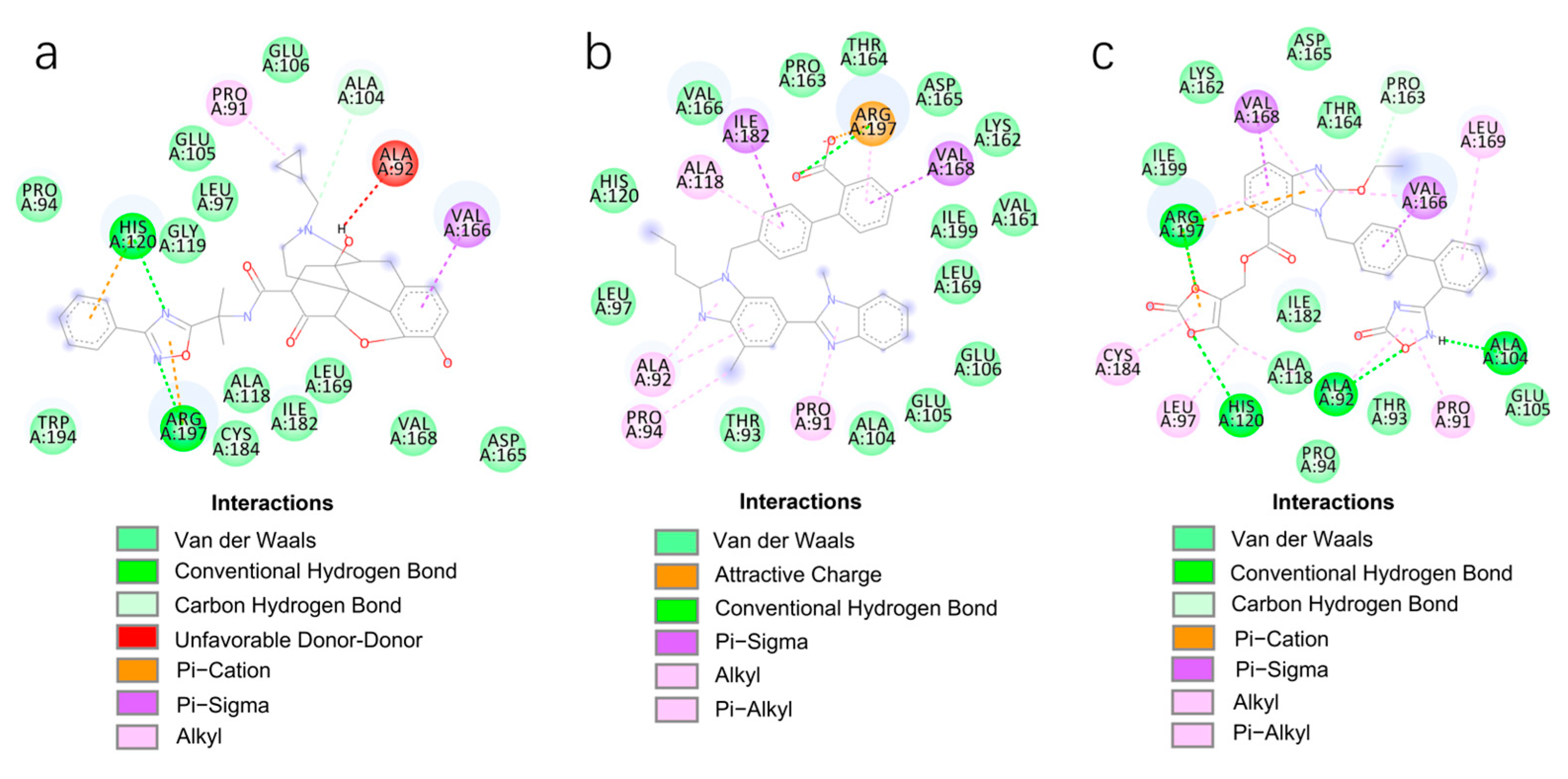
2.2. Virtual Screening using Molecular Docking
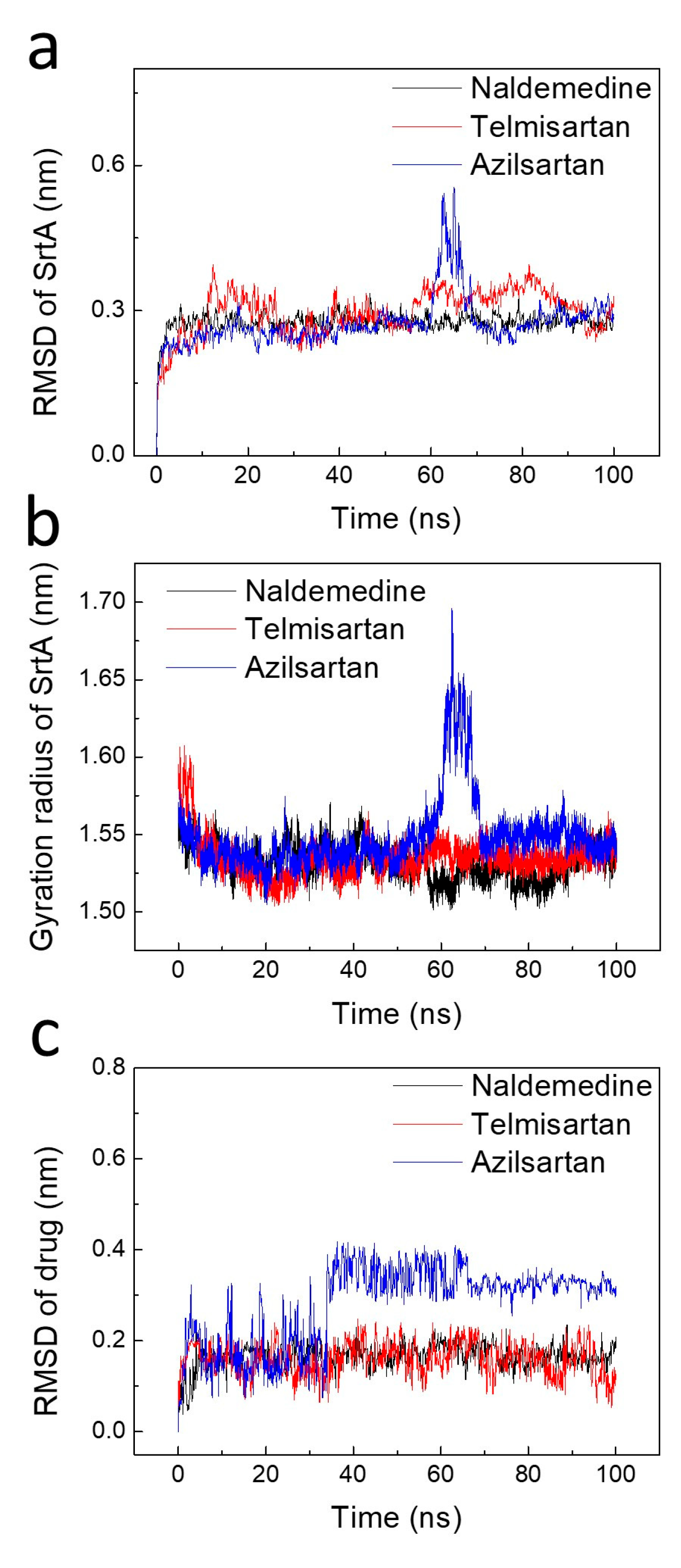
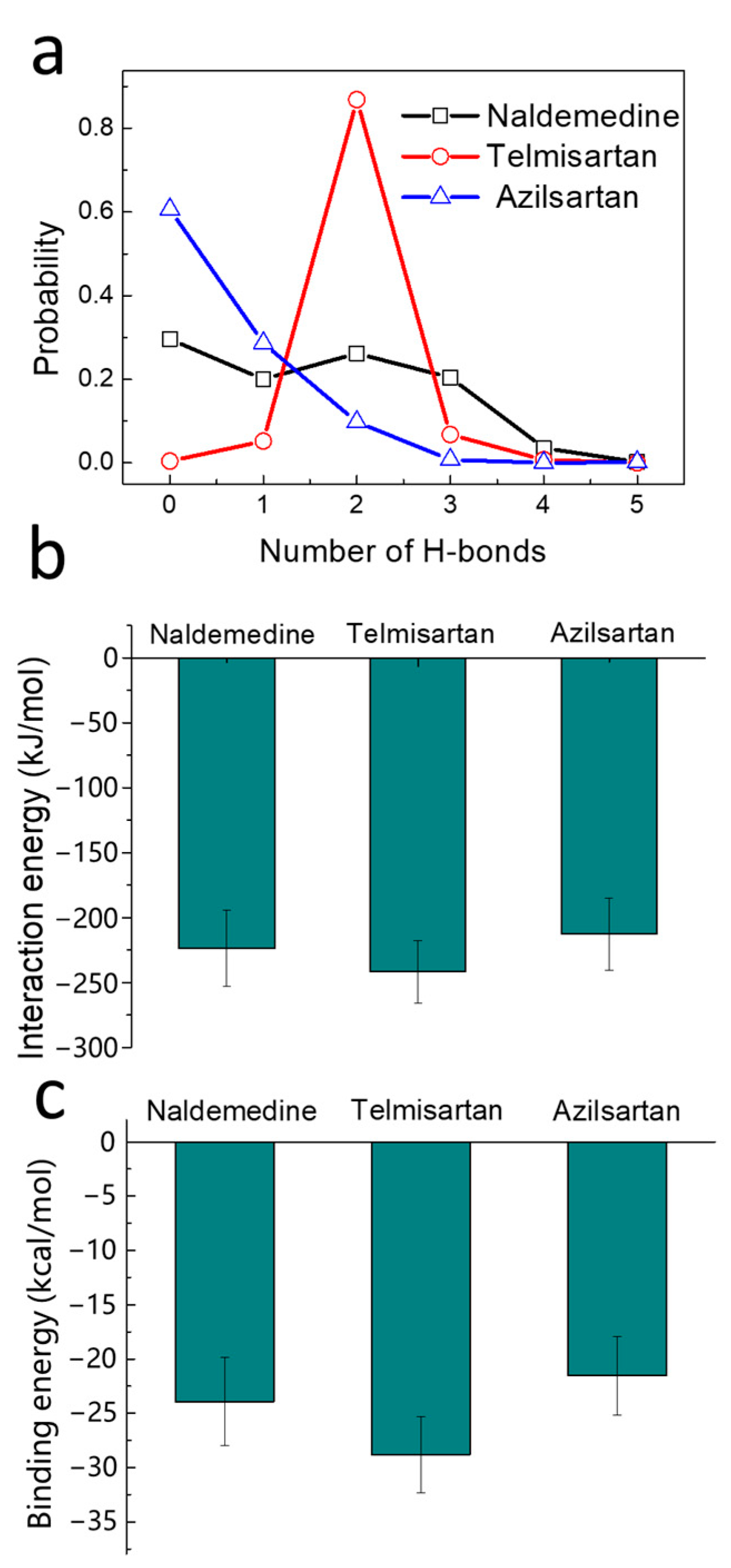
2.3. Stability Analysis of the Binding Poses
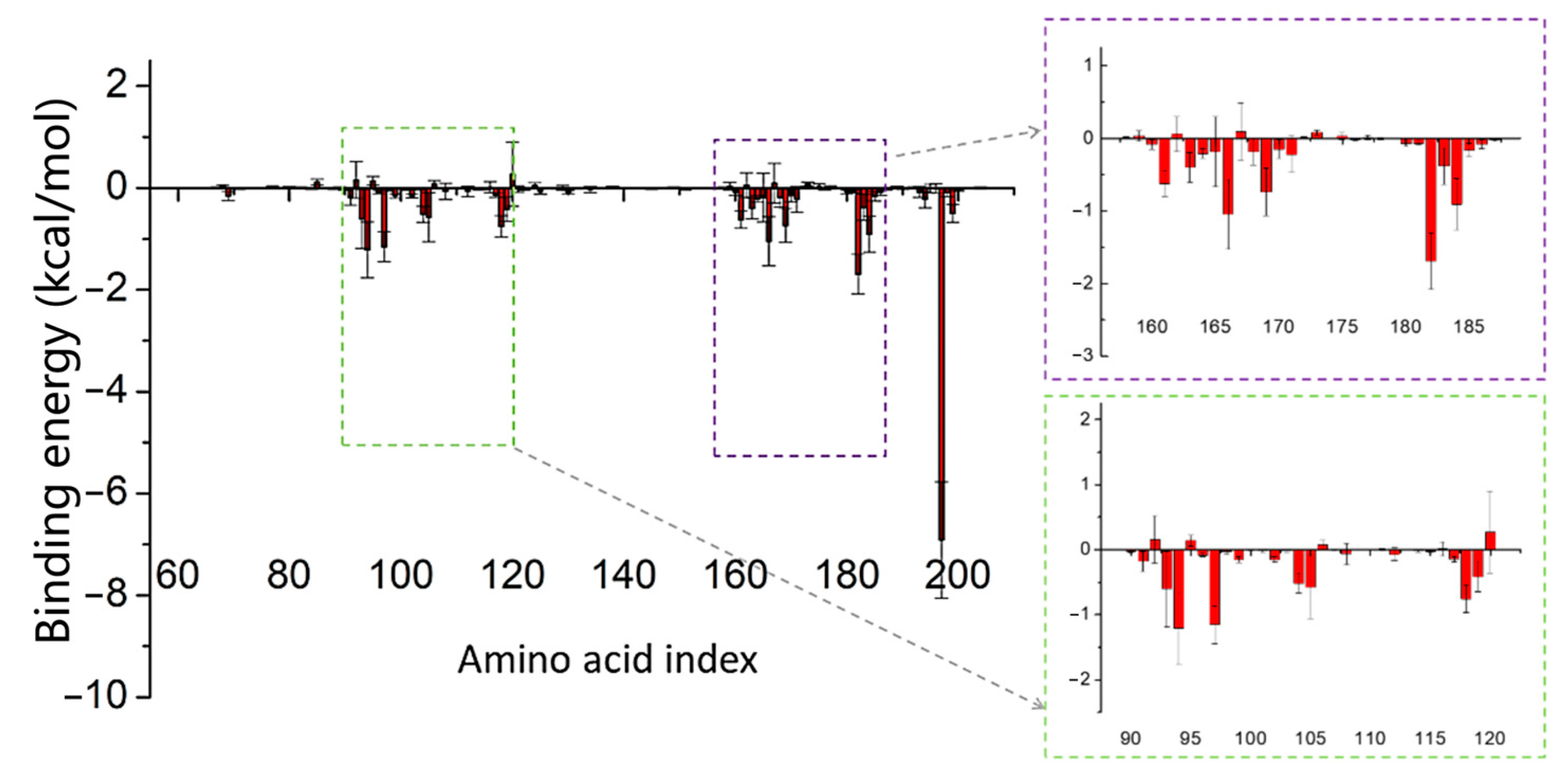
2.4. Binding Strength Assessment
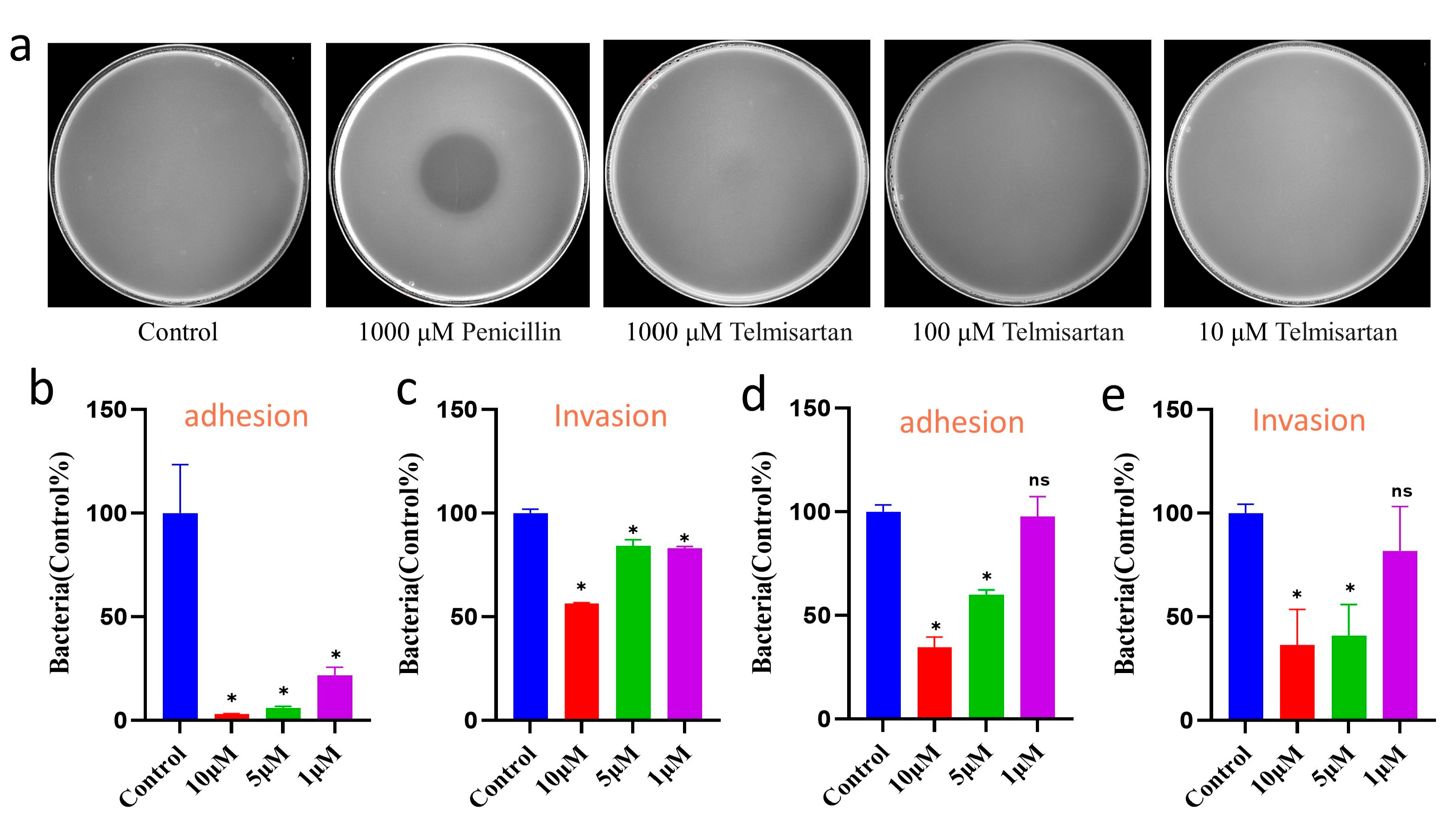
2.5. Efficacy Evaluation of Telmisartan on Bacterial Growth and Infection
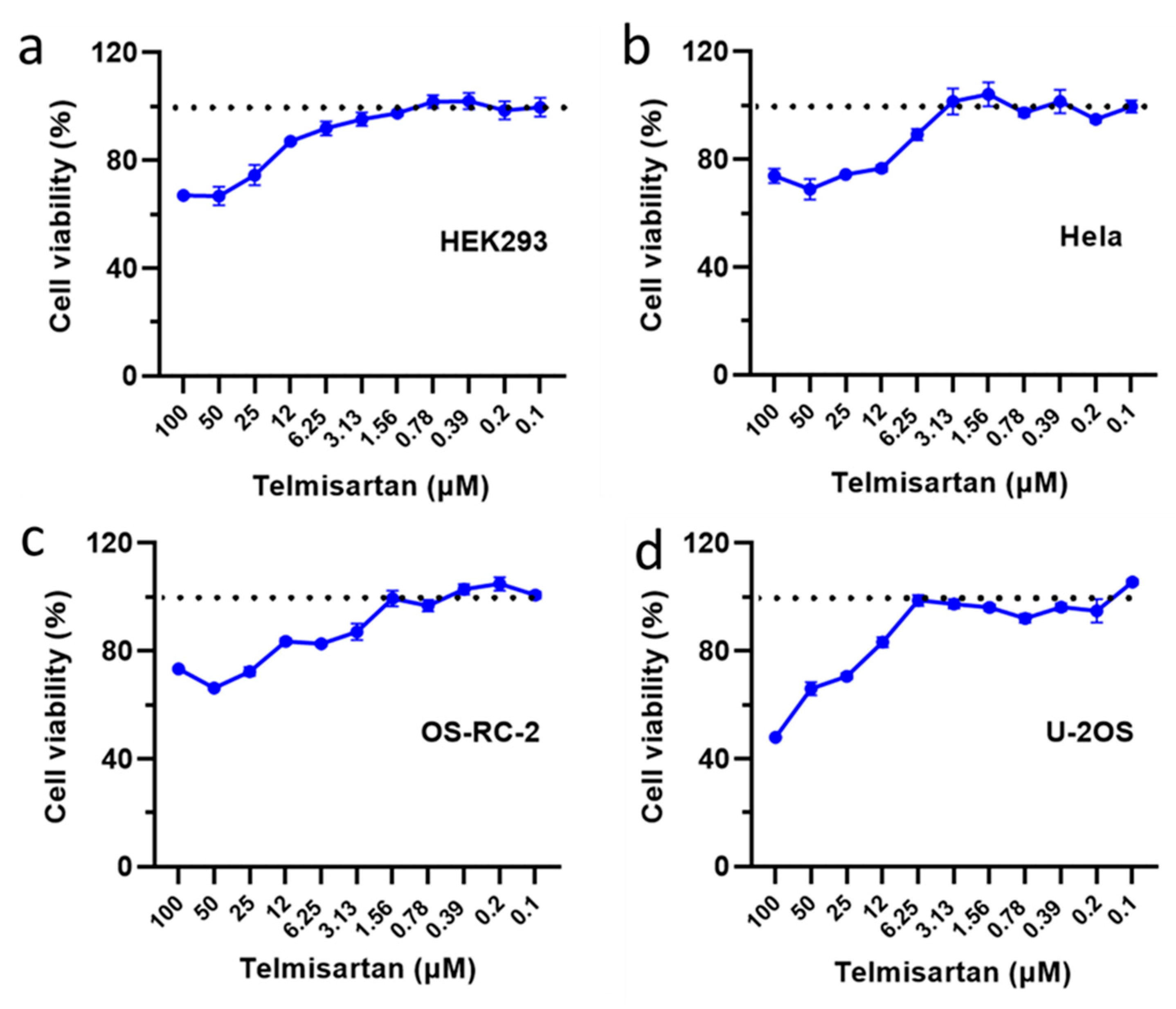
2.6. Toxicity Assay of Telmisartan to Various Cell Types
3. Discussion
4. Materials and Methods
4.1. Structural Preparation for Simulations
4.2. Molecular Docking Simulation
4.3. Molecular Dynamics Simulation
4.4. MM/PBSA Method
4.5. Antibacterial Efficiency Assay
4.6. MIC and Biofilm Formation Tests
4.7. Bacterial Adhesion and Invasion Assay
4.8. Combined Administration Tests
4.9. Drug Toxicity Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ondusko, D.S.; Nolt, D. Staphylococcus aureus. Pediatr. Rev. 2018, 39, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Ahmad-Mansour, N.; Loubet, P.; Pouget, C.; Dunyach-Remy, C.; Sotto, A.; Lavigne, J.-P.; Molle, V. Staphylococcus aureus toxins: An update on their pathogenic properties and potential treatments. Toxins 2021, 13, 677. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-H.; Cohen, T.; Grad, Y.H.; Hanage, W.P.; O’Brien, T.F.; Lipsitch, M. Origin and proliferation of multiple-drug resistance in bacterial pathogens. Microbiol. Mol. Biol. Rev. 2015, 79, 101–116. [Google Scholar] [CrossRef]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- Nandhini, P.; Kumar, P.; Mickymaray, S.; Alothaim, A.S.; Somasundaram, J.; Rajan, M. Recent developments in methicillin-resistant Staphylococcus aureus (MRSA) treatment: A review. Antibiotics 2022, 11, 606. [Google Scholar] [CrossRef]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Ruer, S.; Pinotsis, N.; Steadman, D.; Waksman, G.; Remaut, H. Virulence-targeted antibacterials: Concept, promise, and susceptibility to resistance mechanisms. Chem. Biol. Drug Des. 2015, 86, 379–399. [Google Scholar] [CrossRef]
- Cascioferro, S.; Schillaci, D. The future of antibiotic: From the magic bullet to the smart bullet. J. Microb. Biochem. Technol. 2014, 6, 1000e118. [Google Scholar] [CrossRef]
- Cascioferro, S.; Cusimano, M.G.; Schillaci, D. Antiadhesion agents against Gram-positive pathogens. Future Microbiol. 2014, 9, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Matsumoto, S. Bacterial adhesion: From mechanism to control. Biochem. Eng. J. 2010, 48, 424–434. [Google Scholar] [CrossRef]
- Pishesha, N.; Ingram, J.R.; Ploegh, H.L. Sortase A: A model for transpeptidation and its biological applications. Annu. Rev. Cell Dev. Biol. 2018, 34, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.J.; Lenoy, E.; Murphy, T.; Tardio, L.; Burgio, P.; Projan, S.J.; Schneewind, O.; Alksne, L. Effect of srtA and srtB gene expression on the virulence of Staphylococcus aureus in animal models of infection. J. Antimicrob. Chemother. 2004, 53, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-B.; Oh, M.-N.; Kim, J.-G.; Shin, D.-S.; Shin, J. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 2006, 70, 102–106. [Google Scholar] [CrossRef]
- Sibbald, M.J.; Yang, X.M.; Tsompanidou, E.; Qu, D.; Hecker, M.; Becher, D.; Buist, G.; van Dijl, J.M. Partially overlapping substrate specificities of staphylococcal group A sortases. Proteomics 2012, 12, 3049–3062. [Google Scholar] [CrossRef]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A inhibitors: Recent advances and future perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef]
- Nitulescu, G.; Mihai, D.P.; Nicorescu, I.M.; Olaru, O.T.; Ungurianu, A.; Zanfirescu, A.; Nitulescu, G.M.; Margina, D. Discovery of natural naphthoquinones as sortase A inhibitors and potential anti-infective solutions against Staphylococcus aureus. Drug Dev. Res. 2019, 80, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Thappeta, K.R.V.; Zhao, L.N.; Nge, C.E.; Crasta, S.; Leong, C.Y.; Ng, V.; Kanagasundaram, Y.; Fan, H.; Ng, S.B. In-Silico Identified New Natural Sortase A Inhibitors Disrupt S. aureus Biofilm Formation. Int. J. Mol. Sci. 2020, 21, 8601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A review on applications of computational methods in drug screening and design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, R.; Liu, J.; Wang, S.; Zhang, W.; Feng, K.; Liu, C.; Liu, B.; Miao, Y.; Zhou, S. Virtual Screening-Based Study of Novel Anti-Cancer Drugs Targeting G-Quadruplex. Pharmaceutics 2023, 15, 1414. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Ramazanova, A.G.; Voronin, A.P.; Drozd, K.V.; Churakov, A.V.; Perlovich, G.L. Virtual Screening, Structural Analysis, and Formation Thermodynamics of Carbamazepine Cocrystals. Pharmaceutics 2023, 15, 836. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V. Crystal structures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, R.; Joachimiak, G.; Mazmanian, S.K.; Missiakas, D.M.; Gornicki, P.; Schneewind, O.; Joachimiak, A. Structures of sortase B from Staphylococcus aureus and Bacillus anthracis reveal catalytic amino acid triad in the active site. Structure 2004, 12, 1147–1156. [Google Scholar] [CrossRef]
- Liew, C.K.; Smith, B.T.; Pilpa, R.; Suree, N.; Ilangovan, U.; Connolly, K.M.; Jung, M.E.; Clubb, R.T. Localization and mutagenesis of the sorting signal binding site on sortase A from Staphylococcus aureus. FEBS Lett. 2004, 571, 221–226. [Google Scholar] [CrossRef]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.T.; Nitulescu, G.M. Targeting Bacterial Sortases in Search of Anti-Virulence Therapies with Low Risk of Resistance Development. Pharmaceuticals 2021, 14, 415. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Wallock-Richards, D.J.; Marles-Wright, J.; Clarke, D.J.; Maitra, A.; Dodds, M.; Hanley, B.; Campopiano, D.J. Molecular basis of Streptococcus mutans sortase A inhibition by the flavonoid natural product trans-chalcone. Chem. Commun. 2015, 51, 10483–10485. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Yi, S.W.; Weiner, E.M.; Amer, B.R.; Sue, C.K.; Wereszczynski, J.; Dillen, C.A.; Senese, S.; Torres, J.Z.; McCammon, J.A. NMR structure-based optimization of Staphylococcus aureus sortase A pyridazinone inhibitors. Chem. Biol. Drug Des. 2017, 90, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.W.; Yi, S.W.; Paek, S.-M. Design and synthesis of small molecules as potent staphylococcus aureus sortase a inhibitors. Antibiotics 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R.U.S. FDA approved drugs from 2015–June 2020: A perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef]
- Patridge, E.V.; Gareiss, P.C.; Kinch, M.S.; Hoyer, D.W. An analysis of original research contributions toward FDA-approved drugs. Drug Discov. Today 2015, 20, 1182–1187. [Google Scholar] [CrossRef]
- Bi, C.; Wang, L.; Niu, X.; Cai, H.; Zhong, X.; Deng, X.; Wang, T.; Wang, D. The use of chlorogenic acid and its analogues as inhibitors: An investigation of the inhibition of sortase A of Staphylococcus aureus using molecular docking and dynamic simulation. Biotechnol. Lett. 2016, 38, 1341–1347. [Google Scholar] [CrossRef]
- Smith, D.H.; Matzek, K.M.; Kempthorne-Rawson, J. Dose response and safety of telmisartan in patients with mild to moderate hypertension. J. Clin. Pharmacol. 2000, 40, 1380–1390. [Google Scholar] [CrossRef]
- Zimmermann, S.; Klinger-Strobel, M.; Bohnert, J.A.; Wendler, S.; Rödel, J.; Pletz, M.W.; Löffler, B.; Tuchscherr, L. Clinically Approved Drugs Inhibit the Staphylococcus aureus Multidrug NorA Efflux Pump and Reduce Biofilm Formation. Front. Microbiol. 2019, 10, 2762. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Shanmuganathan, M.V.; Behenna, D.; Stoltz, B.M.; Prasadarao, N.V. Angiotensin II receptor type 1—A novel target for preventing neonatal meningitis in mice by Escherichia coli K1. J. Infect. Dis. 2014, 209, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, L.; Wang, Y. Recent Advances in Application of Computer-Aided Drug Design in Anti-Influenza A Virus Drug Discovery. Int. J. Mol. Sci. 2022, 23, 4738. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Quintal Bojórquez, N.; Campos, M.R.S. Traditional and Novel Computer-Aided Drug Design (CADD) Approaches in the Anticancer Drug Discovery Process. Curr. Cancer Drug Targets. 2023, 23, 333–345. [Google Scholar] [PubMed]
- Alachkar, A.; Ojha, S.K.; Sadeq, A.; Adem, A.; Frank, A.; Stark, H.; Sadek, B. Experimental Models for the Discovery of Novel Anticonvulsant Drugs: Focus on Pentylenetetrazole-Induced Seizures and Associated Memory Deficits. Curr. Pharm. Des. 2020, 26, 1693–1711. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lee, S.; Song, I.H.; Lee, J.H.; Yang, W.Y.; Oh, K.B.; Shin, J. Sortase A inhibitory metabolites from the roots of Pulsatilla koreana. Bioorganic Med. Chem. Lett. 2014, 24, 44–48. [Google Scholar] [CrossRef]
- Oh, K.B.; Kim, S.H.; Lee, J.; Cho, W.J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio. Accelrys, version 2.1; Discovery Studio: Los Angeles, CA, USA, 2008. [Google Scholar]
- Carta, G.; Knox, A.J.; Lloyd, D.G. Unbiasing scoring functions: A new normalization and rescoring strategy. J. Chem. Inf. Model. 2007, 47, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Van, D.S.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701. [Google Scholar]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with th e CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Ding, H.M.; Yin, Y.W.; Sheng, Y.J.; Ma, Y.Q. Accurate evaluation on the interactions of SARS-CoV-2 with its receptor ACE2 and antibodies CR3022/CB6. Chin. Phys. Lett. 2021, 38, 018701. [Google Scholar] [CrossRef]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T.J. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Sheng, Y.J.; Yin, Y.W.; Ma, Y.Q.; Ding, H.M. Improving the Performance of MM/PBSA in Protein–Protein Interactions via the Screening Electrostatic Energy. J. Chem. Inf. Model. 2021, 61, 2454–2462. [Google Scholar] [CrossRef]
- CLSI. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. In Approved Standard CLSI Document M07-A10, 10th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Lopes, L.A.A.; Dos Santos Rodrigues, J.B.; Magnani, M.; de Souza, E.L.; de Siqueira-Júnior, J.P. Inhibitory effects of flavonoids on biofilm formation by Staphylococcus aureus that overexpresses efflux protein genes. Microb. Pathog. 2017, 107, 193–197. [Google Scholar] [CrossRef]
- Sinha, B.; Francois, P.P.; Nusse, O.; Foti, M.; Hartford, O.M.; Vaudaux, P.; Foster, T.J.; Lew, D.P.; Herrmann, M.; Krause, K.-H. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol. 1999, 1, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Juuti, K.M.; Sinha, B.; Werbick, C.; Peters, G.; Kuusela, P.I. Reduced adherence and host cell invasion by methicillin-resistant Staphylococcus aureus expressing the surface protein Pls. J. Infect. Dis. 2004, 189, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Elion, G.B.; Singer, S.; Hitchings, G.H. Antagonists of nucleic acid derivatives. VIII. Synergism in combinations of biochemically related antimetabolites. J. Biol. Chem. 1954, 208, 477–488. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Compound | Structure | Binding Energy (kcal/mol) | corrScore | Ratio of Occupied Sites |
|---|---|---|---|---|---|
| 1 | Trypan Blue | 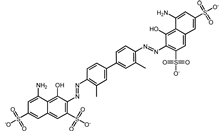 | −9.6 | 0 | 0 |
| 2 | Naldemedine | 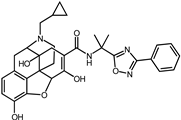 | −9.6 | 0.84 | 71.4% |
| 3 | Lomitapide | 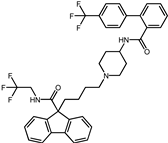 | −9.4 | 0.69 | 64.3% |
| 4 | Norgestrel | 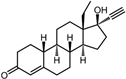 | −9.2 | 0.86 | 0 |
| 5 | Triazolam | 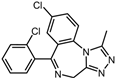 | −9.1 | 0.82 | 50.0% |
| 6 | Flourescein | 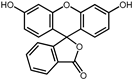 | −9.1 | 0.82 | 57.1% |
| 7 | Midazolam |  | −9.0 | 0.80 | 64.3% |
| 8 | Simeprevir | 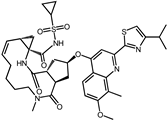 | −9.0 | 0.55 | 50.0% |
| 9 | Alprazolam |  | −9.0 | 0.80 | 50.0% |
| 10 | Telmisartan | 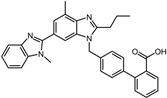 | −9.0 | 0.72 | 71.4% |
| 11 | Nilotinib | 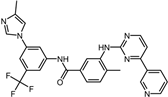 | −9.0 | 0.71 | 64.3% |
| 12 | Azilsartan | 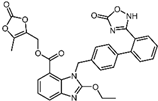 | −9.0 | 0.69 | 85.7% |
| Positive control | Rosmarinic acid | 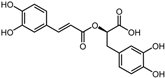 | −7.6 | 0.39 | 64.3% |
| Negative control | 2,3-Bis(4-methoxyphenyl)propanenitrile | 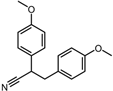 | −6.2 | 0.18 | 35.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Tong, J.; Liu, X.; Liang, D.; Ren, F.; Jiang, N.; Hao, Z.; Li, S.; Wang, Q. The Discovery of Novel Agents against Staphylococcus aureus by Targeting Sortase A: A Combination of Virtual Screening and Experimental Validation. Pharmaceuticals 2024, 17, 58. https://doi.org/10.3390/ph17010058
Liu K, Tong J, Liu X, Liang D, Ren F, Jiang N, Hao Z, Li S, Wang Q. The Discovery of Novel Agents against Staphylococcus aureus by Targeting Sortase A: A Combination of Virtual Screening and Experimental Validation. Pharmaceuticals. 2024; 17(1):58. https://doi.org/10.3390/ph17010058
Chicago/Turabian StyleLiu, Kang, Jiangbo Tong, Xu Liu, Dan Liang, Fangzhe Ren, Nan Jiang, Zhenyu Hao, Shixin Li, and Qiang Wang. 2024. "The Discovery of Novel Agents against Staphylococcus aureus by Targeting Sortase A: A Combination of Virtual Screening and Experimental Validation" Pharmaceuticals 17, no. 1: 58. https://doi.org/10.3390/ph17010058
APA StyleLiu, K., Tong, J., Liu, X., Liang, D., Ren, F., Jiang, N., Hao, Z., Li, S., & Wang, Q. (2024). The Discovery of Novel Agents against Staphylococcus aureus by Targeting Sortase A: A Combination of Virtual Screening and Experimental Validation. Pharmaceuticals, 17(1), 58. https://doi.org/10.3390/ph17010058