

Prominent Neuroprotective Potential of Indole-2-N-methylpropargylamine: High Affinity and Irreversible Inhibition Efficiency towards Monoamine Oxidase B Revealed by Computational Scaffold Analysis



Abstract
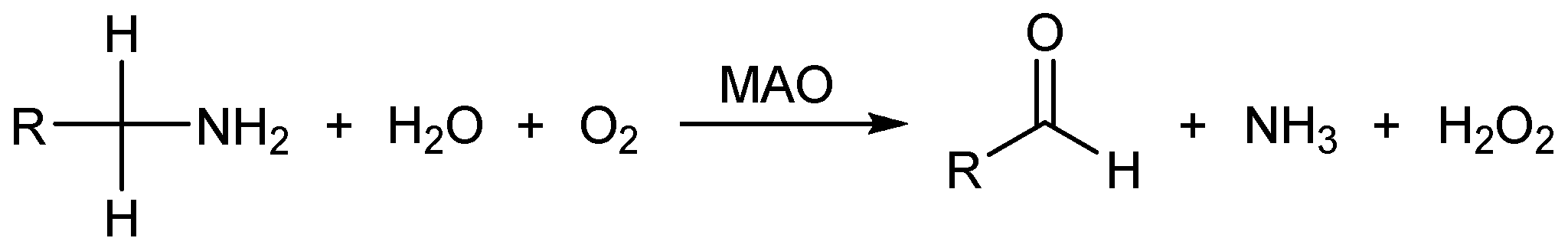
:1. Introduction
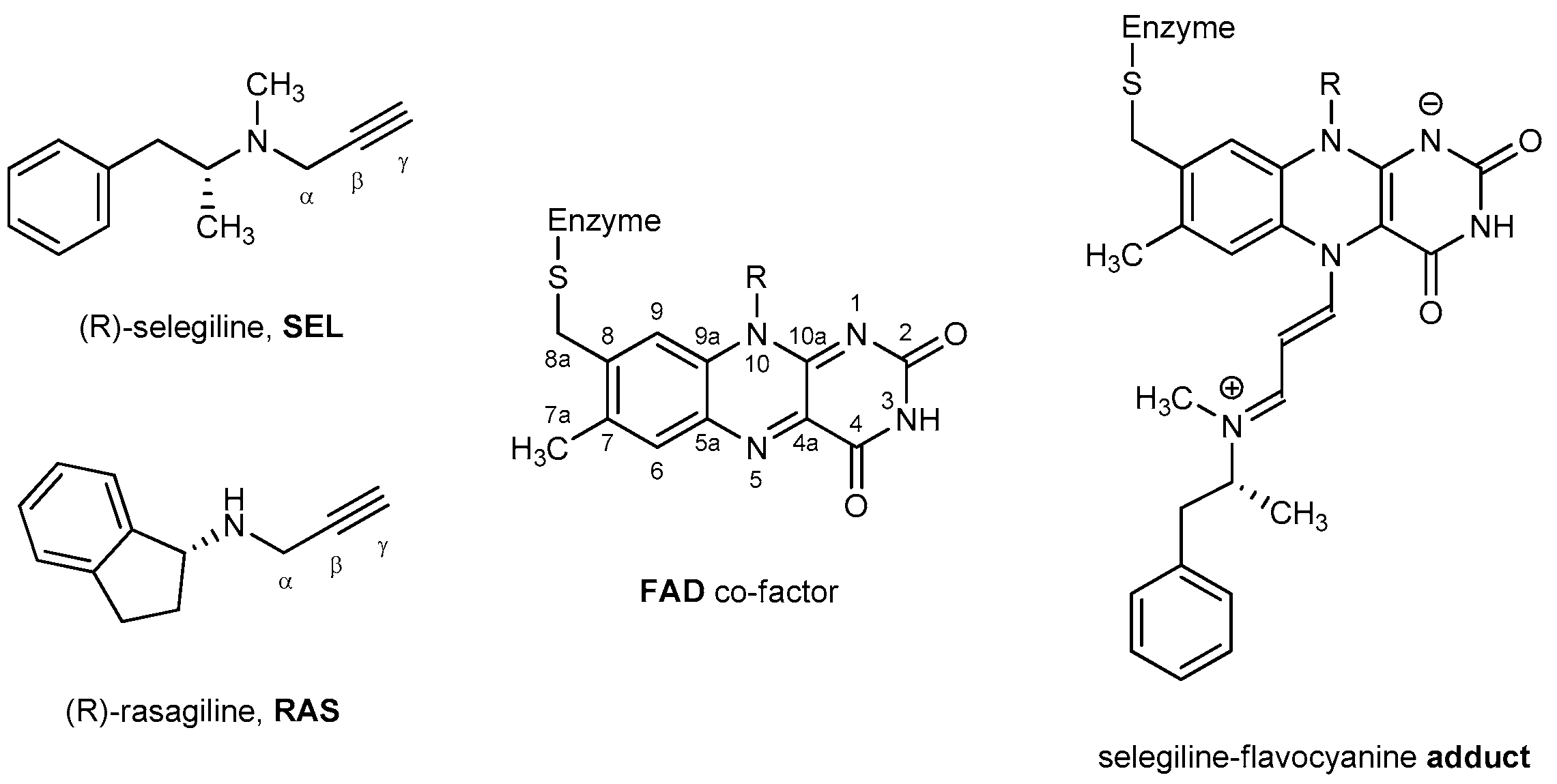
2. Results and Discussion
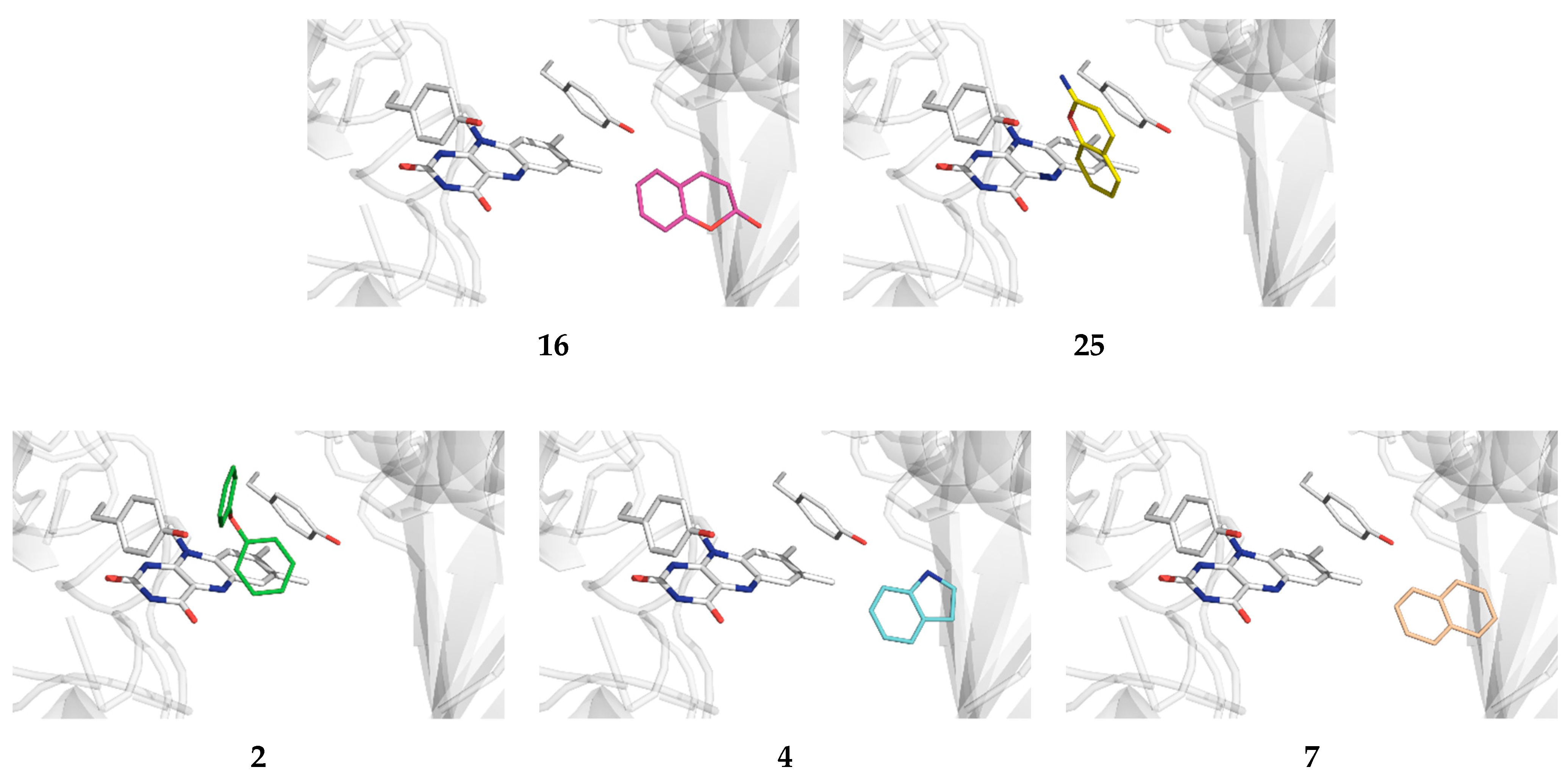
2.1. Docking of Organic Scaffolds into the MAO-B Active Site
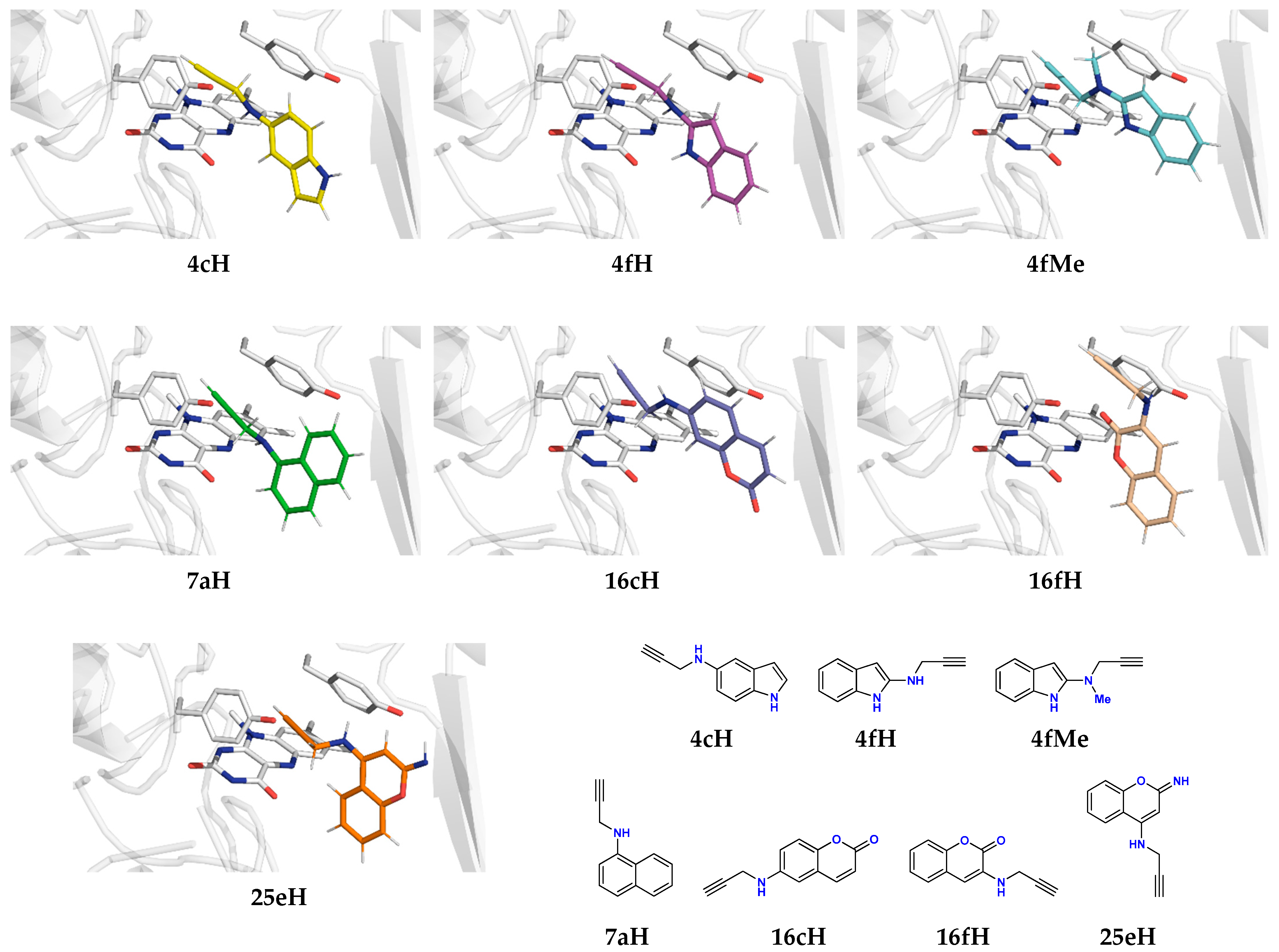
2.2. Docking of Propargylamine Derivatives into the MAO-B Active Site
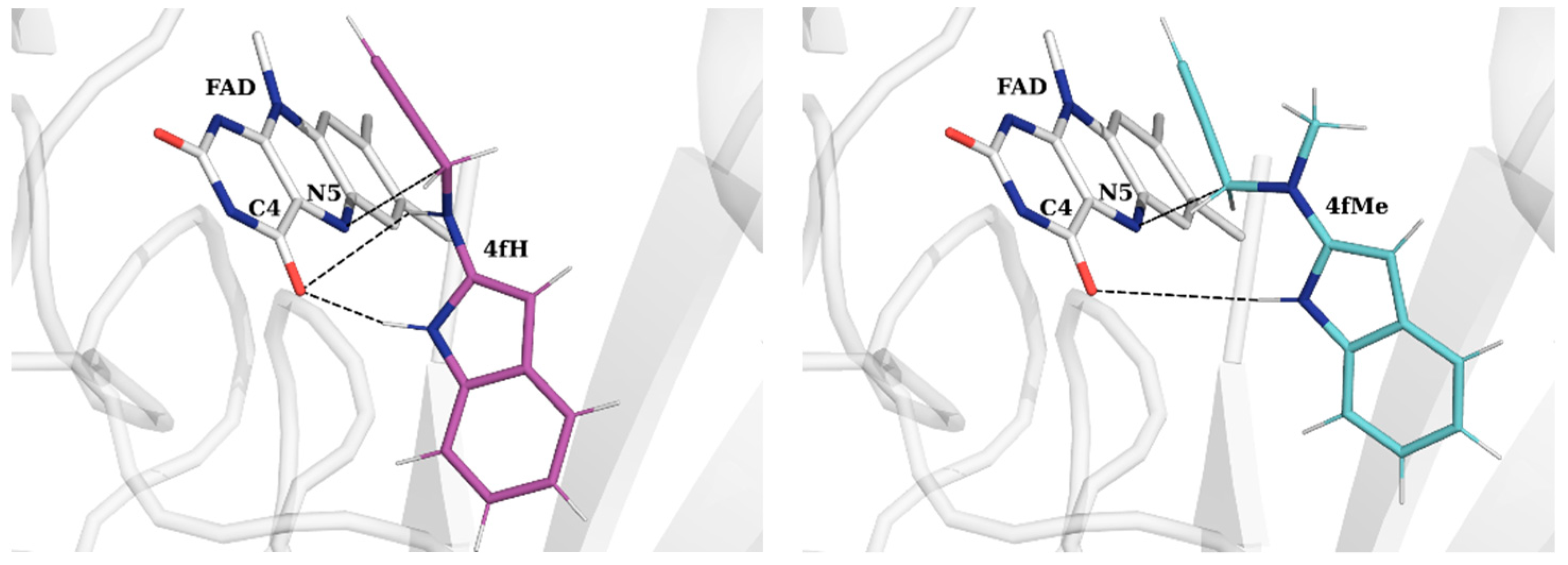
2.3. Molecular Dynamics Simulations of Selected Propargylamines in the MAO-B Active Site
2.4. DFT Analysis of the Inhibition Reaction
3. Materials and Methods
3.1. System Preparation
3.2. Docking Simulations and Cross-Docking Validation
3.3. Molecular Dynamics Simulations
3.4. Binding Free Energy Calculations
3.5. DFT Mechanistic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramsay, R.R. Monoamine oxidases: The biochemistry of the protein as targets in medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2012, 12, 2189–2209. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.E.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Grimsby, J.; Lan, N.C.; Neve, R.; Chen, K.; Shih, J.C. Tissue distribution of human monoamine oxidase A and B mRNA. J. Neurochem. 1990, 55, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Westlund, K.N.; Denney, R.M.; Kochersperger, L.M.; Rose, R.M.; Abell, C.W. Distinct monoamine oxidase A and B populations in primate brain. Science 1985, 230, 181–183. [Google Scholar] [CrossRef] [PubMed]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef] [PubMed]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef]
- Fowler, C.J.; Tipton, K.F. On the substrate specificities of 2 forms of monoamine oxidase. J. Pharm. Pharmacol. 1984, 36, 111–115. [Google Scholar] [CrossRef]
- Yeung, A.W.K.; Georgieva, M.G.; Atanasov, A.G.; Tzvetkov, N.T. Monoamine oxidases (MAOs) as privileged molecular targets in neuroscience: Research literature analysis. Front. Mol. Neurosci. 2019, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Pavlin, M.; Repič, M.; Vianello, R.; Mavri, J. The chemistry of neurodegeneration: Kinetic data and their implications. Mol. Neurobiol. 2016, 53, 3400–3415. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 2012, 7057–7065. [Google Scholar] [CrossRef]
- Repič, M.; Vianello, R.; Purg, M.; Duarte, F.; Bauer, P.; Kamerlin, S.C.L.; Mavri, J. Empirical valence bond simulations of the hydride transfer step in the monoamine oxidase B catalyzed metabolism of dopamine. Proteins 2014, 82, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Poberžnik, M.; Purg, M.; Repič, M.; Mavri, J.; Vianello, R. Empirical valence bond simulations of the hydride-transfer step in the monoamine oxidase A catalyzed metabolism of noradrenaline. J. Phys. Chem. B 2016, 120, 11419–11427. [Google Scholar] [CrossRef] [PubMed]
- Tormos, J.R.; Suarez, M.B.; Fitzpatrick, P.F. 13C kinetic isotope effects on the reaction of a flavin amine oxidase determined from whole molecule isotope effects. Arch. Biochem. Biophys. 2016, 612, 115–119. [Google Scholar] [CrossRef]
- Tararina, M.A.; Allen, K.N. Bioinformatic analysis of the flavin-dependent amine oxidase superfamily: Adaptations for substrate specificity and catalytic diversity. J. Mol. Biol. 2020, 432, 3269–3288. [Google Scholar] [CrossRef]
- Tararina, M.A.; Xue, S.; Smith, L.C.; Muellers, S.N.; Miranda, P.O.; Janda, K.D.; Allen, K.N. Crystallography coupled with kinetic analysis provides mechanistic underpinnings of a nicotine-degrading enzyme. Biochemistry 2018, 57, 3741–3751. [Google Scholar] [CrossRef]
- Zapata-Torres, G.; Fierro, A.; Barriga-González, G.; Salgado, J.C.; Celis-Barros, C. Revealing monoamine oxidase B catalytic mechanisms by means of the quantum chemical cluster approach. J. Chem. Inf. Model. 2015, 55, 1349–1360. [Google Scholar] [CrossRef]
- Atalay, V.; Erdem, S.S. A comparative computational investigation on the proton and hydride transfer mechanisms of monoamine oxidase using model molecules. Comput. Biol. Chem. 2013, 47, 181–191. [Google Scholar] [CrossRef]
- Yildiz, I. Computational insights on the hydride and proton transfer mechanisms of L-proline dehydrogenase. PLoS ONE 2023, 18, e0290901. [Google Scholar] [CrossRef]
- Yildiz, I. Computational analysis of hydride and proton transfer steps in L-lactate oxidase based on QM and QM–MM methods. J. Mol. Struct. 2024, 1295, 136706. [Google Scholar] [CrossRef]
- Sblano, S.; Boccarelli, A.; Mesiti, F.; Purgatorio, R.; de Candia, M.; Catto, M.; Altomare, C.D. A second life for MAO inhibitors? From CNS diseases to anticancer therapy. Eur. J. Med. Chem. 2024, 267, 116180. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Li, H.; Ma, Y.; Zhao, Z.; An, Q.; Zhao, J.; Shi, C. Monoamine oxidase A (MAO A): A promising target for prostate cancer therapy. Cancer Lett. 2023, 563, 216188. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, R.; Alsous, L.; Sabbah, D.A.; Gul, H.I.; Gul, M.; Bardaweel, S.K. Monoamine oxidase (MAO) as a potential target for anticancer drug design and development. Molecules 2021, 26, 6019. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. 2023. Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 13 May 2024).
- Hok, L.; Rimac, H.; Mavri, J.; Vianello, R. COVID-19 infection and neurodegeneration: Computational evidence for interactions between the SARS-CoV-2 spike protein and monoamine oxidase enzymes. Comput. Struct. Biotechnol. J. 2022, 20, 1254–1263. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.; Luo, B. Virus infection participates in the occurrence and development of human diseases through monoamine oxidase. Rev. Med. Virol. 2023, 33, e2465. [Google Scholar] [CrossRef]
- Bouali-Benazzouz, R.; Benazzouz, A. COVID-19 infection and parkinsonism: Is there a link? Mov. Disord. 2021, 36, 1737–1743. [Google Scholar] [CrossRef]
- Fox, H.H.; Gibas, J.T. Synthetic tuberculostats. V. Alkylidene derivatives of isonicotinylhydrazine. J. Org. Chem. 1953, 18, 983–989. [Google Scholar] [CrossRef]
- Asatoor, A.M.; Levi, A.J.; Milne, M.D. Tranylcypromine and cheese. Lancet 1963, 282, 733–734. [Google Scholar] [CrossRef]
- Szökö, É.; Tábi, T.; Riederer, P.; Vécsei, L.; Magyar, K. Pharmacological aspects of the neuroprotective effects of irreversible MAO-B inhibitors, selegiline and rasagiline, in Parkinson’s disease. J. Neural Transm. 2018, 125, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Rasagiline: A novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol. 2010, 92, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Shih, J.C. Behavioral outcomes of monoamine oxidase deficiency: Preclinical and clinical evidence. Int. Rev. Neurobiol. 2011, 100, 13–42. [Google Scholar] [CrossRef]
- Ramsay, R.R. Molecular aspects of monoamine oxidase B. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 69, 81–89. [Google Scholar] [CrossRef] [PubMed]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Zhuo, C.; Zhu, X.; Jiang, R.; Ji, F.; Su, Z.; Xue, R.; Zhou, Y. Comparison for efficacy and tolerability among ten drugs for treatment of Parkinson’s disease: A network meta-analysis. Sci. Rep. 2017, 7, 45865. [Google Scholar] [CrossRef]
- Jost, W.H. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J. Neural Transm. 2022, 129, 723–736. [Google Scholar] [CrossRef]
- Binda, C.; Milczek, E.M.; Bonivento, D.; Wang, J.; Mattevi, A.; Edmondson, D.E. Lights and shadows on monoamine oxidase inhibition in neuroprotective pharmacological therapies. Curr. Top. Med. Chem. 2011, 11, 2788–2796. [Google Scholar] [CrossRef]
- Tang, Y.; Moretti, R.; Meiler, J. Recent advances in automated structure-based de novo drug design. J. Chem. Inf. Model. 2024, 64, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wu, Z.; Wang, Z.; Wang, Y.; Zhou, M.; Li, W.; Liu, G.; Tang, Y. Network-based methods and their applications in drug discovery. J. Chem. Inf. Model. 2024, 64, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K.; Mariam, Z. Computer-aided drug design and drug discovery: A prospective analysis. Pharmaceuticals 2024, 17, 22. [Google Scholar] [CrossRef] [PubMed]
- Bali, N.R.; Salve, P.S. Impact of rasagiline nanoparticles on brain targeting efficiency via gellan gum based transdermal patch: A nanotheranostic perspective for Parkinsonism. Int. J. Biol. Macromol. 2020, 164, 1006–1024. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018, 145, 445–497. [Google Scholar] [CrossRef] [PubMed]
- Monte, C.D.; D’Ascenzio, M.; Guglielmi, P.; Mancini, V.; Carradori, S. Opening new scenarios for human MAO inhibitors. Cent. Nerv. Syst. Agents Med. Chem. 2016, 16, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef]
- Riederer, P.; Müller, T. Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: Clinical-pharmacological aspects. J. Neural Transm. 2018, 125, 1751–1757. [Google Scholar] [CrossRef]
- Tipton, K.F.; Davey, G.P.; McDonald, A.G. Kinetic behavior and reversible inhibition of monoamine oxidases—Enzymes that many want dead. Int. Rev. Neurobiol. 2011, 100, 43–64. [Google Scholar] [CrossRef]
- Zdrazil, B.; Guha, R. The rise and fall of a scaffold: A trend analysis of scaffolds in the medicinal chemistry literature. J. Med. Chem. 2018, 61, 4688–4703. [Google Scholar] [CrossRef]
- Carneiro, A.; Uriarte, E.; Borges, F.; João Matos, M. Propargylamine: An important moiety in drug discovery. Future Med. Chem. 2023, 15, 211–224. [Google Scholar] [CrossRef]
- Zindo, F.T.; Joubert, J.; Malan, S.F. Propargylamine as functional moiety in the design of multifunctional drugs for neurodegenerative disorders: MAO inhibition and beyond. Future Med. Chem. 2015, 7, 609–629. [Google Scholar] [CrossRef]
- Chrienova, Z.; Nepovimova, E.; Andrys, R.; Dolezal, R.; Janockova, J.; Muckova, L.; Fabova, L.; Soukup, O.; Oleksak, P.; Valis, M.; et al. Privileged multi-target directed propargyl-tacrines combining cholinesterase and monoamine oxidase inhibition activities. J. Enzym. Inhib. Med. Chem. 2022, 37, 2605–2620. [Google Scholar] [CrossRef] [PubMed]
- Tandarić, T.; Vianello, R. Computational insight into the mechanism of the irreversible inhibition of monoamine oxidase enzymes by the anti-parkinsonian propargylamine inhibitors rasagiline and selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Tandarić, T.; Prah, A.; Stare, J.; Mavri, J.; Vianello, R. Hydride abstraction as the rate-limiting step of the irreversible inhibition of monoamine oxidase B by rasagiline and selegiline: A computational empirical valence bond study. Int. J. Mol. Sci. 2020, 21, 6151. [Google Scholar] [CrossRef]
- Maršavelski, A.; Vianello, R. What a difference a methyl group makes: The selectivity of monoamine oxidase B towards histamine and N-methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef] [PubMed]
- Beč, A.; Vianello, R.; Hranjec, M. Synthesis and spectroscopic characterization of multifunctional D-π-A benzimidazole derivatives as potential pH sensors. J. Mol. Liq. 2023, 386, 122493. [Google Scholar] [CrossRef]
- Beč, A.; Racane, L.; Žonja, L.; Persoons, L.; Daelemans, D.; Starčević, K.; Vianello, R.; Hranjec, M. Biological evaluation of novel amidino substituted coumarin-benzazole hybrids as promising therapeutic agents. RSC Med. Chem. 2023, 14, 957–968. [Google Scholar] [CrossRef]
- Boček, I.; Hok, L.; Persoons, L.; Daelemans, D.; Vianello, R.; Hranjec, M. Imidazo[4,5-b]pyridine derived tubulin polymerization inhibitors: Design, synthesis, biological activity in vitro and computational analysis. Bioorg. Chem. 2022, 127, 106032. [Google Scholar] [CrossRef]
- Akwu, N.A.; Lekhooa, M.; Deqiang, D.; Aremu, A.O. Antidepressant effects of coumarins and their derivatives: A critical analysis of research advances. Eur. J. Pharmacol. 2023, 956, 175958. [Google Scholar] [CrossRef]
- Pisani, L.; Catto, M.; Muncipinto, G.; Nicolotti, O.; Carrieri, A.; Rullo, M.; Stefanachi, A.; Leonetti, F.; Altomare, C.A. A twenty-year journey exploring coumarin-based derivatives as bioactive molecules. Front. Chem. 2022, 10, 1002547. [Google Scholar] [CrossRef]
- Lv, Y.; Zheng, Z.; Liu, R.; Guo, J.; Zhang, C.; Xie, Y. Monoamine oxidase B inhibitors based on natural privileged scaffolds: A review of systematically structural modification. Int. J. Biol. Macromol. 2023, 251, 126158. [Google Scholar] [CrossRef]
- Kecel-Gunduz, S.; Budama-Kilinc, Y.; Gok, B.; Bicak, B.; Akman, G.; Arvas, B.; Aydogan, F.; Yolacan, C. Computer-aided anticancer drug design: In vitro and in silico studies of new iminocoumarin derivative. J. Mol. Struct. 2021, 1239, 130539. [Google Scholar] [CrossRef]
- Košak, U.; Knez, D.; Coquelle, N.; Brus, B.; Pišlar, A.; Nachon, F.; Brazzolotto, X.; Kos, J.; Colletier, J.-P.; Gobec, S. N-Propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: Potential multifunctional anti-Alzheimer’s agents. Bioorg. Med. Chem. 2017, 25, 633–645. [Google Scholar] [CrossRef]
- Bhawna Kumar, A.; Bhatia, M.; Kapoor, A.; Kumar, P.; Kumar, S. Monoamine oxidase inhibitors: A concise review with special emphasis on structure activity relationship studies. Eur. J. Med. Chem. 2022, 242, 114655. [Google Scholar] [CrossRef]
- Prins, L.H.A.; Petzer, J.P.; Malan, S.F. Inhibition of monoamine oxidase by indole and benzofuran derivatives. Eur. J. Med. Chem. 2021, 45, 4458–4466. [Google Scholar] [CrossRef] [PubMed]
- Elsherbeny, M.H.; Kim, J.; Gouda, N.A.; Gotina, L.; Cho, J.; Pae, A.N.; Lee, K.; Park, K.D.; Elkamhawy, A.; Roh, E.J. Highly potent, selective and competitive indole-based MAO-B inhibitors protect PC12 cells against 6-hydroxydopamine- and rotenone-induced oxidative stress. Antioxidants 2021, 10, 1641. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Jawaid Akhtar, M.; Al Balushi, K.A.; Alam Khan, S. Rational drug design strategies for the development of promising multi-target directed indole hybrids as Anti-Alzheimer agents. Bioorg. Chem. 2022, 127, 105941. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martínez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, as a new multi-target directed propargylamine for Alzheimer’s disease therapy. Front. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, O.M.; Budni, J.; Mina, F.; Behenck Medeiros, E.; Deuther-Conrad, W.; Entrena, J.M.; Moraleda, I.; Iriepa, I.; López-Muñoz, F.; Marco-Contelles, J. Contilisant, a tetratarget small molecule for Alzheimer’s disease therapy combining cholinesterase, monoamine oxidase inhibition, and H3R antagonism with S1R agonism profile. J. Med. Chem. 2018, 61, 6937–6943. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Basile, L.; Maniquet, A.; Hagenow, S.; Pappalardo, M.; Saija, M.C.; Bryant, S.D.; Albreht, A.; Guccione, S. Parameters for irreversible inactivation of monoamine oxidase. Molecules 2020, 25, 5908. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol. Inform. 2021, 31, 114–122. [Google Scholar] [CrossRef]
- Li, Y.; Qiang, X.; Luo, L.; Yang, X.; Xiao, G.; Zheng, Y.; Cao, Z.; Sang, Z.; Su, F.; Deng, Y. Multitarget drug design strategy against Alzheimer’s disease: Homoisoflavonoid Mannich base derivatives serve as acetylcholinesterase and monoamine oxidase B dual inhibitors with multifunctional properties. Bioorg. Med. Chem. 2017, 25, 714–726. [Google Scholar] [CrossRef]
- Jo, G.; Ahn, S.; Kim, B.-G.; Park, H.R.; Kim, Y.H.; Choo, H.A.; Koh, D.; Chong, Y.; Ahn, J.-H.; Lim, Y. Chromenylchalcones with inhibitory effects on monoamine oxidase B. Bioorg. Med. Chem. 2013, 21, 7890–7897. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Binda, C.; Mattevi, A.; Edmondson, D.E. Functional role of the “aromatic cage” in human monoamine oxidase B: Structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 2006, 45, 4775–4784. [Google Scholar] [CrossRef] [PubMed]
- Borštnar, R.; Repič, M.; Kamerlin, S.C.L.; Vianello, R.; Mavri, J. Computational study of the pKa values of potential catalytic residues in the active site of monoamine oxidase B. J. Chem. Theory Comput. 2012, 8, 3864–3870. [Google Scholar] [CrossRef] [PubMed]
- Geha, R.M.; Rebrin, I.; Chen, K.; Shih, J.C. Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J. Biol. Chem. 2001, 276, 9877–9882. [Google Scholar] [CrossRef]
- Ramsay, R.R. Inhibitor design for monoamine oxidases. Curr. Pharm. Des. 2013, 19, 2529–2539. [Google Scholar] [CrossRef]
- Albreht, A.; Vovk, I.; Mavri, J.; Marco-Contelles, J.; Ramsay, R.R. Evidence for a cyanine link between propargylamine drugs and monoamine oxidase clarifies the inactivation mechanism. Front. Chem. 2018, 6, 169. [Google Scholar] [CrossRef]
- Binda, C.; Hubalek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D.E.; Mattevi, A. Binding of rasagiline-related inhibitors to human monoamine oxidases: A kinetic and crystallographic analysis. J. Med. Chem. 2005, 48, 8148–8154. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Prediction of ligand binding mode mong multiple cross-docking poses by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2020, 34, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Mateev, E.; Valkova, I.; Angelov, B.; Georgieva, M.; Zlatkov, A. Validation through Re-Docking, Cross-Docking and Ligand Enrichment in various Well-Resoluted MAO-B Receptors. Int. J. Pharm. Sci. Res. 2022, 13, 1099–1107. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2020.4 Manual. Available online: https://zenodo.org/records/3923644 (accessed on 1 October 2023).
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N∙log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Fierro, A.; Edmondson, D.E.; Celis-Barros, C.; Rebolledo-Fuentes, M.; Zapata-Torres, G. Why p-OMe- and p-Cl-β-methylphenethylamines display distinct activities upon MAO-B binding. PLoS ONE 2016, 11, e0154989. [Google Scholar] [CrossRef]
- Hok, L.; Vianello, R. Selective deuteration improves the affinity of adenosine A2A receptor ligands: A computational case study with istradefylline and caffeine. J. Chem. Inf. Model. 2012, 63, 3138–3149. [Google Scholar] [CrossRef]
- Mehić, E.; Hok, L.; Wang, Q.; Dokli, I.; Svetec Miklenić, M.; Findrik Blažević, Z.; Tang, L.; Vianello, R.; Majerić Elenkov, M. Expanding the scope of enantioselective halohydrin dehalogenases—Group B. Adv. Synth. Catal. 2022, 364, 2576–2588. [Google Scholar] [CrossRef]
- Himo, F.; Sheng, X. The quantum chemical cluster approach in biocatalysis. Acc. Chem. Res. 2023, 56, 938–947. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Brás, N.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. WIREs Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
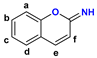
| Scaffold | Position | –N(X)– a | Binding | Mode of Binding | Active Site Binding |
|---|---|---|---|---|---|
 2 | a | N–H | −6.8 | nonproductive | – |
| N–Me | −6.9 | nonproductive | – | ||
| b | N–H | −7.0 | nonproductive | −6.6 | |
| N–Me | −7.4 | nonproductive | −6.9 | ||
| c | N–H | −6.4 | nonproductive | – | |
| N–Me | −5.9 | nonproductive | – | ||
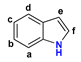 4 | a | N–H | −5.9 | nonproductive | – |
| N–Me | −6.4 | active site | |||
| b | N–H | −6.2 | nonproductive | −6.2 | |
| N–Me | −5.9 | nonproductive | – | ||
| c | N–H | −6.8 | active site | ||
| N–Me | −6.6 | active site | |||
| d | N–H | −6.5 | active site | ||
| N–Me | −6.0 | nonproductive | – | ||
| e | N–H | −5.7 | nonproductive | – | |
| N–Me | −5.9 | nonproductive | – | ||
| f | N–H | −7.5 | active site | ||
| N–Me | −6.3 | active site | |||
 7 | a | N–H | −7.8 | active site | |
| N–Me | −6.4 | nonproductive | – | ||
| b | N–H | −6.4 | nonproductive | −6.3 | |
| N–Me | −6.4 | nonproductive | – | ||
 16 | a | N–H | −6.0 | nonproductive | – |
| N–Me | −6.3 | nonproductive | – | ||
| b | N–H | −6.2 | active site | – | |
| N–Me | −6.1 | nonproductive | – | ||
| c | N–H | −6.9 | active site | – | |
| N–Me | −5.7 | nonproductive | – | ||
| d | N–H | −6.3 | nonproductive | – | |
| N–Me | −6.0 | nonproductive | – | ||
| e | N–H | −6.6 | active site | – | |
| N–Me | −6.1 | nonproductive | – | ||
| f | N–H | −6.2 | nonproductive | – | |
| N–Me | −5.8 | nonproductive | – | ||
 25 | a | N–H | −6.6 | nonproductive | – |
| N–Me | −6.2 | nonproductive | – | ||
| b | N–H | −6.3 | nonproductive | – | |
| N–Me | −6.8 | nonproductive | – | ||
| c | N–H | −7.1 | nonproductive | −6.9 | |
| N–Me | −6.5 | nonproductive | – | ||
| d | N–H | −6.7 | nonproductive | – | |
| N–Me | −6.1 | nonproductive | – | ||
| e | N–H | −7.1 | active site | ||
| N–Me | −6.2 | nonproductive | −5.4 | ||
| f | N–H | −6.2 | nonproductive | – | |
| N–Me | −6.3 | nonproductive | – | ||
| Rasagiline RAS | −5.9 | active site | |||
| Selegiline SEL | −6.2 | active site |
| Ligand | SEL | RAS | 4fH | 4fMe | 16cH |
|---|---|---|---|---|---|
| ΔGBIND | −20.1 | −18.1 | −20.0 | −21.6 | −19.7 |
| FAD | −2.87 | −3.00 | −4.63 | −4.84 | −3.13 |
| Tyr398 | −2.43 | −1.70 | −2.07 | −2.13 | −2.44 |
| Tyr435 | −2.26 | −2.43 | −2.31 | −2.25 | −2.15 |
| Gln206 | −1.81 | −1.62 | −2.21 | −2.56 | −1.37 |
| Leu171 | −1.15 | −1.81 | −0.96 | −0.89 | −0.96 |
| Cys172 | −1.15 | −0.99 | −0.59 | −0.76 | −0.83 |
| Ile199 | −1.00 | −0.57 | −0.98 | −1.06 | −1.45 |
| Phe343 | −1.00 | −0.80 | −0.98 | −1.19 | −0.28 |
| Tyr326 | −0.80 | −0.90 | −0.74 | −0.69 | −2.52 |
| Ile198 | −0.63 | −0.75 | −0.18 | −0.34 | −0.29 |
| Tyr60 | −0.56 | −0.19 | −0.75 | −1.01 | −0.27 |
| Tyr188 | −0.51 | −0.40 | −0.22 | −0.29 | −0.19 |
| Gly434 | −0.48 | −0.35 | −0.44 | −0.32 | −0.41 |
| Leu328 | −0.32 | −0.17 | −0.37 | −0.39 | −0.43 |
| Trp432 | −0.20 | −0.13 | −0.16 | −0.15 | −0.14 |
| Val173 | −0.19 | −0.11 | −0.14 | −0.18 | −0.10 |
| Thr399 | −0.19 | −0.09 | −0.14 | −0.10 | −0.06 |
| Cys192 | −0.18 | −0.08 | −0.05 | −0.06 | −0.08 |
| Gly205 | −0.14 | −0.10 | −0.25 | −0.21 | −0.13 |
| Met341 | −0.13 | −0.07 | −0.10 | −0.30 | −0.06 |
| Phe168 | −0.11 | −0.31 | −0.07 | −0.09 | −0.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrban, L.; Vianello, R. Prominent Neuroprotective Potential of Indole-2-N-methylpropargylamine: High Affinity and Irreversible Inhibition Efficiency towards Monoamine Oxidase B Revealed by Computational Scaffold Analysis. Pharmaceuticals 2024, 17, 1292. https://doi.org/10.3390/ph17101292
Vrban L, Vianello R. Prominent Neuroprotective Potential of Indole-2-N-methylpropargylamine: High Affinity and Irreversible Inhibition Efficiency towards Monoamine Oxidase B Revealed by Computational Scaffold Analysis. Pharmaceuticals. 2024; 17(10):1292. https://doi.org/10.3390/ph17101292
Chicago/Turabian StyleVrban, Lucija, and Robert Vianello. 2024. "Prominent Neuroprotective Potential of Indole-2-N-methylpropargylamine: High Affinity and Irreversible Inhibition Efficiency towards Monoamine Oxidase B Revealed by Computational Scaffold Analysis" Pharmaceuticals 17, no. 10: 1292. https://doi.org/10.3390/ph17101292