

An Antagonist Antibody That Inhibits Cancer Cell Growth In Vitro through RACK1

Abstract
:1. Introduction
2. Results
2.1. Selection System for Inducing G0/G1 Tumor Cell Cycle Arrest
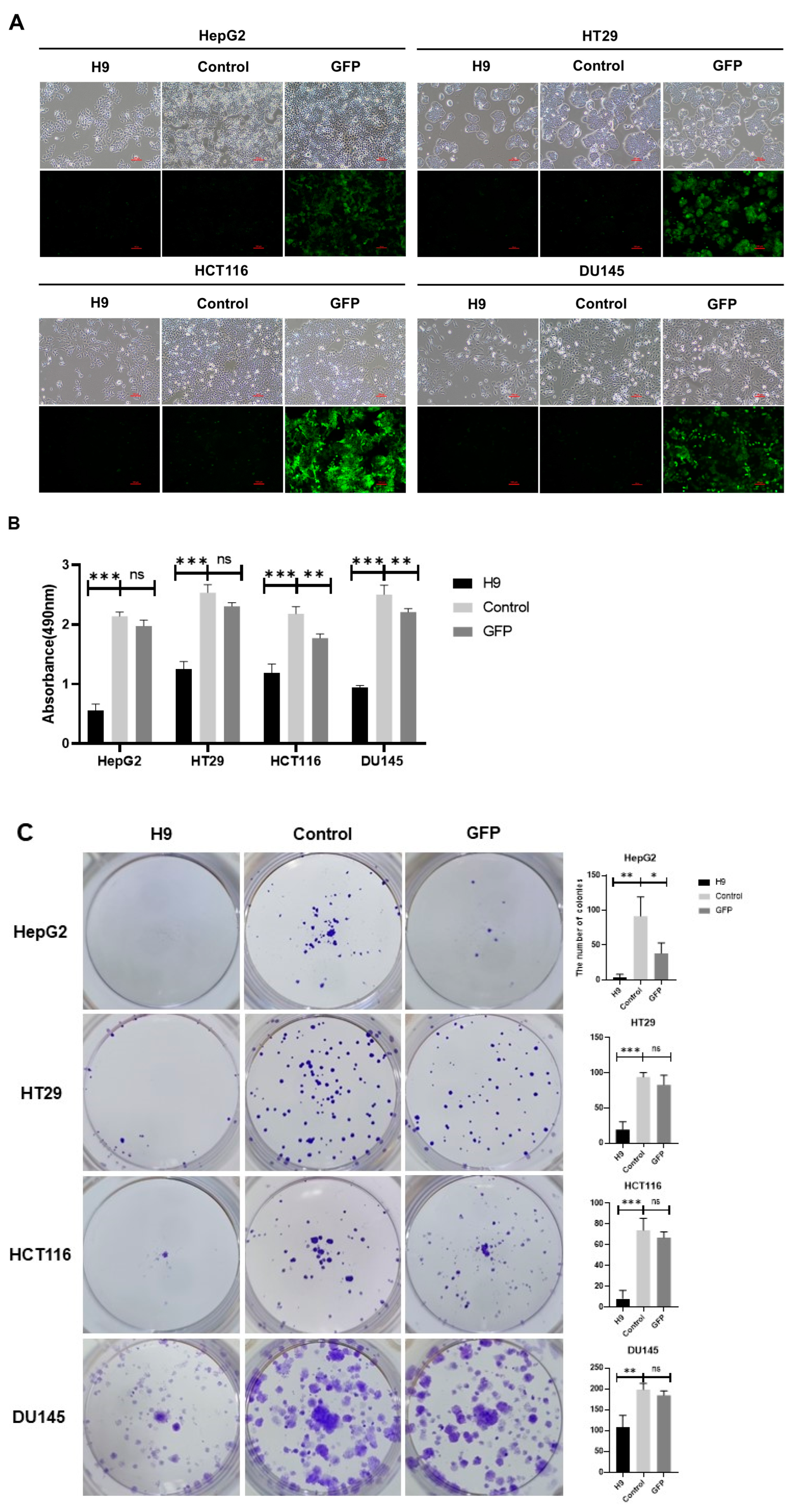
2.2. Selected H9 Antibody Inhibits Proliferation and Migration of Four Cancer Cell Lines
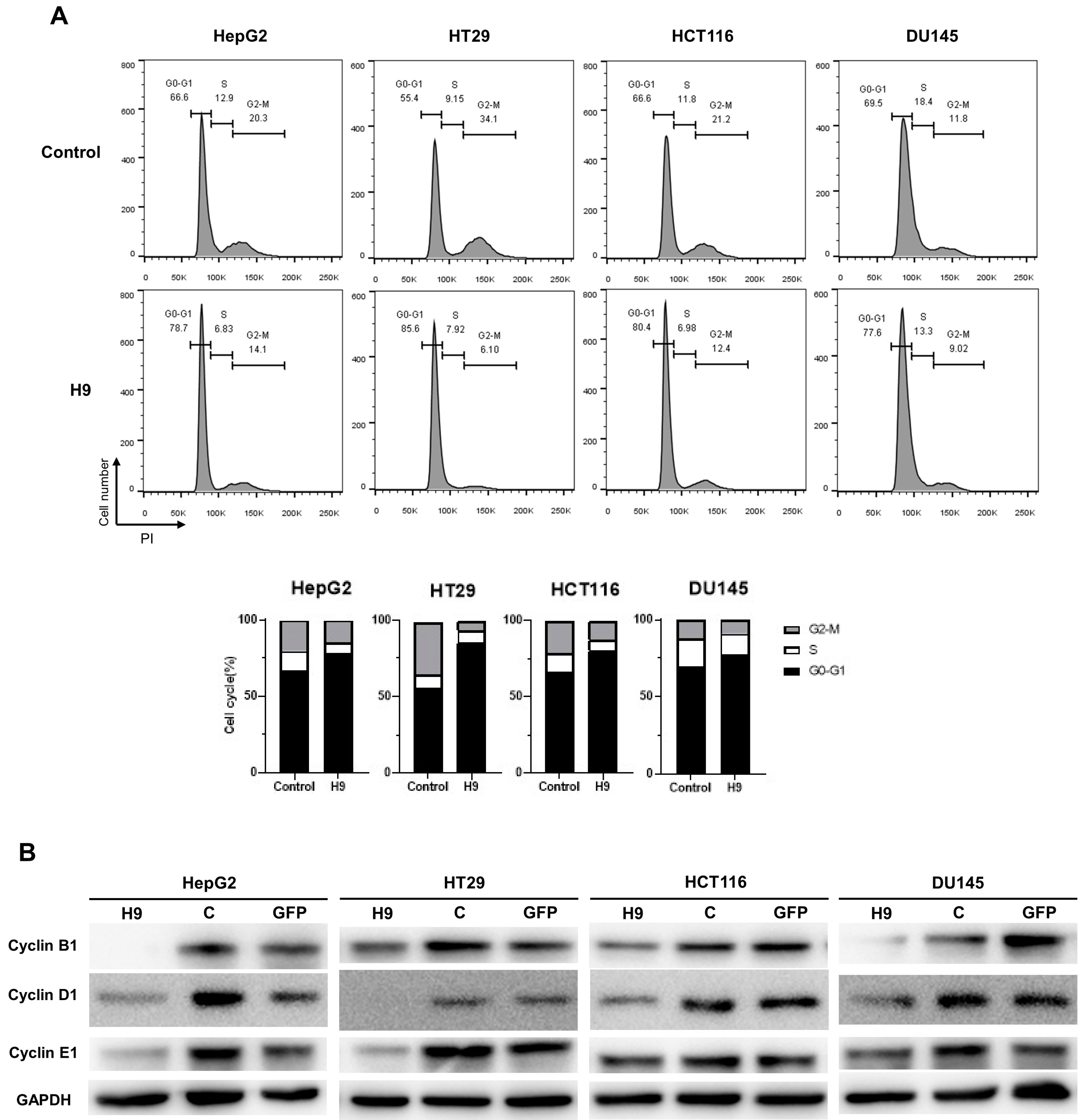
2.3. Selected H9 Antibody Induces G0/G1 Phase Cell Cycle Arrest
2.4. Selected H9 Antibody Induces Apoptosis
2.5. Identification of a Target Protein
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Media
4.2. Expression and Purification of ScFv-Fc Antibodies
4.3. Lentivirus Transfection
4.4. RNA Interference
4.5. Cell Viability Assay
4.6. Colony Formation Assay
4.7. Wound Healing Assay
4.8. Immunoblotting
4.9. Flow Cytometry
4.10. Immunoprecipitation and Liquid Chromatography–Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Kang, M.J.; Park, E.H.; Yun, E.H.; Kim, H.J.; Kong, H.J.; Im, J.S.; Seo, H.G. Prediction of Cancer Incidence and Mortality in Korea, 2023. Cancer Res. Treat. 2023, 55, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Kim, H.C.; Choi, C.M. Recent Trends of Lung Cancer in Korea. Tuberc. Respir. Dis. 2021, 84, 89–95. [Google Scholar] [CrossRef]
- Zordan, R.; Manitta, V.; Nandurkar, H.; Cole-Sinclair, M.; Philip, J. Prevalence and predictors of fatigue in haemo-oncological patients. Intern. Med. J. 2014, 44, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Majidpoor, J.; Toolee, H.; Mortezaee, K. The current knowledge concerning solid cancer and therapy. J. Biochem. Mol. Toxicol. 2021, 35, e22900. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Park, H.W.; Han, K.H. Antibodies Regulate Dual-Function Enzyme IYD to Induce Functional Synergy between Metabolism and Thermogenesis. Int. J. Mol. Sci. 2022, 23, 7834. [Google Scholar] [CrossRef]
- Han, K.H.; Arlian, B.M.; Lin, C.W.; Jin, H.Y.; Kang, G.H.; Lee, S.; Lee, P.C.; Lerner, R.A. Agonist Antibody Converts Stem Cells into Migrating Brown Adipocyte-Like Cells in Heart. Cells 2020, 9, 256. [Google Scholar] [CrossRef]
- Han, K.H.; Arlian, B.M.; Macauley, M.S.; Paulson, J.C.; Lerner, R.A. Migration-based selections of antibodies that convert bone marrow into trafficking microglia-like cells that reduce brain amyloid beta. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Gonzalez-Quintial, R.; Peng, Y.; Baccala, R.; Theofilopoulos, A.N.; Lerner, R.A. An agonist antibody that blocks autoimmunity by inducing anti-inflammatory macrophages. FASEB J. 2016, 30, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Lerner, R.A. Antibody Libraries as Tools to Discover Functional Antibodies and Receptor Pleiotropism. Int. J. Mol. Sci. 2021, 22, 4123. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Baek, S.H.; Song, C.H.; Choi, Y.H.; Han, K.H. Agonist (P1) Antibody Converts Stem Cells into Migrating Beta-Like Cells in Pancreatic Islets. J. Microbiol. Biotechnol. 2022, 32, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Chih-Wei Lin, K.H.H. Antibody Libraries as Platforms to Exploring Target and Receptor Pleiotropy. Isr. J. Chem. 2023, 63. [Google Scholar] [CrossRef]
- Levy, A.; Albiges-Sauvin, L.; Massard, C.; Soria, J.C.; Deutsch, E. Cell cycle, mitosis and therapeutic applications. Bull. Cancer 2011, 98, 1037–1045. [Google Scholar] [CrossRef]
- Nakanishi, M. Cell cycle checkpoints and cancer. Tanpakushitsu Kakusan Koso 2009, 54, 556–560. [Google Scholar] [PubMed]
- Alfieri, R.; Merelli, I.; Mosca, E.; Milanesi, L. The cell cycle DB: A systems biology approach to cell cycle analysis. Nucleic Acids Res. 2008, 36, D641–D645. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P.; Masui, Y.; Hartwell, L. Understanding the cell cycle. Nat. Med. 1998, 4, 1103–1106. [Google Scholar] [CrossRef]
- Al-Ali, H.N.; Crichton, S.J.; Fabian, C.; Pepper, C.; Butcher, D.R.; Dempsey, F.C.; Parris, C.N. A therapeutic antibody targeting annexin-A1 inhibits cancer cell growth in vitro and in vivo. Oncogene 2024, 43, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Song, S.; Yan, A.; Guo, X.; Chang, L.; Xu, L.; Hu, L.; Kuang, M.; Liu, B.; He, D.; et al. RACK1 promotes the invasive activities and lymph node metastasis of cervical cancer via galectin-1. Cancer Lett. 2020, 469, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Hua, H.; Gu, L.; Cao, S.; Cai, H.; Yao, X.; Chen, X. Overexpression of RACK1 Predicts Poor Prognosis in Melanoma. J. Cancer 2020, 11, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lv, Y.; Rong, Y.; Chen, W.; Fang, Y.; Mao, W.; Lou, W.; Jin, D.; Xu, X. Downregulated expression of RACK1 results in pancreatic cancer growth and metastasis. Onco Targets Ther. 2019, 12, 1007–1020. [Google Scholar] [CrossRef]
- Xiao, T.; Zhu, W.; Huang, W.; Lu, S.S.; Li, X.H.; Xiao, Z.Q.; Yi, H. RACK1 promotes tumorigenicity of colon cancer by inducing cell autophagy. Cell Death Dis. 2018, 9, 1148. [Google Scholar] [CrossRef]
- Duff, D.; Long, A. Roles for RACK1 in cancer cell migration and invasion. Cell Signal 2017, 35, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Yong-Zheng, X.; Wan-Li, M.; Ji-Ming, M.; Xue-Qun, R. Receptor for activated protein kinase C 1 suppresses gastric tumor progression through nuclear factor-kB pathway. Indian. J. Cancer 2015, 52 (Suppl. 3), E172–E175. [Google Scholar] [CrossRef]
- Shen, F.; Yan, C.; Liu, M.; Feng, Y.; Chen, Y. RACK1 promotes prostate cancer cell proliferation, invasion and metastasis. Mol. Med. Rep. 2013, 8, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Z.; Yao, F.; Li, J.J.; Mao, Z.F.; Hu, P.T.; Long, L.Y.; Li, G.; Ji, X.D.; Shi, S.; Guan, D.X.; et al. RACK1 suppresses gastric tumorigenesis by stabilizing the beta-catenin destruction complex. Gastroenterology 2012, 142, 812–823.e15. [Google Scholar] [CrossRef] [PubMed]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T therapy beyond cancer: The evolution of a living drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef]
- Hamdan, F.; Fusciello, M.; Cerullo, V. Personalizing Oncolytic Virotherapy. Hum. Gene Ther. 2023, 34, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Lin, E.Z.; Anekoji, M.; Ichim, T.E.; Hu, J.; Marincola, F.M.; Jones, L.D.; Kesari, S.; Ashili, S. Advancing personalized medicine in brain cancer: Exploring the role of mRNA vaccines. J. Transl. Med. 2023, 21, 830. [Google Scholar] [CrossRef] [PubMed]
- Plana, D.; Palmer, A.C.; Sorger, P.K. Independent Drug Action in Combination Therapy: Implications for Precision Oncology. Cancer Discov. 2022, 12, 606–624. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Majidpoor, J. Checkpoint inhibitor/interleukin-based combination therapy of cancer. Cancer Med. 2022, 11, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Ettrich, T.J.; Seufferlein, T. Systemic Therapy for Metastatic Pancreatic Cancer. Curr. Treat. Options Oncol. 2021, 22, 106. [Google Scholar] [CrossRef]
- Moloudi, K.; Abrahamse, H.; George, B.P. Photodynamic therapy induced cell cycle arrest and cancer cell synchronization: Review. Front. Oncol. 2023, 13, 1225694. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Song, J.; Tang, Z.; Wei, S.; Chen, L.; Zhou, R. Hypericin-mediated photodynamic therapy inhibits growth of colorectal cancer cells via inducing S phase cell cycle arrest and apoptosis. Eur. J. Pharmacol. 2021, 900, 174071. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wang, X.; Wang, H.; Zhong, L.; Zou, D. Advances in ATM, ATR, WEE1, and CHK1/2 inhibitors in the treatment of PARP inhibitor-resistant ovarian cancer. Cancer Biol. Med. 2024, 20, 915–921. [Google Scholar] [CrossRef]
- Sofianidi, A.; Dumbrava, E.E.; Syrigos, K.N.; Nasrazadani, A. Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers 2024, 16, 1139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Cowan, C.M.; Coutts, J.A.; Christy, D.D.; Saraph, A.; Hsueh, S.C.C.; Plotkin, S.S.; Mackenzie, I.R.; Kaplan, J.M.; Cashman, N.R. Targeting RACK1 to alleviate TDP-43 and FUS proteinopathy-mediated suppression of protein translation and neurodegeneration. Acta Neuropathol. Commun. 2023, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Han, Y.; Wang, T. RACK1 modulates polyglutamine-induced neurodegeneration by promoting ERK degradation in Drosophila. PLoS Genet. 2021, 17, e1009558. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J. Innate immune genes of the chicken MHC and related regions. Immunogenetics 2022, 74, 167–177. [Google Scholar] [CrossRef]
- Shen, C.; Hua, H.; Gu, L.; Cao, S.; Cai, H.; Yao, X.; Chen, X. miR-124 Functions As A Melanoma Tumor Suppressor By Targeting RACK1. Onco Targets Ther. 2019, 12, 9975–9986. [Google Scholar] [CrossRef] [PubMed]
- Kershner, L.; Welshhans, K. RACK1 regulates neural development. Neural Regen. Res. 2017, 12, 1036–1039. [Google Scholar] [CrossRef]
- Guillemot, F.; Billault, A.; Auffray, C. Physical linkage of a guanine nucleotide-binding protein-related gene to the chicken major histocompatibility complex. Proc. Natl. Acad. Sci. USA 1989, 86, 4594–4598. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Wu, H.; Jia, D.; Wu, W.; Ren, S.; Wang, L.; Song, S.; Guo, X.; Liu, F.; Ruan, Y.; et al. O-GlcNAcylation of RACK1 promotes hepatocellular carcinogenesis. J. Hepatol. 2018, 68, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Xu, J.D.; Liu, X.L.; Xu, J.W.; Wang, W.J.; Li, Q.Q.; Chen, Q.; Xu, Z.D.; Liu, X.P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int. J. Cancer 2010, 127, 1172–1179. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| RACK1#1 | 5′ CCAUCAAGCUAUGGAAUACTT 3′ |
| RACK1#2 | 5′ GCUAUGGAAUACCCUGGGUTT 3′ |
| RACK1#3 | 5′ CCUUUACACGCUAGAUGGUTT 3′ |
| RACK1#4 | 5′ GACAUCAUCAUGUGGAAGCTT 3′ |
| siNC | 5′ GACCAUCAUCAUGUGGAAGCTT 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.H.; Lee, E.J.; Han, K.H. An Antagonist Antibody That Inhibits Cancer Cell Growth In Vitro through RACK1. Pharmaceuticals 2024, 17, 1303. https://doi.org/10.3390/ph17101303
Kim JH, Lee EJ, Han KH. An Antagonist Antibody That Inhibits Cancer Cell Growth In Vitro through RACK1. Pharmaceuticals. 2024; 17(10):1303. https://doi.org/10.3390/ph17101303
Chicago/Turabian StyleKim, Ji Hoe, Eun Ji Lee, and Kyung Ho Han. 2024. "An Antagonist Antibody That Inhibits Cancer Cell Growth In Vitro through RACK1" Pharmaceuticals 17, no. 10: 1303. https://doi.org/10.3390/ph17101303