

GL-V9 Promotes Autophagy-Mediated YAP1 Degradation and Activates Mitochondrial Apoptosis in PDAC Cells


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
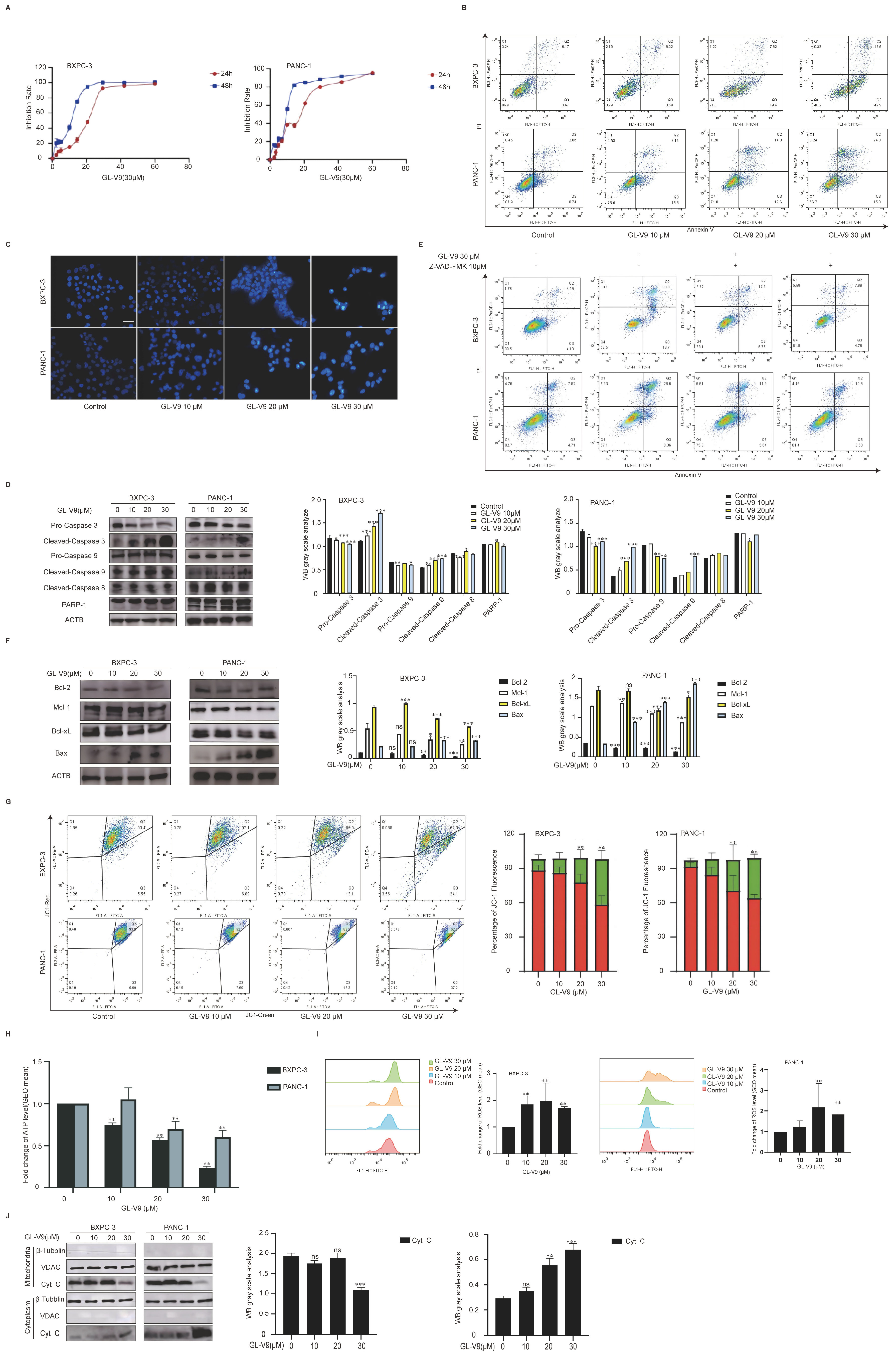
2.1. GL-V9 Exhibits Potent Anticancer Activity in PDAC Cells by Inducing Mitochondrial-Mediated Apoptosis
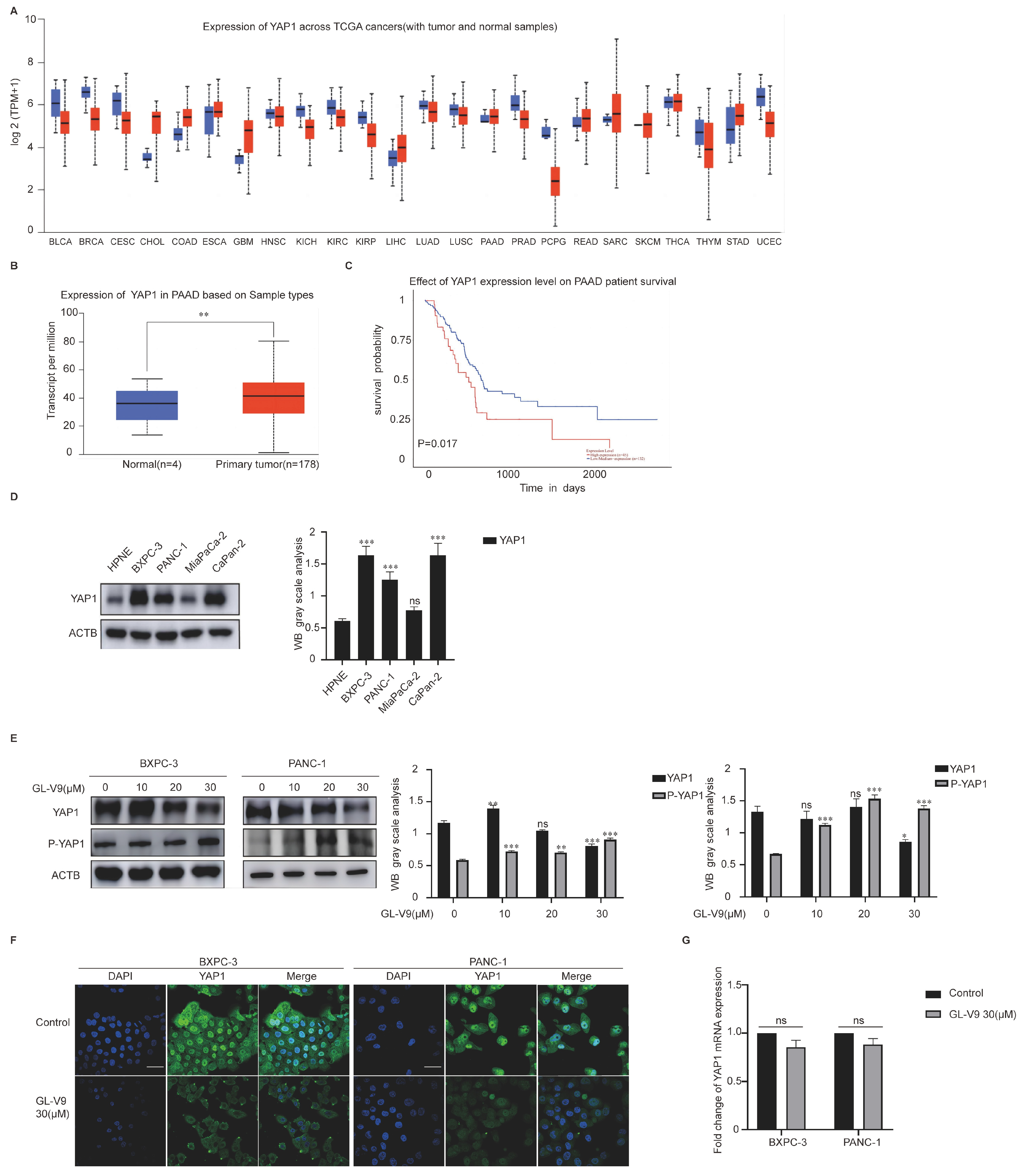
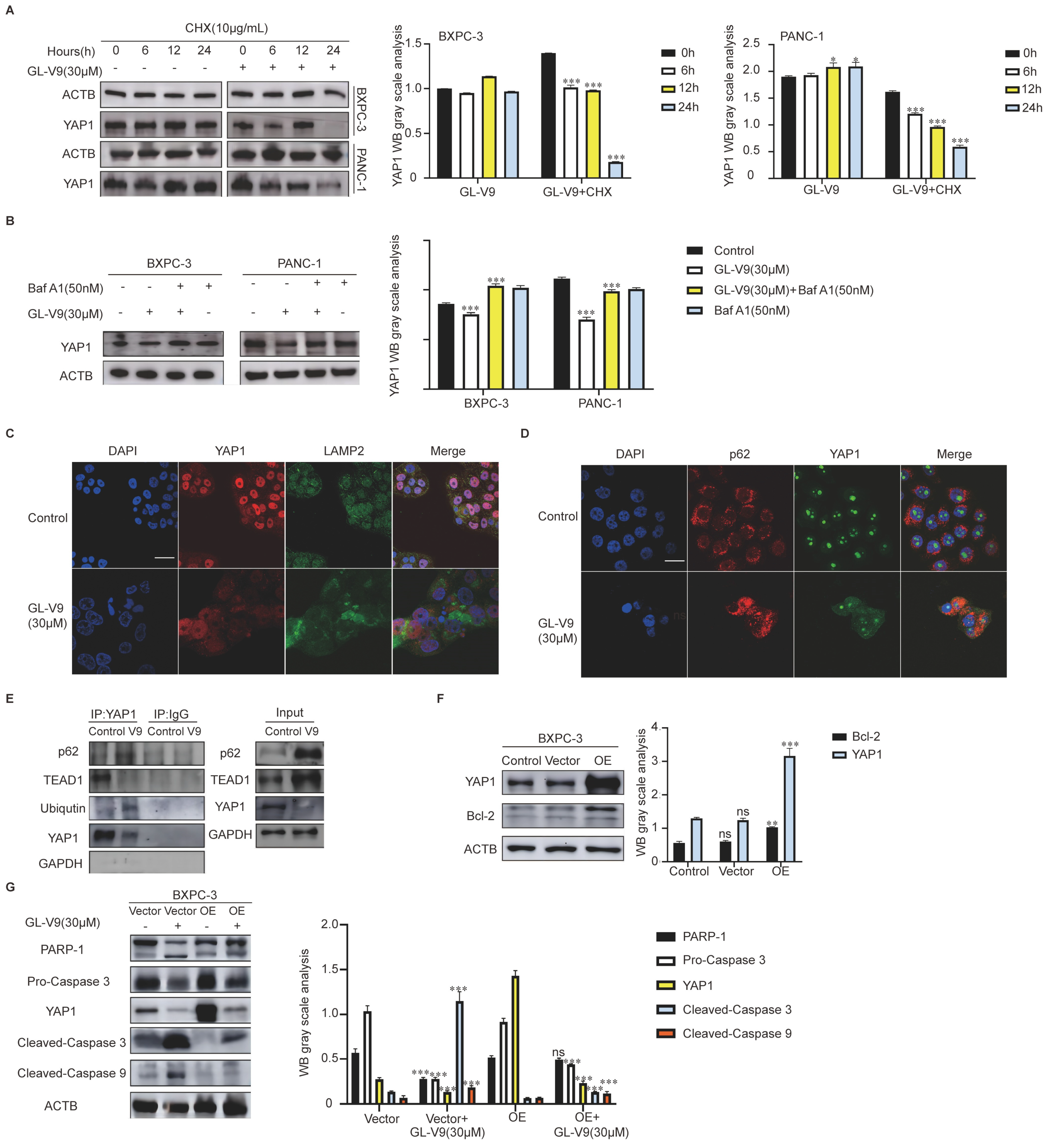
2.2. GL-V9 Induces Apoptosis via Autophagy-Mediated Degradation of YAP1 in PDAC Cells
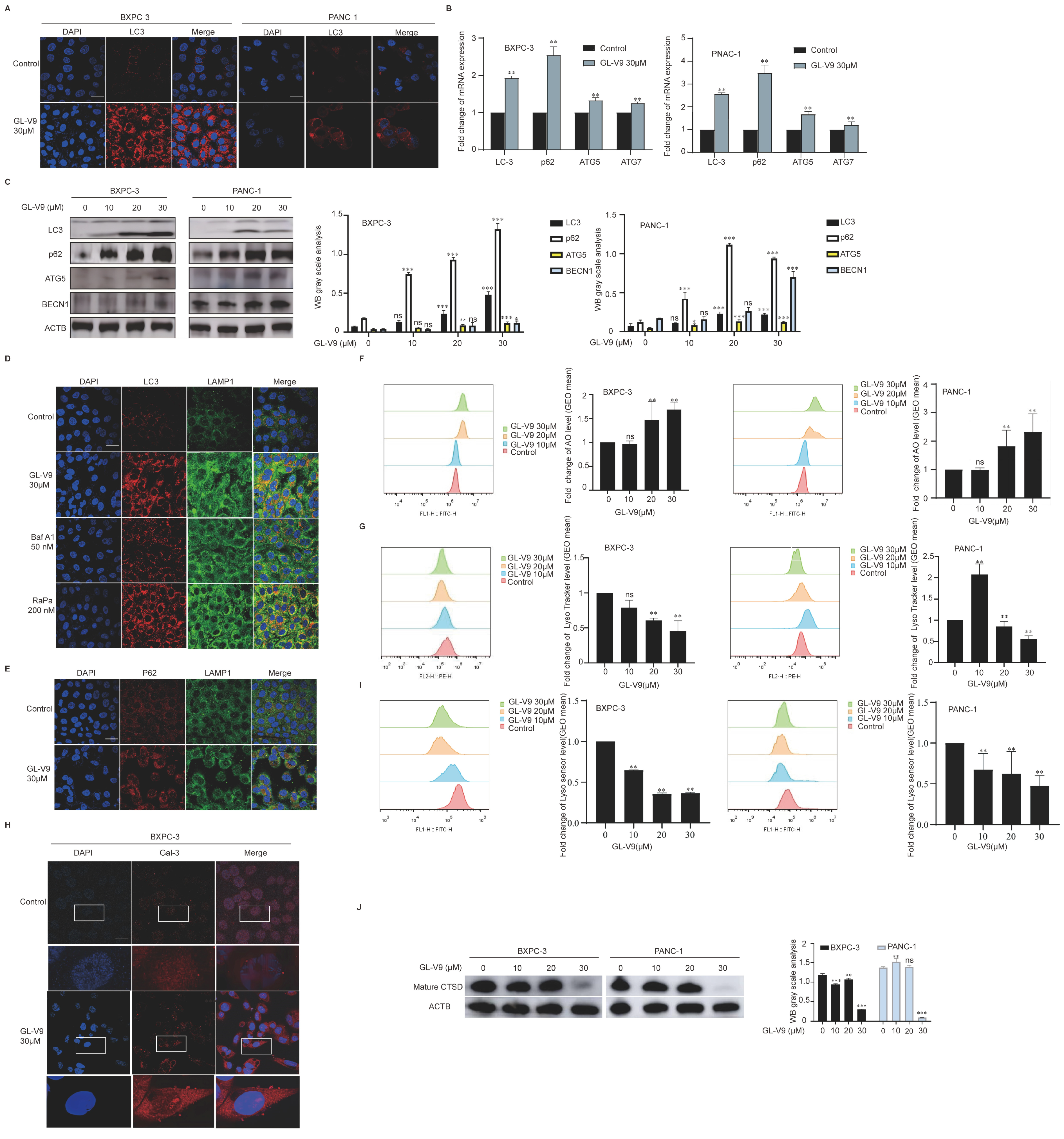
2.3. GL-V9 Induces Autophagosome–Lysosome Fusion and Lysosomal Repair in PDAC Cells
2.4. GL-V9 Induces Mitochondrial Apoptosis by Promoting AKT/mTOR/TFEB-Mediated Autophagy
2.5. GL-V9 Inhibits PDAC Growth In Vivo by Activating Autophagy and Inducing Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Reagents
4.3. PDAC Graft Tumor Model
4.4. Western Blot
4.5. Annexin V/PI Staining Assay
4.6. Mitochondrial Isolation and Protein Extraction
4.7. ROS Measurement
4.8. Detection of the Loss of Mitochondrial Membrane Potential
4.9. ATP Assay
4.10. Real-Time PCR Analysis
| Name | Sequence |
| LC3 forward | AACATGAGCGAGTTGGTCAAG |
| LC3 reverse | GCTCGTAGATGTCCGCGAT |
| p62 forward | GCACCCCAATGTGATCTGC |
| p62 reverse | CGCTACACAAGTCGTAGTCTGG |
| ATG5 forward | AAAGATGTGCTTCGAGATGTGT |
| ATG5 reverse | CACTTTGTCAGTTACCAACGTCA |
| ATG7 forward | ATGATCCCTGTAACTTAGCCCA |
| ATG7 reverse | CACGGAAGCAAACAACTTCAAC |
| GAPDH forward | CGGAGTCAACGGATTTGGTCGTAT |
| GAPDH reverse | AGCCTTCTCCATGGTGGTGAAGAC |
| YAP1 forward | GGATTTCTGCCTTCCCTGAA |
| YAP1 reverse | GATAGCAGGGCGTGAGGAAC |
4.11. Immunofluorescence (IF)
4.12. Tissue Staining
4.13. Co-Immunoprecipitation
4.14. Immunohistochemistry
4.15. Tissue Protein Extraction
4.16. Construction of Stable Transient Cells
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Del Chiaro, M.; Sugawara, T.; Karam, S.D.; Messersmith, W.A. Advances in the management of pancreatic cancer. BMJ 2023, 383, e073995. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Tischner, D.; Manzl, C.; Soratroi, C.; Villunger, A.; Krumschnabel, G. Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis Int. J. Program. Cell Death 2012, 17, 1197–1209. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Jain, V.; Singh, M.P.; Amaravadi, R.K. Recent advances in targeting autophagy in cancer. Trends Pharmacol. Sci. 2023, 44, 290–302. [Google Scholar] [CrossRef]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2022, 80, 1–17. [Google Scholar] [CrossRef]
- Mohamed, E.; Kumar, A.; Zhang, Y.; Wang, A.S.; Chen, K.; Lim, Y.; Shai, A.; Taylor, J.W.; Clarke, J.; Hilz, S.; et al. PI3K/AKT/mTOR signaling pathway activity in IDH-mutant diffuse glioma and clinical implications. Neuro-Oncology 2022, 24, 1471–1481. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yue, H.; Huang, X.; Wang, J.; Li, Z.; Deng, X. Novel Platinum Nanoclusters Activate PI3K/AKT/mTOR Signaling Pathway-Mediated Autophagy for Cisplatin-Resistant Ovarian Cancer Therapy. ACS Appl. Mater. Interfaces 2022, 14, 48502–48514. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yuan, W.; Li, Y.; Luo, J.; Hou, N. Role of Hippo-YAP1/TAZ pathway and its crosstalk in cardiac biology. Int. J. Biol. Sci. 2020, 16, 2454–2463. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Franklin, J.M.; Wu, Z.; Guan, K.L. Insights into recent findings and clinical application of YAP and TAZ in cancer. Nat. Rev. Cancer 2023, 23, 512–525. [Google Scholar] [CrossRef]
- Eibl, G.; Rozengurt, E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin. Cancer Biol. 2019, 54, 50–62. [Google Scholar] [CrossRef]
- Morvaridi, S.; Dhall, D.; Greene, M.I.; Pandol, S.J.; Wang, Q. Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci. Rep. 2015, 5, 16759. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Liu, J.; Guo, Y.; Zhang, R.; Xu, Y.; Luo, C.; Wang, R.; Xu, S.; Wei, L. Inhibition of TRPV4 remodels single cell polarity and suppresses the metastasis of hepatocellular carcinoma. Cell Death Dis. 2023, 14, 379. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, L.; Zhou, Y.; Lu, N.; Tang, X.; Li, Z.; Wang, X. Flavonoid GL-V9 induces apoptosis and inhibits glycolysis of breast cancer via disrupting GSK-3β-modulated mitochondrial binding of HKII. Free Radic. Biol. Med. 2020, 146, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Li, H.; Yu, X.; Liu, X.; Wang, X.; Qing, Y.; Wang, Z.; Wang, H.; Zhu, M.; Xu, J.; et al. GL-V9 exerts anti-T cell malignancies effects via promoting lysosome-dependent AKT1 degradation and activating AKT1/FOXO3A/BIM axis. Free Radic. Biol. Med. 2019, 145, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; He, Z.; Yao, J.; Tan, R.; Zhu, Y.; Li, Z.; Guo, Q.; Wei, L. Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells. Redox Biol. 2018, 17, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, N.; Dai, Q.; Wei, L.; Zhao, Q.; Li, Z.; He, Q.; Dai, Y.; Guo, Q. GL-V9, a newly synthetic flavonoid derivative, induces mitochondrial-mediated apoptosis and G2/M cell cycle arrest in human hepatocellular carcinoma HepG2 cells. Eur. J. Pharmacol. 2011, 670, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kang, Y.; Huo, T.; Tao, R.; Wang, X.; Li, Z.; Guo, Q.; Zhao, L. GL-V9 induced upregulation and mitochondrial localization of NAG-1 associates with ROS generation and cell death in hepatocellular carcinoma cells. Free Radic. Biol. Med. 2017, 112, 49–59. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Zhao, K.; Zhou, Y.; Li, W.; Pan, C.; Qiang, L.; Li, Z.; Lu, N. Small molecule GL-V9 protects against colitis-associated colorectal cancer by limiting NLRP3 inflammasome through autophagy. Oncoimmunology 2017, 7, e1375640. [Google Scholar] [CrossRef]
- Winter, J.M.; Yadav, T.; Rutter, J. Stressed to death: Mitochondrial stress responses connect respiration and apoptosis in cancer. Mol. Cell 2022, 82, 3321–3332. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Fukunaga, Y.; Fukuda, A.; Omatsu, M.; Namikawa, M.; Sono, M.; Masuda, T.; Araki, O.; Nagao, M.; Yoshikawa, T.; Ogawa, S.; et al. Loss of Arid1a and Pten in Pancreatic Ductal Cells Induces Intraductal Tubulopapillary Neoplasm via the YAP/TAZ Pathway. Gastroenterology 2022, 163, 466–480.e6. [Google Scholar] [CrossRef]
- Strnadel, J.; Choi, S.; Fujimura, K.; Wang, H.; Zhang, W.; Wyse, M.; Wright, T.; Gross, E.; Peinado, C.; Park, H.W.; et al. eIF5A-PEAK1 Signaling Regulates YAP1/TAZ Protein Expression and Pancreatic Cancer Cell Growth. Cancer Res. 2017, 77, 1997–2007. [Google Scholar] [CrossRef]
- Pavel, M.; Park, S.J.; Frake, R.A.; Son, S.M.; Manni, M.M.; Bento, C.F.; Renna, M.; Ricketts, T.; Menzies, F.M.; Tanasa, R.; et al. α-Catenin levels determine direction of YAP/TAZ response to autophagy perturbation. Nat. Commun. 2021, 12, 1703. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.; Yao, J.; Ferri-Borgogno, S.; Zhao, J.; Chen, S.; Wang, Q.; Yan, L.; Zhou, X.; Zhu, C.; Bang, S.; et al. YAP1 oncogene is a context-specific driver for pancreatic ductal adenocarcinoma. JCI Insight 2019, 4, e130811. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Castelli, S.; Dorard, C.; Toifl, S.; Grazi, G.L.; Ciriolo, M.R.; Baccarini, M. Impaired degradation of YAP1 and IL6ST by chaperone-mediated autophagy promotes proliferation and migration of normal and hepatocellular carcinoma cells. Autophagy 2023, 19, 152–162. [Google Scholar] [CrossRef]
- Jin, L.; Chen, Y.; Cheng, D.; He, Z.; Shi, X.; Du, B.; Xi, X.; Gao, Y.; Guo, Y. YAP inhibits autophagy and promotes progression of colorectal cancer via upregulating Bcl-2 expression. Cell Death Dis. 2021, 12, 457. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Chen, R.; Zou, J.; Zhong, X.; Li, J.; Kang, R.; Tang, D. HMGB1 in the interplay between autophagy and apoptosis in cancer. Cancer Lett. 2024, 581, 216494. [Google Scholar] [CrossRef]
- Choudhury, D.; Ghosh, D.; Mondal, M.; Singha, D.; Pothuraju, R.; Malakar, P. Polyploidy and mTOR signaling: A possible molecular link. Cell Commun. Signal. 2024, 22, 196. [Google Scholar] [CrossRef]
- Tang, H.; Hou, H.; Song, L.; Tian, Z.; Liu, W.; Xia, T.; Wang, A. The role of mTORC1/TFEB axis mediated lysosomal biogenesis and autophagy impairment in fluoride neurotoxicity and the intervention effects of resveratrol. J. Hazard. Mater. 2024, 467, 133634. [Google Scholar] [CrossRef]
- Seufferlein, T.; Uhl, W.; Kornmann, M.; Algül, H.; Friess, H.; König, A.; Ghadimi, M.; Gallmeier, E.; Bartsch, D.K.; Lutz, M.P.; et al. Perioperative or only adjuvant gemcitabine plus nab-paclitaxel for resectable pancreatic cancer (NEONAX)-a randomized phase II trial of the AIO pancreatic cancer group. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2023, 34, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Rohila, D.; Park, I.H.; Pham, T.V.; Weitz, J.; Hurtado de Mendoza, T.; Madheswaran, S.; Ishfaq, M.; Beaman, C.; Tapia, E.; Sun, S.; et al. Syk Inhibition Reprograms Tumor-Associated Macrophages and Overcomes Gemcitabine-Induced Immunosuppression in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2023, 83, 2675–2689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xie, Y.; Hou, X.; Bai, W.; Li, X.; Liu, Z.; Man, Q.; Sun, J.; Fu, D.; Yan, J.; et al. Irbesartan overcomes gemcitabine resistance in pancreatic cancer by suppressing stemness and iron metabolism via inhibition of the Hippo/YAP1/c-Jun axis. J. Exp. Clin. Cancer Res. 2023, 42, 111. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, X.; Zhang, R.; Sun, Y.; Liu, J.; Luo, C.; Yang, J.; Fang, W.; Guo, Q.; Wei, L. AFP deletion leads to anti-tumorigenic but pro-metastatic roles in liver cancers with concomitant CTNNB1 mutations. Cancer Lett. 2023, 566, 216240. [Google Scholar] [CrossRef]
- Yue, M.; Hu, B.; Li, J.; Chen, R.; Yuan, Z.; Xiao, H.; Chang, H.; Jiu, Y.; Cai, K.; Ding, B. Coronaviral ORF6 protein mediates inter-organelle contacts and modulates host cell lipid flux for virus production. EMBO J. 2023, 42, e112542. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Lin, Z.; Guo, Y.; Zhou, Y.; Li, W. GL-V9 Promotes Autophagy-Mediated YAP1 Degradation and Activates Mitochondrial Apoptosis in PDAC Cells. Pharmaceuticals 2024, 17, 1352. https://doi.org/10.3390/ph17101352
Liu H, Lin Z, Guo Y, Zhou Y, Li W. GL-V9 Promotes Autophagy-Mediated YAP1 Degradation and Activates Mitochondrial Apoptosis in PDAC Cells. Pharmaceuticals. 2024; 17(10):1352. https://doi.org/10.3390/ph17101352
Chicago/Turabian StyleLiu, Hao, Zhangxing Lin, Yongjian Guo, Yuxin Zhou, and Wei Li. 2024. "GL-V9 Promotes Autophagy-Mediated YAP1 Degradation and Activates Mitochondrial Apoptosis in PDAC Cells" Pharmaceuticals 17, no. 10: 1352. https://doi.org/10.3390/ph17101352
APA StyleLiu, H., Lin, Z., Guo, Y., Zhou, Y., & Li, W. (2024). GL-V9 Promotes Autophagy-Mediated YAP1 Degradation and Activates Mitochondrial Apoptosis in PDAC Cells. Pharmaceuticals, 17(10), 1352. https://doi.org/10.3390/ph17101352