
ATP-Binding Cassette and Solute Carrier Transporters: Understanding Their Mechanisms and Drug Modulation Through Structural and Modeling Approaches

 , , , and
, , , and 
Abstract
1. Introduction
2. Membrane Transporters: Key Biological Roles and Implication in Medical Conditions
2.1. ABC Transporters
2.2. SLC Transporters
3. Structure–Function Relationships in Membrane Transporters Based on Structural and Modeling Studies
3.1. Structures, Functions, and Transport Mechanisms of ABC Transporters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter | Substrates | Functions | References |
|---|---|---|---|
| ABCA1 | phospholipids | phospholipid transfer to apolipoproteins | [165,166,167] |
| ABCA3 | phospholipids | transport of phospholipids from the cytoplasm into the lumen side of lamellar bodies; participates in the lamellar bodies biogenesis and homeostasis of pulmonary surfactant | [168] |
| ABCA4 | retinal-phosphatidylethanolamine conjugates | transport of retinal-phosphatidylethanolamine conjugates from the lumen to the cytoplasmic leaflet of photoreceptor outer segment disk membranes | [169,170,171] |
| ABCA7 | phosphatidylserine | lipid homeostasis and macrophage-mediated phagocytosis | [172] |
| ABCB1 | phosphatidylcholine, diverse compounds, xenobiotics, drugs, … | efflux pump responsible for decreased drug accumulation in multidrug-resistant cells | [173,174,175,176,177] |
| ABCB3 | peptide antigens | mediates unidirectional translocation of peptide antigens from cytosol to endoplasmic reticulum | [178] |
| ABCB4 | phosphatidylcholine | floppase translocating phosphatidylcholine from the inner to the outer leaflet of the canalicular membrane bilayer into the canaliculi of hepatocytes | [179,180] |
| ABCB6 | porphyrins | importer of porphyrins from the cytoplasm into the mitochondria | [181,182,183,184] |
| ABCB7 | glutathione-coordinated iron–sulfur cluster | allows assembly of the cytosolic iron-sulfur (Fe/S) cluster-containing proteins and participates in iron homeostasis | [185] |
| ABCB8 | potassium | subunit of the mitochondrial ATP-gated potassium channel (mitoK (ATP)) | [186] |
| ABCB10 | mitochondrial biliverdin | export of substrate from the mitochondrial matrix to the cytosol | [187,188] |
| ABCB11 | bile salts | transport of bile salts across the canalicular membrane of hepatocytes, hepatic bile acid homeostasis | [189,190,191] |
| ABCC2 | conjugated organic anions, various substrates, drugs | active transport of various substrates including many drugs, toxicants and endogenous compound across cell membranes | [192] |
| ABCC3 | bile acids, glucuronides, various drugs | transports various substrates including many drugs, toxicants, and endogenous compound across cell membrane, transports glucuronide conjugates and also various bile salts | [193] |
| ABCC4 | cAMP and cGMP, bile acids, steroid conjugates, urate, prostaglandins, xenobiotics, drugs | extrudes physiological compounds and xenobiotics from cells, transports endogenous molecules that have a key role in cellular communication and signaling | [194,195] |
| ABCC7 | chloride, bicarbonate | ion channel that plays an important role in the regulation of epithelial ion and water transport and fluid homeostasis | [68,196,197,198,199,200,201,202] |
| ABCC8 | potassium | subunit of the beta-cell ATP-sensitive potassium channel (KATP), regulator of ATP-sensitive K+ channels and insulin release | [203,204] |
| ABCD1 | (VLCFA)-CoA | transport of very-long-chain fatty acid (VLCFA)-CoA from the cytosol to the peroxisome lumen | [205,206,207,208] |
| ABCD4 | cobalamin (vitamin B12) | transports cobalamin (vitamin B12) from the lysosomal lumen to the cytosol | [209] |
| ABCG1 | phospholipids, cholesterol | efflux of phospholipids, active component of the macrophage lipid export complex | [210,211,212] |
| AGBCG2 | diverse compounds, xenobiotics, drugs, … | extrudes a wide variety of physiological compounds, dietary toxins, and xenobiotics from cells | [213,214,215,216,217,218,219,220] |
| ABCG5-G8 | cholesterol | obligates heterodimer mediating sterol transport across cell membrane, selective transport of sterols/cholesterol in and out of the enterocytes and in selective sterol excretion by the liver into bile | [212,221,222,223] |
3.2. Structures, Functions, and Transport Mechanisms of SLC Transporters
| Transporter | Substrates | Functions | References |
|---|---|---|---|
| GLUT1/SLC2A1 | glucose | facilitative glucose transporter, which is responsible for constitutive or basal glucose uptake | [278,279,280] |
| GLUT3/SLC2A3 | glucose | mediates the uptake of glucose, 2-deoxyglucose, galactose, mannose, xylose and fructose | [281,282] |
| GLUT4/SLC2A4 | glucose | insulin-regulated facilitative glucose transporter, which plays a key role in removal of glucose from circulation | [283] |
| SGLT1/SLC5A1 | glucose/Na+ | electrogenic Na+-coupled sugar symporter that actively transports D-glucose or D-galactose at the plasma membrane, driven by a transmembrane Na+ electrochemical gradient set by the Na+/K+ pump | [284] |
| SGLT2/SLC5A2 | glucose/Na+ | electrogenic Na+-coupled sugar symporter that actively transports D-glucose at the plasma membrane, driven by a transmembrane Na+ electrochemical gradient set by the Na+/K+ pump | [285,286,287] |
| HsPepT1/SLC15A1 | oligopeptides | electrogenic proton-coupled amino-acid transporter that transports oligopeptides, primarily responsible for the absorption of dietary di- and tripeptides from the small intestinal lumen | [110] |
| HsPepT2/SLC15A2 | oligopeptides | electrogenic proton-coupled amino-acid transporter that transports oligopeptides | [110] |
| PHT2/SLC15A3 | peptide histidine | proton-coupled amino-acid transporter that transports free histidine and certain di- and tripeptides | |
| HPHT1/SLC15A4 | L-histidine GlySar dipeptide | proton-coupled amino-acid transporter that mediates the transmembrane transport of L-histidine and some di- and tripeptides from inside the lysosome to the cytosol, and plays a key role in innate immune response | [288] |
| MCT1/SLC16A1 | monocarboxylate | transport across the plasma membrane of many monocarboxylates; contributes to the maintenance of intracellular pH | [289,290] |
| MCT2/SLC16A7 | monocarboxylate | proton-coupled monocarboxylate symporter; transport across the plasma membrane of monocarboxylates | [291] |
| SIALIN/SLC17A5 | nitrates | anion transporter that operates via 2 distinct transport mechanisms: proton-coupled anion cotransport and membrane potential-dependent anion transport; exports glucuronic acid and free sialic acid derived from sialoglycoconjugate degradation out of lysosomes | [292] |
| VAT1/VMAT1/SLC18A1 | H+/monoamine | electrogenic antiporter that exchanges one cationic monoamine with two intravesicular protons across the membrane of secretory and synaptic vesicles; transports catecholamines and indolamines with higher affinity for serotonin | [293] |
| VAT2/VMAT2/SLC18A2 | H+/monoamine | electrogenic antiporter that exchanges one cationic monoamine with two intravesicular protons across the membrane of secretory and synaptic vesicles; transports a variety of catecholamines such as dopamine, adrenaline and noradrenaline, histamine, and indolamines such as serotonin | [294,295,296] |
| OATP1B1/SLCO1B1/LST-1/OATP-C/SLC21A6 | organic anion | mediates the uptake of organic anions; broad substrate specificity, can transport both organic anions and conjugated steroids | [297,298] |
| OATP1B3 | organic anion | mediates the uptake of organic anions; broad substrate specificity, can transport both organic anions and conjugated steroids | [297] |
| OCT1/SLC22A1 | organic cation | transport of a variety of organic cations such as endogenous bioactive amines, cationic drugs and xenobiotics; functions as a pH- and Na+-independent, bidirectional transporter | [265,266,267] |
| OCT2/SLC22A2 | organic cation | transport of a variety of organic cations such as endogenous bioactive amines, cationic drugs and xenobiotics | [265] |
| OCT3/SLC22A3 | organic cation | transport of a variety of organic cations such as endogenous bioactive amines, cationic drugs and xenobiotics; functions as a Na+- and Cl−-independent, bidirectional uniporter | [268] |
| SPNS2 | sphingosine-1-phosphate | exports S1P via facilitated diffusion; required for the egress of T-cells from lymph nodes during an immune response by mediating S1P secretion | [299,300,301] |
| FLVCR1 | heme | heme b transporter that mediates heme efflux from the cytoplasm to the extracellular compartment | [302,303] |
| FLVCR2 | heme | putative heme b importer involved in heme homeostasis in response to the metabolic state of the cell | [303] |
| FPN1/SLC40A1 | iron (Fe2+) | transports Fe2+ from the inside of a cell to the outside of the cell, playing a key role for maintaining systemic iron homeostasis | [137,275] |
4. Conclusions, Challenges, and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Alam, A.; Locher, K.P. Structure and Mechanism of Human ABC Transporters. Annu. Rev. Biophys. 2023, 52, 275–300. [Google Scholar] [CrossRef]
- Colas, C.; Ung, P.M.-U.; Schlessinger, A. SLC transporters: Structure, function, and drug discovery. MedChemComm 2016, 7, 1069–1081. [Google Scholar] [CrossRef]
- Schlessinger, A.; Zatorski, N.; Hutchinson, K.; Colas, C. Targeting SLC transporters: Small molecules as modulators and therapeutic opportunities. Trends Biochem. Sci. 2023, 48, 801–814. [Google Scholar] [CrossRef]
- Türková, A.; Zdrazil, B. Current Advances in Studying Clinically Relevant Transporters of the Solute Carrier (SLC) Family by Connecting Computational Modeling and Data Science. Comput. Struct. Biotechnol. J. 2019, 17, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Galetin, A.; Brouwer, K.L.R.; Tweedie, D.; Yoshida, K.; Sjöstedt, N.; Aleksunes, L.; Chu, X.; Evers, R.; Hafey, M.J.; Lai, Y.; et al. Membrane transporters in drug development and as determinants of precision medicine. Nat. Rev. Drug Discov. 2024, 23, 255–280. [Google Scholar] [CrossRef]
- Dudas, B.; Miteva, M.A. Computational and artificial intelligence-based approaches for drug metabolism and transport prediction. Trends Pharmacol. Sci. 2023, 45, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef]
- Huang, J.; Ecker, G.F. A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance. Molecules 2023, 28, 495. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, D.C.; Vergani, P.; Csanády, L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006, 440, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Bodzioch, M.; Orsó, E.; Klucken, J.; Langmann, T.; Böttcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Büchler, C.; Porsch-Özcürümez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Sun, K.; Meng, Z.; Chen, L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ayka, A.; Şehirli, A.Ö. The Role of the SLC Transporters Protein in the Neurodegenerative Disorders. Clin. Psychopharmacol. Neurosci. 2020, 18, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yu, F.; Sun, T.; Hu, Z. Transporter-mediated drug–drug interactions—Study design, data analysis, and implications for in vitro evaluations. Med. Drug Discov. 2021, 11, 100096. [Google Scholar] [CrossRef]
- Xie, T.; Chi, X.; Huang, B.; Ye, F.; Zhou, Q.; Huang, J. Rational exploration of fold atlas for human solute carrier proteins. Structure 2022, 30, 1321–1330.e5. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and General Transport Mechanisms by the Major Facilitator Superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Novič, M. Recent Advances on P-Glycoprotein (ABCB1) Transporter Modelling with In Silico Methods. Int. J. Mol. Sci. 2022, 23, 14804. [Google Scholar] [CrossRef]
- Grandits, M.; Ecker, G.F. Ligand- and Structure-based Approaches for Transmembrane Transporter Modeling. Curr. Drug Res. Rev. 2023, 16, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Tampé, R. Structural and Mechanistic Principles of ABC Transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef]
- Szöllősi, D.; Rose-Sperling, D.; Hellmich, U.A.; Stockner, T. Comparison of mechanistic transport cycle models of ABC exporters. Biochim. Biophys. Acta BBA Biomembr. 2018, 1860, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Szöllősi, D.; Chiba, P.; Szakacs, G.; Stockner, T. Conversion of chemical to mechanical energy by the nucleotide binding domains of ABCB1. Sci. Rep. 2020, 10, 2589. [Google Scholar] [CrossRef]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME–Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef]
- Benadiba, M.; Maor, Y. Importance of ABC Transporters in Drug Development. Curr. Pharm. Des. 2016, 22, 5817–5829. [Google Scholar] [CrossRef]
- Sun, H.; Scott, D.O. Structure-based Drug Metabolism Predictions for Drug Design. Chem. Biol. Drug Des. 2010, 75, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. The Spectrum of Liver Diseases Related to ABCB4 Gene Mutations: Pathophysiology and Clinical Aspects. Semin. Liver Dis. 2010, 30, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Stoeltje, L.; Luc, J.K.; Haddad, T.; Schrankel, C.S. The roles of ABCB1/P-glycoprotein drug transporters in regulating gut microbes and inflammation: Insights from animal models, old and new. Philos. Trans. R. Soc. B Biol. Sci. 2024, 379, 20230074. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta BBA-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef]
- Dalton, W.S.; Grogan, T.M.; Meltzer, P.S.; Scheper, R.J.; Durie, B.G.; Taylor, C.W.; Miller, T.P.; Salmon, S.E. Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: Detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J. Clin. Oncol. 1989, 7, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Pajeva, I.K.; Wiese, M. Structure–Activity Relationships of Tariquidar Analogs as Multidrug Resistance Modulators. AAPS J. 2009, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.J.M.; Schinkel, A.H.; Elferink, R.P.J.O.; Groen, A.K.; Wagenaar, E.; Van Deemter, L.; Mol, C.A.A.M.; Ottenhoff, R.; Van Der Lugt, N.M.T.; Van Roon, M.A.; et al. Homozygous disruption of the murine MDR2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993, 75, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M.; Lammert, F. ABCB4 Gene Aberrations in Human Liver Disease: An Evolving Spectrum. Semin. Liver Dis. 2018, 38, 299–307. [Google Scholar] [CrossRef]
- Jacquemin, E.; Bernard, O.; Hadchouel, M.; Cresteil, D.; De Vree, J.M.L.; Paul, M.; Elferink, R.P.J.O.; Bosma, P.J.; Sokal, E.M.; Sturm, E.; et al. The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001, 120, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Hermelin, B.; Boelle, P.Y.; Parc, R.; Taboury, J.; Poupon, R. ABCB4 gene mutation—Associated cholelithiasis in adults. Gastroenterology 2003, 125, 452–459. [Google Scholar] [CrossRef]
- Floreani, A.; Carderi, I.; Paternoster, D.; Soardo, G.; Azzaroli, F.; Esposito, W.; Montagnani, M.; Marchesoni, D.; Variola, A.; Rosa Rizzotto, E.; et al. Hepatobiliary phospholipid transporter ABCB4, MDR3 gene variants in a large cohort of Italian women with intrahepatic cholestasis of pregnancy. Dig. Liver Dis. 2008, 40, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, J.; Durand-Schneider, A.; Dossier, C.; Falguières, T.; Gautherot, J.; Davit-Spraul, A.; Aït-Slimane, T.; Housset, C.; Jacquemin, E.; Maurice, M. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016, 63, 1620–1631. [Google Scholar] [CrossRef]
- Trauner, M.; Boyer, J.L. Bile Salt Transporters: Molecular Characterization, Function, and Regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef]
- Knisely, A.S.; Portmann, B.C. Deficiency of BSEP in PFIC with hepatocellular malignancy. Pediatr. Transplant. 2006, 10, 644–645. [Google Scholar] [CrossRef]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet J. Rare Dis. 2009, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Apte, U. Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer. Adv. Pharmacol. 2015, 74, 263–302. [Google Scholar]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085.e9. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.C. Targeting Multidrug Resistance Protein 1 (MRP1, ABCC1): Past, Present, and Future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Hanssen, K.M.; Haber, M.; Fletcher, J.I. Targeting multidrug resistance-associated protein 1 (MRP1)-expressing cancers: Beyond pharmacological inhibition. Drug Resist. Updat. 2021, 59, 100795. [Google Scholar] [CrossRef]
- Poku, V.O.; Iram, S.H. A critical review on modulators of Multidrug Resistance Protein 1 in cancer cells. PeerJ 2022, 10, e12594. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Yang, Y.; Cai, C.-Y.; Teng, Q.-X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.-S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist. Updat. 2021, 54, 100743. [Google Scholar] [CrossRef] [PubMed]
- Wada, M. Mutations in the canilicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Hum. Mol. Genet. 1998, 7, 203–207. [Google Scholar] [CrossRef]
- Kool, M.; Van Der Linden, M.; De Haas, M.; Scheffer, G.L.; De Vree, J.M.L.; Smith, A.J.; Jansen, G.; Peters, G.J.; Ponne, N.; Scheper, R.J.; et al. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc. Natl. Acad. Sci. USA 1999, 96, 6914–6919. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Wielinga, P.; Zelcer, N.; De Haas, M.; Van Deemter, L.; Wijnholds, J.; Balzarini, J.; Borst, P. Characterization of the Transport of Nucleoside Analog Drugs by the Human Multidrug Resistance Proteins MRP4 and MRP5. Mol. Pharmacol. 2003, 63, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Bergen, A.A.B.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; Van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Thakur, S.; Ankita; Dash, S.; Verma, R.; Kaur, C.; Kumar, R.; Mazumder, A.; Singh, G. Understanding CFTR Functionality: A Comprehensive Review of Tests and Modulator Therapy in Cystic Fibrosis. Cell Biochem. Biophys. 2023, 82, 15–34. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.-S.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Mall, M.A. The future of cystic fibrosis treatment: From disease mechanisms to novel therapeutic approaches. Lancet Lond. Engl. 2023, 402, 1185–1198. [Google Scholar] [CrossRef]
- Du, K.; Lukacs, G.L. Cooperative Assembly and Misfolding of CFTR Domains In Vivo. Mol. Biol. Cell 2009, 20, 1903–1915. [Google Scholar] [CrossRef]
- Amaral, M.D.; Hutt, D.M.; Tomati, V.; Botelho, H.M.; Pedemonte, N. CFTR processing, trafficking and interactions. J. Cyst. Fibros. 2020, 19, S33–S36. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Jih, K.-Y.; Hwang, T.-C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef]
- Habib, A.-R.R.; Kajbafzadeh, M.; Desai, S.; Yang, C.L.; Skolnik, K.; Quon, B.S. A Systematic Review of the Clinical Efficacy and Safety of CFTR Modulators in Cystic Fibrosis. Sci. Rep. 2019, 9, 7234. [Google Scholar] [CrossRef]
- Pedemonte, N. Small-molecule correctors of defective F508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, S.; Gees, M.; Hwang, T.-C. Physiological and pharmacological characterization of the N1303K mutant CFTR. J. Cyst. Fibros. 2018, 17, 573–581. [Google Scholar] [CrossRef]
- Phuan, P.-W.; Tan, J.-A.; Rivera, A.A.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Haggie, P.M.; Verkman, A.S. Nanomolar-potency ‘co-potentiator’ therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants. Sci. Rep. 2019, 9, 17640. [Google Scholar] [CrossRef]
- Phuan, P.-W.; Son, J.-H.; Tan, J.-A.; Li, C.; Musante, I.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Kurth, M.J.; Galietta, L.J.; et al. Combination potentiator (‘co-potentiator’) therapy for CF caused by CFTR mutants, including N1303K, that are poorly responsive to single potentiators. J. Cyst. Fibros. 2018, 17, 595–606. [Google Scholar] [CrossRef]
- Verkman, A.S.; Synder, D.; Tradtrantip, L.; Thiagarajah, J.R.; Anderson, M.O. CFTR Inhibitors. Curr. Pharm. Des. 2013, 19, 3529–3541. [Google Scholar] [CrossRef]
- Delaunay, J.-L.; Elbahnsi, A.; Bruneau, A.; Madry, C.; Durand-Schneider, A.-M.; Stary, A.; Housset, C.; Gautheron, J.; Callebaut, I.; Aït-Slimane, T. Ivacaftor-Mediated Potentiation of ABCB4 Missense Mutations Affecting Critical Motifs of the NBDs: Repositioning Perspectives for Hepatobiliary Diseases. Int. J. Mol. Sci. 2023, 24, 1236. [Google Scholar] [CrossRef]
- Mareux, E.; Lapalus, M.; Amzal, R.; Almes, M.; Aït-Slimane, T.; Delaunay, J.L.; Adnot, P.; Collado-Hilly, M.; Davit-Spraul, A.; Falguières, T.; et al. Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int. 2020, 40, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, J.L.; Bruneau, A.; Hoffmann, B.; Durand-Schneider, A.M.; Barbu, V.; Jacquemin, E.; Maurice, M.; Housset, C.; Callebaut, I.; Aït-Slimane, T. Functional defect of variants in the adenosine triphosphate–binding sites of ABCB4 and their rescue by the cystic fibrosis transmembrane conductance regulator potentiator, ivacaftor (VX-770). Hepatology 2017, 65, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Mareux, E.; Lapalus, M.; Ben-Saad, A.; Callebaut, I.; Falguières, T.; Gonzales, E.; Jacquemin, E. In vitro functional rescue by ivacaftor of an ABCB11 variant involved in PFIC2 and intrahepatic cholestasis of pregnancy. Orphanet J. Rare Dis. 2021, 16, 484. [Google Scholar] [CrossRef] [PubMed]
- Mareux, E.; Lapalus, M.; Ben Saad, A.; Zelli, R.; Lakli, M.; Riahi, Y.; Almes, M.; Banet, M.; Callebaut, I.; Decout, J.-L.; et al. In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. Int. J. Mol. Sci. 2022, 23, 10758. [Google Scholar] [CrossRef]
- Suzuki, M.; Suzuki, H.; Sugimoto, Y.; Sugiyama, Y. ABCG2 Transports Sulfated Conjugates of Steroids and Xenobiotics. J. Biol. Chem. 2003, 278, 22644–22649. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the Breast Cancer Resistance Protein (BCRP/ABCG2) in Drug Transport—An Update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van De Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar]
- Gillet, J.-P.; Gottesman, M.M. Advances in the Molecular Detection of ABC Transporters Involved in Multidrug Resistance in Cancer. Curr. Pharm. Biotechnol. 2011, 12, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Yu, L.; Li-Hawkins, J.; Hammer, R.E.; Berge, K.E.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J. Clin. Investig. 2002, 110, 671–680. [Google Scholar] [CrossRef]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of Dietary Cholesterol in Sitosterolemia Caused by Mutations in Adjacent ABC Transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef]
- Lee, M.-H.; Lu, K.; Hazard, S.; Yu, H.; Shulenin, S.; Hidaka, H.; Kojima, H.; Allikmets, R.; Sakuma, N.; Pegoraro, R.; et al. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat. Genet. 2001, 27, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, M.K.; Alavi, M.S.; Roohbakhsh, A. The role of ATP-binding cassette transporter G1 (ABCG1) in Alzheimer’s disease: A review of the mechanisms. Basic Clin. Pharmacol. Toxicol. 2024, 134, 423–438. [Google Scholar] [CrossRef]
- Hediger, M.A.; Romero, M.F.; Peng, J.-B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins. Pflügers Arch. Eur. J. Physiol. 2004, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Mehta, V.; Gulati, A.; Drew, D. Structure and electromechanical coupling of a voltage-gated Na+/H+ exchanger. Nature 2023, 623, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kalienkova, V.; Peter, M.F.; Rheinberger, J.; Paulino, C. Structures of a sperm-specific solute carrier gated by voltage and cAMP. Nature 2023, 623, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Superti-Furga, G.; Lackner, D.; Wiedmer, T.; Ingles-Prieto, A.; Barbosa, B.; Girardi, E.; Goldmann, U.; Gürtl, B.; Klavins, K.; Klimek, C.; et al. The RESOLUTE consortium: Unlocking SLC transporters for drug discovery. Nat. Rev. Drug Discov. 2020, 19, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, V.; Superti-Furga, G. Structural and functional annotation of solute carrier transporters: Implication for drug discovery. Expert Opin. Drug Discov. 2023, 18, 1099–1115. [Google Scholar] [CrossRef]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A guide to plasma membrane solute carrier proteins. FEBS J. 2021, 288, 2784–2835. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.W.; Giacomini, K.M. Emerging Roles of the Human Solute Carrier 22 Family. Drug Metab. Dispos. 2022, 50, 1193–1210. [Google Scholar] [CrossRef]
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Asp. Med. 2013, 34, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K. The SLC22 Transporter Family: A Paradigm for the Impact of Drug Transporters on Metabolic Pathways, Signaling, and Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 663–687. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Kuroda, T.; Tsuchiya, T. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta BBA-Proteins Proteom. 2009, 1794, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Omote, H.; Hiasa, M.; Matsumoto, T.; Otsuka, M.; Moriyama, Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 2006, 27, 587–593. [Google Scholar] [CrossRef]
- Terada, T.; Inui, K. Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A). Biochem. Pharmacol. 2008, 75, 1689–1696. [Google Scholar] [CrossRef]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and Functional Characterization of a New Human Kidney–Specific H+/Organic Cation Antiporter, Kidney-Specific Multidrug and Toxin Extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflügers Arch. Eur. J. Physiol. 2004, 447, 653–665. [Google Scholar] [CrossRef]
- Stieger, B.; Hagenbuch, B. Organic Anion-Transporting Polypeptides. Curr. Top. Membr. 2014, 73, 205–232. [Google Scholar] [PubMed]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Interactions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef] [PubMed]
- Adibi, S.A.; Morse, E.L.; Masilamani, S.S.; Amin, P.M. Evidence for two different modes of tripeptide disappearance in human intestine. Uptake by peptide carrier systems and hydrolysis by peptide hydrolases. J. Clin. Investig. 1975, 56, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Killer, M.; Wald, J.; Pieprzyk, J.; Marlovits, T.C.; Löw, C. Structural snapshots of human PepT1 and PepT2 reveal mechanistic insights into substrate and drug transport across epithelial membranes. Sci. Adv. 2021, 7, eabk3259. [Google Scholar] [CrossRef]
- Ganapathy, M.E.; Brandsch, M.; Prasad, P.D.; Ganapathy, V.; Leibach, F.H. Differential Recognition of β-Lactam Antibiotics by Intestinal and Renal Peptide Transporters, PEPT 1 and PEPT 2. J. Biol. Chem. 1995, 270, 25672–25677. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Smith, D.E.; Ma, K.; Jappar, D.; Thomas, W.; Hillgren, K.M. Targeted Disruption of Peptide Transporter Pept1 Gene in Mice Significantly Reduces Dipeptide Absorption in Intestine. Mol. Pharm. 2008, 5, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Yao, S.Y.M.; Sun, L.; Cass, C.E.; Baldwin, S.A. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 2008, 38, 995–1021. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.; Beaumont, N.; Yao, S.Y.M.; Sundaram, M.; Boumah, C.E.; Davies, A.; Kwong, F.Y.P.; Coe, I.; Cass, C.E.; Young, J.D.; et al. Cloning of a human nucleoside transporter implicated in the Cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 1997, 3, 89–93. [Google Scholar] [CrossRef]
- Boswell-Casteel, R.C.; Hays, F.A. Equilibrative nucleoside transporters—A review. Nucleosides Nucleotides Nucleic Acids 2017, 36, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.J.; Lee, S.-Y. Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat. Struct. Mol. Biol. 2019, 26, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Döring, B.; Lütteke, T.; Geyer, J.; Petzinger, E. The SLC10 Carrier Family. Curr. Top. Membr. 2012, 70, 105–168. [Google Scholar]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Goutam, K.; Ielasi, F.S.; Pardon, E.; Steyaert, J.; Reyes, N. Structural basis of sodium-dependent bile salt uptake into the liver. Nature 2022, 606, 1015–1020. [Google Scholar] [CrossRef]
- Oelkers, P.; Kirby, L.C.; Heubi, J.E.; Dawson, P.A. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Investig. 1997, 99, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B.; Dawson, P. The sodium bile salt cotransport family SLC10. Pflügers Arch. Eur. J. Physiol. 2004, 447, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.-J.; Iwata, S.; Cameron, A.D.; Drew, D. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 2011, 478, 408–411. [Google Scholar] [CrossRef]
- Marchant, J.S.; Subramanian, V.S.; Parker, I.; Said, H.M. Intracellular Trafficking and Membrane Targeting Mechanisms of the Human Reduced Folate Carrier in Mammalian Epithelial Cells. J. Biol. Chem. 2002, 277, 33325–33333. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Cordova, A.F.; Hess, G.T.; Bassik, M.C.; Li, L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 2019, 75, 372–381.e5. [Google Scholar] [CrossRef]
- Luteijn, R.D.; Zaver, S.A.; Gowen, B.G.; Wyman, S.K.; Garelis, N.E.; Onia, L.; McWhirter, S.M.; Katibah, G.E.; Corn, J.E.; Woodward, J.J.; et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 2019, 573, 434–438. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, X.; Zhu, Y.; Sun, P.; Zhang, L.; Ma, J.; Zhang, Y.; Zeng, L.; Nie, X.; Gao, Y.; et al. Recognition of cyclic dinucleotides and folates by human SLC19A1. Nature 2022, 612, 170–176. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Uguen, K.; Ka, C.; Collod-Béroud, G.; Le Tertre, M.; Guellec, J.; Férec, C.; Béroud, C.; Callebaut, I.; Le Gac, G. The Spectra of Disease-Causing Mutations in the Ferroportin 1 (SLC40A1) Encoding Gene and Related Iron Overload Phenotypes (Hemochromatosis Type 4 and Ferroportin Disease). Hum. Mutat. 2023, 2023, 5162256. [Google Scholar] [CrossRef]
- Uguen, K.; Le Tertre, M.; Tchernitchko, D.; Elbahnsi, A.; Maestri, S.; Gourlaouen, I.; Férec, C.; Ka, C.; Callebaut, I.; Le Gac, G. The dual loss and gain of function of the FPN1 iron exporter results in the ferroportin disease phenotype. Hum. Genet. Genom. Adv. 2024, 5, 100335. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haematologica 2017, 102, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, E.F.; Liziczai, M.; Drożdżyk, K.; Altermatt, P.; Langini, C.; Manolova, V.; Sundstrom, H.; Dürrenberger, F.; Dutzler, R.; Manatschal, C. Structures of ferroportin in complex with its specific inhibitor vamifeport. eLife 2023, 12, e83053. [Google Scholar] [CrossRef]
- Pilo, F.; Angelucci, E. Vamifeport: Monography of the First Oral Ferroportin Inhibitor. J. Clin. Med. 2024, 13, 5524. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.P.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Reyes, C.L.; Yu, J.; Roth, C.B.; Chang, G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA 2007, 104, 19005–19010. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.-W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef]
- Mora Lagares, L.; Minovski, N.; Caballero Alfonso, A.Y.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int. J. Mol. Sci. 2020, 21, 4058. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Pérez-Castillo, Y.; Novič, M. Exploring the dynamics of the ABCB1 membrane transporter P-glycoprotein in the presence of ATP and active/non-active compounds through molecular dynamics simulations. Toxicology 2024, 502, 153732. [Google Scholar] [CrossRef] [PubMed]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.-J.U.; Gillet, J.-P.; Cordeiro, M.N.D.S.; Dos Santos, D.J.V.A. Theoretical insights on helix repacking as the origin of P-glycoprotein promiscuity. Sci. Rep. 2020, 10, 9823. [Google Scholar] [CrossRef]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.-J.U.; Durães, F.; Sousa, E.; Gillet, J.-P.; Cordeiro, M.N.D.S.; Dos Santos, D.J.V.A. Probing the Allosteric Modulation of P-Glycoprotein: A Medicinal Chemistry Approach Toward the Identification of Noncompetitive P-Gp Inhibitors. ACS Omega 2023, 8, 11281–11287. [Google Scholar] [CrossRef]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.-J.U.; Gillet, J.-P.; Cordeiro, M.N.D.S.; Dos Santos, D.J.V.A. Long-range communication between transmembrane- and nucleotide-binding domains does not depend on drug binding to mutant P-glycoprotein. J. Biomol. Struct. Dyn. 2023, 41, 14428–14437. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Chen, L.; Li, Q.; Nasif, K.F.A.; Xie, Y.; Deng, B.; Niu, S.; Pouriyeh, S.; Dai, Z.; Chen, J.; Xie, C.Y. AI-Driven Deep Learning Techniques in Protein Structure Prediction. Int. J. Mol. Sci. 2024, 25, 8426. [Google Scholar] [CrossRef]
- Behmard, E.; Barzegari, E.; Najafipour, S.; Kouhpayeh, A.; Ghasemi, Y.; Asadi-Pooya, A.A. Efflux dynamics of the antiseizure drug, levetiracetam, through the P-glycoprotein channel revealed by advanced comparative molecular simulations. Sci. Rep. 2022, 12, 13674. [Google Scholar] [CrossRef]
- Nagy, T.; Tóth, Á.; Telbisz, Á.; Sarkadi, B.; Tordai, H.; Tordai, A.; Hegedűs, T. The transport pathway in the ABCG2 protein and its regulation revealed by molecular dynamics simulations. Cell. Mol. Life Sci. 2021, 78, 2329–2339. [Google Scholar] [CrossRef]
- Elbahnsi, A.; Dudas, B.; Cisternino, S.; Declèves, X.; Miteva, M.A. Mechanistic insights into P-glycoprotein ligand transport and inhibition revealed by enhanced molecular dynamics simulations. Comput. Struct. Biotechnol. J. 2024, 23, 2548–2564. [Google Scholar] [CrossRef]
- Dudas, B.; Decleves, X.; Cisternino, S.; Perahia, D.; Miteva, M.A. ABCG2/BCRP transport mechanism revealed through kinetically excited targeted molecular dynamics simulations. Comput. Struct. Biotechnol. J. 2022, 20, 4195–4205. [Google Scholar] [CrossRef]
- Verhalen, B.; Dastvan, R.; Thangapandian, S.; Peskova, Y.; Koteiche, H.A.; Nakamoto, R.K.; Tajkhorshid, E.; Mchaourab, H.S. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 2017, 543, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, D.; McComas, S.E.; Alleva, C.; Bonaccorsi, M.; Drew, D.; Delemotte, L. Reconstructing the transport cycle in the sugar porter superfamily using coevolution-powered machine learning. eLife 2023, 12, e84805. [Google Scholar] [CrossRef]
- Yee, S.W.; Macdonald, C.B.; Mitrovic, D.; Zhou, X.; Koleske, M.L.; Yang, J.; Buitrago Silva, D.; Rockefeller Grimes, P.; Trinidad, D.D.; More, S.S.; et al. The full spectrum of SLC22 OCT1 mutations illuminates the bridge between drug transporter biophysics and pharmacogenomics. Mol. Cell 2024, 84, 1932–1947.e10. [Google Scholar] [CrossRef]
- Furuta, T. Structural dynamics of ABC transporters: Molecular simulation studies. Biochem. Soc. Trans. 2021, 49, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kodan, A.; Kimura, Y.; Ueda, K.; Nakatsu, T.; Kato, H. Functional role of the linker region in purified human P-glycoprotein. FEBS J. 2009, 276, 3504–3516. [Google Scholar] [CrossRef]
- Ford, R.C.; Marshall-Sabey, D.; Schuetz, J. Linker Domains: Why ABC Transporters ‘Live in Fragments no Longer’. Trends Biochem. Sci. 2020, 45, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, T.; Aleksandrov, A.; Mengos, A.; Cui, L.; Jensen, T.J.; Riordan, J.R. Role of individual R domain phosphorylation sites in CFTR regulation by protein kinase A. Biochim. Biophys. Acta BBA-Biomembr. 2009, 1788, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e10. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, X. Cholesterol Efflux Mechanism Revealed by Structural Analysis of Human ABCA1 Conformational States. Nat. Cardiovasc. Res. 2022, 1, 238–245. [Google Scholar] [CrossRef]
- Plummer-Medeiros, A.M.; Culbertson, A.T.; Morales-Perez, C.L.; Liao, M. Activity and Structural Dynamics of Human ABCA1 in a Lipid Membrane. J. Mol. Biol. 2023, 435, 168038. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Zhang, Z.; Yue, J.; Fang, Q.; Gong, X. Cryo-EM Structures of the Human Surfactant Lipid Transporter ABCA3. Sci. Adv. 2022, 8, eabn3727. [Google Scholar] [CrossRef]
- Scortecci, J.F.; Molday, L.L.; Curtis, S.B.; Garces, F.A.; Panwar, P.; Van Petegem, F.; Molday, R.S. Cryo-EM Structures of the ABCA4 Importer Reveal Mechanisms Underlying Substrate Binding and Stargardt Disease. Nat. Commun. 2021, 12, 5902. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, Z.; Fang, Q.; Du, B.; Gong, X. Structural Basis of Substrate Recognition and Translocation by Human ABCA4. Nat. Commun. 2021, 12, 3853. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.; Chen, J. Molecular Structures of the Eukaryotic Retinal Importer ABCA4. eLife 2021, 10, e63524. [Google Scholar] [CrossRef]
- Le, L.T.M.; Thompson, J.R.; Dehghani-Ghahnaviyeh, S.; Pant, S.; Dang, P.X.; French, J.B.; Kanikeyo, T.; Tajkhorshid, E.; Alam, A. Cryo-EM Structures of Human ABCA7 Provide Insights into Its Phospholipid Translocation Mechanisms. EMBO J. 2023, 42, e111065. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Alam, A.; Küng, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.N.; Smolinski, M.P. Discovery and Characterization of Potent Dual P-Glycoprotein and CYP3A4 Inhibitors: Design, Synthesis, Cryo-EM Analysis, and Biological Evaluations. J. Med. Chem. 2022, 65, 191–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.L.; Grigorieff, N.; Chen, J. Structure of the Transporter Associated with Antigen Processing Trapped by Herpes Simplex Virus. eLife 2016, 5, e21829. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Bang-Sørensen, R.; Irobalieva, R.N.; Erramilli, S.K.; Stieger, B.; Kossiakoff, A.A.; Locher, K.P. Structures of ABCB4 Provide Insight into Phosphatidylcholine Translocation. Proc. Natl. Acad. Sci. USA 2021, 118, e2106702118. [Google Scholar] [CrossRef]
- Olsen, J.A.; Alam, A.; Kowal, J.; Stieger, B.; Locher, K.P. Structure of the Human Lipid Exporter ABCB4 in a Lipid Environment. Nat. Struct. Mol. Biol. 2020, 27, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Park, J.G.; Jang, E.; Choi, S.H.; Kim, S.; Kim, J.W.; Jin, M.S. W546 Stacking Disruption Traps the Human Porphyrin Transporter ABCB6 in an Outward-Facing Transient State. Commun. Biol. 2023, 6, 960. [Google Scholar] [CrossRef]
- Wang, C.; Cao, C.; Wang, N.; Wang, X.; Wang, X.; Zhang, X.C. Cryo- Electron Microscopy Structure of Human ABCB6 Transporter. Protein Sci. 2020, 29, 2363–2374. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.S.; Park, J.G.; Kim, J.W.; Ju, S.; Choi, S.H.; Kim, S.; Kim, N.J.; Hong, S.; Kang, J.Y.; et al. Structural Insights into Porphyrin Recognition by the Human ATP-Binding Cassette Transporter ABCB6. Mol. Cells 2022, 45, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhang, S.; Tian, M.; Zhang, L.; Guo, R.; Zhuo, W.; Yang, M. Molecular Insights into the Human ABCB6 Transporter. Cell Discov. 2021, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Shen, Y.; Yang, X. Cryo-EM Structure of AMP-PNP-Bound Human Mitochondrial ATP-Binding Cassette Transporter ABCB7. J. Struct. Biol. 2022, 214, 107832. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ren, Y.; Lu, X.; Shen, Y.; Yang, X. Cryo-EM Structure of Human ABCB8 Transporter in Nucleotide Binding State. Biochem. Biophys. Res. Commun. 2021, 557, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Shintre, C.A.; Pike, A.C.W.; Li, Q.; Kim, J.-I.; Barr, A.J.; Goubin, S.; Shrestha, L.; Yang, J.; Berridge, G.; Ross, J.; et al. Structures of ABCB10, a Human ATP-Binding Cassette Transporter in Apo- and Nucleotide-Bound States. Proc. Natl. Acad. Sci. USA 2013, 110, 9710–9715. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Yang, Y.; He, L.; Hang, Y.; Yan, X.; Shi, H.; Wu, J.; Ouyang, Z. Cryo-EM Structures of Mitochondrial ABC Transporter ABCB10 in Apo and Biliverdin-Bound Form. Nat. Commun. 2023, 14, 2030. [Google Scholar] [CrossRef]
- Wang, L.; Hou, W.-T.; Chen, L.; Jiang, Y.-L.; Xu, D.; Sun, L.; Zhou, C.-Z.; Chen, Y. Cryo-EM Structure of Human Bile Salts Exporter ABCB11. Cell Res. 2020, 30, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hou, W.-T.; Wang, J.; Xu, D.; Guo, C.; Sun, L.; Ruan, K.; Zhou, C.-Z.; Chen, Y. Structures of Human Bile Acid Exporter ABCB11 Reveal a Transport Mechanism Facilitated by Two Tandem Substrate-Binding Pockets. Cell Res. 2022, 32, 501–504. [Google Scholar] [CrossRef]
- Liu, H.; Irobalieva, R.N.; Kowal, J.; Ni, D.; Nosol, K.; Bang-Sørensen, R.; Lancien, L.; Stahlberg, H.; Stieger, B.; Locher, K.P. Structural Basis of Bile Salt Extrusion and Small-Molecule Inhibition in Human BSEP. Nat. Commun. 2023, 14, 7296. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.-X.; Chen, Z.-P.; Wang, L.; Wang, J.; Zhou, C.-Z.; Hou, W.-T.; Chen, Y. Transport Mechanism of Human Bilirubin Transporter ABCC2 Tuned by the Inter-Module Regulatory Domain. Nat. Commun. 2024, 15, 1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.; Wang, F.; Cheng, M.; Mao, Y.; Fang, S.; Wang, L.; Zhou, C.; Hou, W.; Chen, Y. Placing Steroid Hormones within the Human ABCC3 Transporter Reveals a Compatible Amphiphilic Substrate-binding Pocket. EMBO J. 2023, 42, e113415. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.; Raj, I.; Pape, T.; Taylor, N.M.I. Structural and Mechanistic Basis of Substrate Transport by the Multidrug Transporter MRP4. Structure 2023, 31, 1407–1418.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Hou, W.-T.; Zha, Z.; Xu, K.; Zhou, C.-Z.; Li, Q.; Chen, Y. Structural Insights into Human ABCC4-Mediated Transport of Platelet Agonist and Antagonist. Nat. Cardiovasc. Res. 2023, 2, 693–701. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Levring, J.; Terry, D.S.; Kilic, Z.; Fitzgerald, G.; Blanchard, S.C.; Chen, J. CFTR function, pathology and pharmacology at single-molecule resolution. Nature 2023, 616, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Young, P.G.; Levring, J.; Fiedorczuk, K.; Blanchard, S.C.; Chen, J. Structural basis for CFTR inhibition by CFTR inh-172. Proc. Natl. Acad. Sci. USA 2024, 121, e2316675121. [Google Scholar] [CrossRef] [PubMed]
- Levring, J.; Chen, J. Structural identification of a selectivity filter in CFTR. Proc. Natl. Acad. Sci. USA 2024, 121, e2316673121. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; MacKinnon, R. Molecular Structure of an Open Human KATP Channel. Proc. Natl. Acad. Sci. USA 2021, 118, e2112267118. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular Structure of Human KATP in Complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef]
- Xiong, C.; Jia, L.-N.; Xiong, W.-X.; Wu, X.-T.; Xiong, L.-L.; Wang, T.-H.; Zhou, D.; Hong, Z.; Liu, Z.; Tang, L. Structural Insights into Substrate Recognition and Translocation of Human Peroxisomal ABC Transporter ALDP. Signal Transduct. Target. Ther. 2023, 8, 74. [Google Scholar] [CrossRef]
- Le, L.T.M.; Thompson, J.R.; Dang, P.X.; Bhandari, J.; Alam, A. Structures of the Human Peroxisomal Fatty Acid Transporter ABCD1 in a Lipid Environment. Commun. Biol. 2022, 5, 7. [Google Scholar] [CrossRef]
- Wang, R.; Qin, Y.; Li, X. Structural Basis of Acyl-CoA Transport across the Peroxisomal Membrane by Human ABCD1. Cell Res. 2022, 32, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, Y.; Wang, W.; Lei, J.; Ying, Z.; Yang, G. Structural and Functional Insights of the Human Peroxisomal ABC Transporter ALDP. eLife 2022, 11, e75039. [Google Scholar] [CrossRef]
- Xu, D.; Feng, Z.; Hou, W.-T.; Jiang, Y.-L.; Wang, L.; Sun, L.; Zhou, C.-Z.; Chen, Y. Cryo-EM Structure of Human Lysosomal Cobalamin Exporter ABCD4. Cell Res. 2019, 29, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Skarda, L.; Kowal, J.; Locher, K.P. Structure of the Human Cholesterol Transporter ABCG1. J. Mol. Biol. 2021, 433, 167218. [Google Scholar] [CrossRef]
- Xu, D.; Li, Y.; Yang, F.; Sun, C.-R.; Pan, J.; Wang, L.; Chen, Z.-P.; Fang, S.-C.; Yao, X.; Hou, W.-T.; et al. Structure and Transport Mechanism of the Human Cholesterol Transporter ABCG1. Cell Rep. 2022, 38, 110298. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, J.; Long, T.; Qi, X.; Donnelly, L.; Elghobashi-Meinhardt, N.; Esparza, L.; Cohen, J.C.; Xie, X.-S.; Hobbs, H.H.; et al. Molecular Basis of Cholesterol Efflux via ABCG Subfamily Transporters. Proc. Natl. Acad. Sci. USA 2021, 118, e2110483118. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural Basis of Small-Molecule Inhibition of Human Multidrug Transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM Structures of a Human ABCG2 Mutant Trapped in ATP-Bound and Substrate-Bound States. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Liao, M. ABCG2 Transports Anticancer Drugs via a Closed-to-Open Switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Kowal, J.; Ni, D.; Jackson, S.M.; Manolaridis, I.; Stahlberg, H.; Locher, K.P. Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under Turnover Conditions Reveal a Key Step in the Drug Transport Mechanism. Nat. Commun. 2021, 12, 4376. [Google Scholar] [CrossRef]
- Rasouli, A.; Yu, Q.; Dehghani-Ghahnaviyeh, S.; Wen, P.-C.; Kowal, J.; Locher, K.P.; Tajkhorshid, E. Differential Dynamics and Direct Interaction of Bound Ligands with Lipids in Multidrug Transporter ABCG2. Proc. Natl. Acad. Sci. USA 2023, 120, e2213437120. [Google Scholar] [CrossRef]
- Irobalieva, R.N.; Manolaridis, I.; Jackson, S.M.; Ni, D.; Pardon, E.; Stahlberg, H.; Steyaert, J.; Locher, K.P. Structural Basis of the Allosteric Inhibition of Human ABCG2 by Nanobodies. J. Mol. Biol. 2023, 435, 168234. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kinch, L.N.; Borek, D.M.; Wang, J.; Wang, J.; Urbatsch, I.L.; Xie, X.-S.; Grishin, N.V.; Cohen, J.C.; Otwinowski, Z.; et al. Crystal Structure of the Human Sterol Transporter ABCG5/ABCG8. Nature 2016, 533, 561–564. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, C.-S.; Yu, X.; Lee, J.; Vaish, A.; Chen, Q.; Zhou, M.; Wang, Z.; Min, X. Cryo-EM Structure of ABCG5/G8 in Complex with Modulating Antibodies. Commun. Biol. 2021, 4, 526. [Google Scholar] [CrossRef] [PubMed]
- Farhat, D.; Rezaei, F.; Ristovski, M.; Yang, Y.; Stancescu, A.; Dzimkova, L.; Samnani, S.; Couture, J.-F.; Lee, J.-Y. Structural Analysis of Cholesterol Binding and Sterol Selectivity by ABCG5/G8. J. Mol. Biol. 2022, 434, 167795. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, A.; Thonghin, N.; Shafi, T.; Prince, S.M.; Collins, R.F.; Ford, R.C. Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes 2021, 11, 923. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 23, 362. [Google Scholar] [CrossRef]
- Xing, J.; Huang, S.; Heng, Y.; Mei, H.; Pan, X. Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding. Molecules 2020, 25, 6006. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, W.; Wang, Y.; Liu, Y.; Li, C. Exploring movement and energy in human P-glycoprotein conformational rearrangement. J. Biomol. Struct. Dyn. 2019, 37, 1104–1119. [Google Scholar] [CrossRef]
- Barreto-Ojeda, E.; Corradi, V.; Gu, R.-X.; Tieleman, D.P. Coarse-grained molecular dynamics simulations reveal lipid access pathways in P-glycoprotein. J. Gen. Physiol. 2018, 150, 417–429. [Google Scholar] [CrossRef]
- Chambers, T.C.; Pohl, J.; Glass, D.B.; Kuo, J.F. Phosphorylation by protein kinase C and cyclic AMP-dependent protein kinase of synthetic peptides derived from the linker region of human P-glycoprotein. Biochem. J. 1994, 299, 309–315. [Google Scholar] [CrossRef]
- Szabó, K.; Bakos, É.; Welker, E.; Müller, M.; Goodfellow, H.R.; Higgins, C.F.; Váradi, A.; Sarkadi, B. Phosphorylation Site Mutations in the Human Multidrug Transporter Modulate Its Drug-stimulated ATPase Activity. J. Biol. Chem. 1997, 272, 23165–23171. [Google Scholar] [CrossRef]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.U.; Dos Santos, D.J.V.A. Assessing the Stabilization of P-Glycoprotein’s Nucleotide-Binding Domains by the Linker, Using Molecular Dynamics. Mol. Inform. 2013, 32, 529–540. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; Dos Santos, D.J.V.A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Klepsch, F.; Chiba, P.; Ecker, G.F. Exhaustive Sampling of Docking Poses Reveals Binding Hypotheses for Propafenone Type Inhibitors of P-Glycoprotein. PLoS Comput. Biol. 2011, 7, e1002036. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wong, I.L.K.; Sang, J.; Liu, F.; Yan, C.S.W.; Kan, J.W.Y.; Chan, T.H.; Chow, L.M.C. Identification of Binding Sites in the Nucleotide-Binding Domain of P-Glycoprotein for a Potent and Nontoxic Modulator, the Amine-Containing Monomeric Flavonoid FM04. J. Med. Chem. 2023, 66, 6160–6183. [Google Scholar] [CrossRef]
- Xu, J.; Liu, Y.; Yang, Y.; Bates, S.; Zhang, J.-T. Characterization of Oligomeric Human Half-ABC Transporter ATP-binding Cassette G2. J. Biol. Chem. 2004, 279, 19781–19789. [Google Scholar] [CrossRef] [PubMed]
- László, L.; Sarkadi, B.; Hegedűs, T. Jump into a New Fold—A Homology Based Model for the ABCG2/BCRP Multidrug Transporter. PLoS ONE 2016, 11, e0164426. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Wang, Y.-J.; Lei, Z.-N.; Zhang, G.-N.; Zhang, X.-Y.; Wang, D.; Al-Rihani, S.B.; Shukla, S.; Ambudkar, S.V.; Kaddoumi, A.; et al. Regorafenib antagonizes BCRP-mediated multidrug resistance in colon cancer. Cancer Lett. 2019, 442, 104–112. [Google Scholar] [CrossRef]
- Zattoni, I.F.; Kronenberger, T.; Kita, D.H.; Guanaes, L.D.; Guimarães, M.M.; De Oliveira Prado, L.; Ziasch, M.; Vesga, L.C.; Gomes De Moraes Rego, F.; Picheth, G.; et al. A new porphyrin as selective substrate-based inhibitor of breast cancer resistance protein (BCRP/ABCG2). Chem. Biol. Interact. 2022, 351, 109718. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Mekhemer, G.A.H.; Shawky, A.M.; Moustafa, M.F.; Atia, M.A.M. In Silico Targeting Human Multidrug Transporter ABCG2 in Breast Cancer: Database Screening, Molecular Docking, and Molecular Dynamics Study. Mol. Inform. 2022, 41, 2060039. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Shawky, A.M.; Mekhemer, G.A.H.; Alrumaihi, F.; Moustafa, M.F.; Atia, M.A.M. Prospective Drug Candidates as Human Multidrug Transporter ABCG2 Inhibitors: An In Silico Drug Discovery Study. Cell Biochem. Biophys. 2021, 79, 189–200. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Bonito, C.A.; Cordeiro, M.N.D.S.; Ferreira, M.-J.U.; Dos Santos, D.J.V.A. Structure-function relationships in ABCG2: Insights from molecular dynamics simulations and molecular docking studies. Sci. Rep. 2017, 7, 15534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016, 167, 1586–1597.e9. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell 2017, 170, 483–491.e8. [Google Scholar] [CrossRef]
- Gao, X.; Yeh, H.-I.; Yang, Z.; Fan, C.; Jiang, F.; Howard, R.J.; Lindahl, E.; Kappes, J.C.; Hwang, T.-C. Allosteric inhibition of CFTR gating by CFTRinh-172 binding in the pore. Nat. Commun. 2024, 15, 6668. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.F.; Aleksandrov, L.A.; Jensen, T.J.; Cui, L.L.; Kousouros, J.N.; He, L.; Aleksandrov, A.A.; Gingerich, D.S.; Riordan, J.R.; Chen, J.Z. Cryo-EM Visualization of an Active High Open Probability CFTR Anion Channel. Biochemistry 2018, 57, 6234–6246. [Google Scholar] [CrossRef]
- Mornon, J.-P.; Hoffmann, B.; Jonic, S.; Lehn, P.; Callebaut, I. Full-open and closed CFTR channels, with lateral tunnels from the cytoplasm and an alternative position of the F508 region, as revealed by molecular dynamics. Cell. Mol. Life Sci. 2015, 72, 1377–1403. [Google Scholar] [CrossRef]
- Cui, G.; Freeman, C.S.; Knotts, T.; Prince, C.Z.; Kuang, C.; McCarty, N.A. Two Salt Bridges Differentially Contribute to the Maintenance of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channel Function. J. Biol. Chem. 2013, 288, 20758–20767. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Zhang, Z.-R.; O’Brien, A.R.W.; Song, B.; McCarty, N.A. Mutations at arginine 352 alter the pore architecture of CFTR. J. Membr. Biol. 2008, 222, 91–106. [Google Scholar] [CrossRef]
- El Hiani, Y.; Negoda, A.; Linsdell, P. Cytoplasmic pathway followed by chloride ions to enter the CFTR channel pore. Cell. Mol. Life Sci. CMLS 2016, 73, 1917–1925. [Google Scholar] [CrossRef]
- Negoda, A.; El Hiani, Y.; Cowley, E.A.; Linsdell, P. Contribution of a lysine residue in the first transmembrane segment to the selectivity filter region in the CFTR chloride channel. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1049–1058. [Google Scholar] [CrossRef]
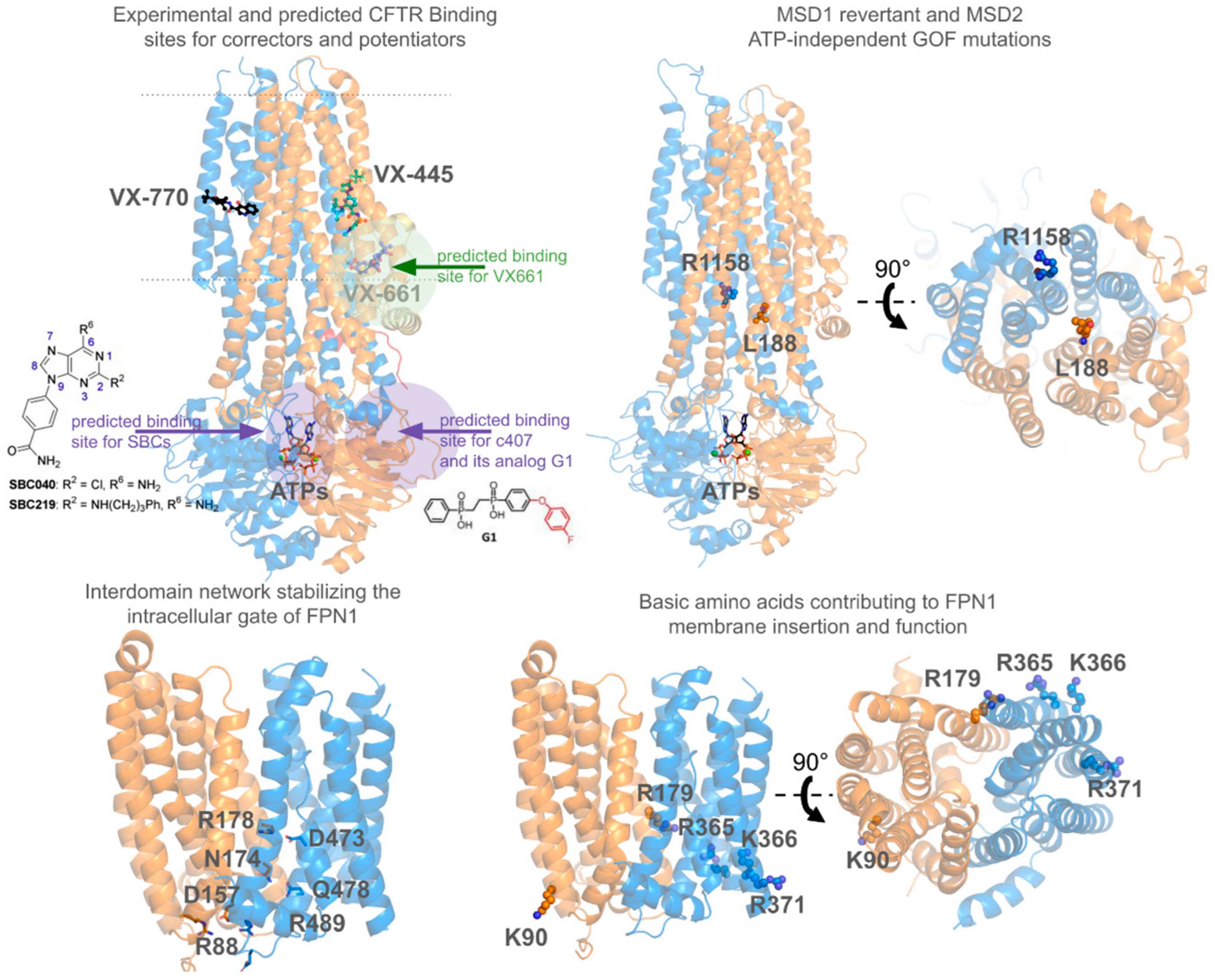
- Hoffmann, B.; Elbahnsi, A.; Lehn, P.; Décout, J.-L.; Pietrucci, F.; Mornon, J.-P.; Callebaut, I. Combining theoretical and experimental data to decipher CFTR 3D structures and functions. Cell. Mol. Life Sci. 2018, 75, 3829–3855. [Google Scholar] [CrossRef] [PubMed]
- Baatallah, N.; Elbahnsi, A.; Mornon, J.-P.; Chevalier, B.; Pranke, I.; Servel, N.; Zelli, R.; Décout, J.-L.; Edelman, A.; Sermet-Gaudelus, I.; et al. Pharmacological chaperones improve intra-domain stability and inter-domain assembly via distinct binding sites to rescue misfolded CFTR. Cell. Mol. Life Sci. 2021, 78, 7813–7829. [Google Scholar] [CrossRef] [PubMed]
- Baatallah, N.; Elbahnsi, A.; Chevalier, B.; Castanier, S.; Mornon, J.-P.; Pranke, I.; Edelman, A.; Sermet-Gaudelus, I.; Callebaut, I.; Hinzpeter, A. Acting on the CFTR Membrane-Spanning Domains Interface Rescues Some Misfolded Mutants. Int. J. Mol. Sci. 2022, 23, 16225. [Google Scholar] [CrossRef] [PubMed]
- Castanier, S.; Elbahnsi, A.; Chevalier, B.; Baatallah, N.; Pranke, I.; Berri, L.; Edelman, A.; Sermet-Gaudelus, I.; Mornon, J.-P.; Callebaut, I.; et al. Novel gain-of-function mutants identify a critical region within CFTR membrane-spanning domain 2 controlling cAMP-dependent and ATP-independent channel activation. Cell. Mol. Life Sci. 2024, 81, 426. [Google Scholar] [CrossRef] [PubMed]
- Froux, L.; Elbahnsi, A.; Boucherle, B.; Billet, A.; Baatallah, N.; Hoffmann, B.; Alliot, J.; Zelli, R.; Zeinyeh, W.; Haudecoeur, R.; et al. Targeting different binding sites in the CFTR structures allows to synergistically potentiate channel activity. Eur. J. Med. Chem. 2020, 190, 112116. [Google Scholar] [CrossRef]
- Bitam, S.; Elbahnsi, A.; Creste, G.; Pranke, I.; Chevalier, B.; Berhal, F.; Hoffmann, B.; Servel, N.; Baatalah, N.; Tondelier, D.; et al. New insights into structure and function of bis-phosphinic acid derivatives and implications for CFTR modulation. Sci. Rep. 2021, 11, 6842. [Google Scholar]
- McDonald, E.F.; Woods, H.; Smith, S.T.; Kim, M.; Schoeder, C.T.; Plate, L.; Meiler, J. Structural Comparative Modeling of Multi-Domain F508del CFTR. Biomolecules 2022, 12, 471. [Google Scholar] [CrossRef]
- Farkas, B.; Tordai, H.; Padányi, R.; Tordai, A.; Gera, J.; Paragi, G.; Hegedűs, T. Discovering the chloride pathway in the CFTR channel. Cell. Mol. Life Sci. 2020, 77, 765–778. [Google Scholar] [CrossRef]
- Zeng, Z.W.; Linsdell, P.; Pomès, R. Molecular dynamics study of Cl− permeation through cystic fibrosis transmembrane conductance regulator (CFTR). Cell. Mol. Life Sci. 2023, 80, 51. [Google Scholar] [CrossRef]
- Guellec, J.; Elbahnsi, A.; Tertre, M.L.; Uguen, K.; Gourlaouen, I.; Férec, C.; Ka, C.; Callebaut, I.; Gac, G.L. Molecular model of the ferroportin intracellular gate and implications for the human iron transport cycle and hemochromatosis type 4A. FASEB J. 2019, 33, 14625–14635. [Google Scholar] [CrossRef] [PubMed]
- Debbiche, R.; Elbahnsi, A.; Uguen, K.; Ka, C.; Callebaut, I.; Le Gac, G. Insights into the role of glycerophospholipids on the iron export function of SLC40A1 and the molecular mechanisms of ferroportin disease. FASEB J. 2024, 38, e23725. [Google Scholar] [CrossRef]
- Quistgaard, E.M.; Löw, C.; Guettou, F.; Nordlund, P. Understanding transport by the major facilitator superfamily (MFS): Structures pave the way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. [Google Scholar] [CrossRef]
- Reis, R.; Moraes, I. Structural biology and structure–function relationships of membrane proteins. Biochem. Soc. Trans. 2019, 47, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Suo, Y.; Wright, N.J.; Guterres, H.; Fedor, J.G.; Butay, K.J.; Borgnia, M.J.; Im, W.; Lee, S.-Y. Molecular basis of polyspecific drug and xenobiotic recognition by OCT1 and OCT2. Nat. Struct. Mol. Biol. 2023, 30, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, A.; Kong, F.; Chen, J.; Lan, B.; He, G.; Gao, K.; Cheng, L.; Sun, X.; Yan, C.; et al. Structural insights into human organic cation transporter 1 transport and inhibition. Cell Discov. 2024, 10, 30. [Google Scholar] [CrossRef]
- Zeng, Y.C.; Sobti, M.; Quinn, A.; Smith, N.J.; Brown, S.H.J.; Vandenberg, J.I.; Ryan, R.M.; O’Mara, M.L.; Stewart, A.G. Structural basis of promiscuous substrate transport by Organic Cation Transporter 1. Nat. Commun. 2023, 14, 6374. [Google Scholar] [CrossRef] [PubMed]
- Khanppnavar, B.; Maier, J.; Herborg, F.; Gradisch, R.; Lazzarin, E.; Luethi, D.; Yang, J.-W.; Qi, C.; Holy, M.; Jäntsch, K.; et al. Structural basis of organic cation transporter-3 inhibition. Nat. Commun. 2022, 13, 6714. [Google Scholar] [CrossRef] [PubMed]
- Dou, T.; Lian, T.; Shu, S.; He, Y.; Jiang, J. The substrate and inhibitor binding mechanism of polyspecific transporter OAT1 revealed by high-resolution cryo-EM. Nat. Struct. Mol. Biol. 2023, 30, 1794–1805. [Google Scholar] [CrossRef]
- Parker, J.L.; Kato, T.; Kuteyi, G.; Sitsel, O.; Newstead, S. Molecular basis for selective uptake and elimination of organic anions in the kidney by OAT1. Nat. Struct. Mol. Biol. 2023, 30, 1786–1793. [Google Scholar] [CrossRef]
- Janaszkiewicz, A.; Tóth, Á.; Faucher, Q.; Martin, M.; Chantemargue, B.; Barin-Le Guellec, C.; Marquet, P.; Di Meo, F. Insights into the structure and function of the human organic anion transporter 1 in lipid bilayer membranes. Sci. Rep. 2022, 12, 7057. [Google Scholar] [CrossRef] [PubMed]
- Janaszkiewicz, A.; Tóth, Á.; Faucher, Q.; Arnion, H.; Védrenne, N.; Barin-Le Guellec, C.; Marquet, P.; Di Meo, F. Substrate binding and lipid-mediated allostery in the human organic anion transporter 1 at the atomic-scale. Biomed. Pharmacother. 2023, 160, 114342. [Google Scholar] [CrossRef]
- Taniguchi, R.; Kato, H.E.; Font, J.; Deshpande, C.N.; Wada, M.; Ito, K.; Ishitani, R.; Jormakka, M.; Nureki, O. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun. 2015, 6, 8545. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, C.N.; Ruwe, T.A.; Shawki, A.; Xin, V.; Vieth, K.R.; Valore, E.V.; Qiao, B.; Ganz, T.; Nemeth, E.; Mackenzie, B.; et al. Calcium is an essential cofactor for metal efflux by the ferroportin transporter family. Nat. Commun. 2018, 9, 3075. [Google Scholar] [CrossRef]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural basis of ion transport and inhibition in ferroportin. Nat. Commun. 2020, 11, 5686. [Google Scholar] [CrossRef]
- Le Tertre, M.; Elbahnsi, A.; Ka, C.; Callebaut, I.; Le Gac, G. Insights into the Role of the Discontinuous TM7 Helix of Human Ferroportin through the Prism of the Asp325 Residue. Int. J. Mol. Sci. 2021, 22, 6412. [Google Scholar] [CrossRef] [PubMed]
- Custódio, T.F.; Paulsen, P.A.; Frain, K.M.; Pedersen, B.P. Structural Comparison of GLUT1 to GLUT3 Reveal Transport Regulation Mechanism in Sugar Porter Family. Life Sci. Alliance 2021, 4, e202000858. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Müller, T.; Siebeneicher, H.; et al. Mechanism of Inhibition of Human Glucose Transporter GLUT1 Is Conserved between Cytochalasin B and Phenylalanine Amides. Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal Structure of the Human Glucose Transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Deng, D.; Sun, P.; Yan, C.; Ke, M.; Jiang, X.; Xiong, L.; Ren, W.; Hirata, K.; Yamamoto, M.; Fan, S.; et al. Molecular Basis of Ligand Recognition and Transport by Glucose Transporters. Nature 2015, 526, 391–396. [Google Scholar] [CrossRef]
- Huang, J.; Yuan, Y.; Zhao, N.; Pu, D.; Tang, Q.; Zhang, S.; Luo, S.; Yang, X.; Wang, N.; Xiao, Y.; et al. Orthosteric–Allosteric Dual Inhibitors of PfHT1 as Selective Antimalarial Agents. Proc. Natl. Acad. Sci. USA 2021, 118, e2017749118. [Google Scholar] [CrossRef]
- Yuan, Y.; Kong, F.; Xu, H.; Zhu, A.; Yan, N.; Yan, C. Cryo-EM Structure of Human Glucose Transporter GLUT4. Nat. Commun. 2022, 13, 2671. [Google Scholar] [CrossRef]
- Niu, Y.; Cui, W.; Liu, R.; Wang, S.; Ke, H.; Lei, X.; Chen, L. Structural Mechanism of SGLT1 Inhibitors. Nat. Commun. 2022, 13, 6440. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Niu, Y.; Sun, Z.; Liu, R.; Chen, L. Structures of Human SGLT in the Occluded State Reveal Conformational Changes during Sugar Transport. Nat. Commun. 2023, 14, 2920. [Google Scholar] [CrossRef]
- Niu, Y.; Liu, R.; Guan, C.; Zhang, Y.; Chen, Z.; Hoerer, S.; Nar, H.; Chen, L. Structural Basis of Inhibition of the Human SGLT2–MAP17 Glucose Transporter. Nature 2022, 601, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Hiraizumi, M.; Akashi, T.; Murasaki, K.; Kishida, H.; Kumanomidou, T.; Torimoto, N.; Nureki, O.; Miyaguchi, I. Transport and Inhibition Mechanism of the Human SGLT2–MAP17 Glucose Transporter. Nat. Struct. Mol. Biol. 2024, 31, 159–169. [Google Scholar] [CrossRef]
- Boeszoermenyi, A.; Bernaleau, L.; Chen, X.; Kartnig, F.; Xie, M.; Zhang, H.; Zhang, S.; Delacrétaz, M.; Koren, A.; Hopp, A.-K.; et al. A Conformation-Locking Inhibitor of SLC15A4 with TASL Proteostatic Anti-Inflammatory Activity. Nat. Commun. 2023, 14, 6626. [Google Scholar] [CrossRef]
- Wang, N.; Jiang, X.; Zhang, S.; Zhu, A.; Yuan, Y.; Xu, H.; Lei, J.; Yan, C. Structural Basis of Human Monocarboxylate Transporter 1 Inhibition by Anti-Cancer Drug Candidates. Cell 2021, 184, 370–383.e13. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, M.; Zhang, B.; Chi, W.; Ma, X.; Zhang, W.; Dong, M.; Sheng, L.; Zhang, Y.; Jiao, W.; et al. Embigin Facilitates Monocarboxylate Transporter 1 Localization to the Plasma Membrane and Transition to a Decoupling State. Cell Rep. 2022, 40, 111343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jin, Q.; Xu, L.; Li, N.; Meng, Y.; Chang, S.; Zheng, X.; Wang, J.; Chen, Y.; Neculai, D.; et al. Cooperative Transport Mechanism of Human Monocarboxylate Transporter 2. Nat. Commun. 2020, 11, 2429. [Google Scholar] [CrossRef]
- Hu, W.; Chi, C.; Song, K.; Zheng, H. The Molecular Mechanism of Sialic Acid Transport Mediated by Sialin. Sci. Adv. 2023, 9, eade8346. [Google Scholar] [CrossRef]
- Ye, J.; Chen, H.; Wang, K.; Wang, Y.; Ammerman, A.; Awasthi, S.; Xu, J.; Liu, B.; Li, W. Structural Insights into Vesicular Monoamine Storage and Drug Interactions. Nature 2024, 629, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chen, Q.; Yu, Z.; Huang, B.; Zhao, J.; Wang, Y.; Su, J.; Zhou, F.; Yan, R.; Li, N.; et al. Transport and Inhibition Mechanisms of Human VMAT2. Nature 2024, 626, 427–434. [Google Scholar] [CrossRef]
- Pidathala, S.; Liao, S.; Dai, Y.; Li, X.; Long, C.; Chang, C.-L.; Zhang, Z.; Lee, C.-H. Mechanisms of Neurotransmitter Transport and Drug Inhibition in Human VMAT2. Nature 2023, 623, 1086–1092. [Google Scholar] [CrossRef]
- Dalton, M.P.; Cheng, M.H.; Bahar, I.; Coleman, J.A. Structural Mechanisms for VMAT2 Inhibition by Tetrabenazine. eLife 2024, 12, RP91973. [Google Scholar] [CrossRef]
- Ciută, A.-D.; Nosol, K.; Kowal, J.; Mukherjee, S.; Ramírez, A.S.; Stieger, B.; Kossiakoff, A.A.; Locher, K.P. Structure of Human Drug Transporters OATP1B1 and OATP1B3. Nat. Commun. 2023, 14, 5774. [Google Scholar] [CrossRef]
- Shan, Z.; Yang, X.; Liu, H.; Yuan, Y.; Xiao, Y.; Nan, J.; Zhang, W.; Song, W.; Wang, J.; Wei, F.; et al. Cryo-EM Structures of Human Organic Anion Transporting Polypeptide OATP1B1. Cell Res. 2023, 33, 940–951. [Google Scholar] [CrossRef]
- Chen, H.; Ahmed, S.; Zhao, H.; Elghobashi-Meinhardt, N.; Dai, Y.; Kim, J.H.; McDonald, J.G.; Li, X.; Lee, C.-H. Structural and Functional Insights into Spns2-Mediated Transport of Sphingosine-1-Phosphate. Cell 2023, 186, 2644–2655.e16. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Leong, N.C.P.; Zhao, J.; Zhang, Y.; Nguyen, D.T.; Ha, H.T.T.; Wang, N.; Xia, R.; Xu, Z.; Ma, Z.; et al. Structural Basis of Sphingosine-1-Phosphate Transport via Human SPNS2. Cell Res. 2023, 34, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Yu, L.; Li, T.; Jiao, H.; Wu, X.; Wang, J.; He, R.; Zhang, Y.; Wang, J.; Hu, H.; et al. Molecular Basis of Spns2-Facilitated Sphingosine-1-Phosphate Transport. Cell Res. 2023, 34, 173–176. [Google Scholar] [CrossRef]
- Son, Y.; Kenny, T.C.; Khan, A.; Birsoy, K.; Hite, R.K. Structural Basis of Lipid Head Group Entry to the Kennedy Pathway by FLVCR1. Nature 2024, 629, 710–716. [Google Scholar] [CrossRef]
- Ri, K.; Weng, T.-H.; Claveras Cabezudo, A.; Jösting, W.; Zhang, Y.; Bazzone, A.; Leong, N.C.P.; Welsch, S.; Doty, R.T.; Gursu, G.; et al. Molecular Mechanism of Choline and Ethanolamine Transport in Humans. Nature 2024, 630, 501–508. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Hénin, J.; Lelièvre, T.; Shirts, M.R.; Valsson, O.; Delemotte, L. Enhanced Sampling Methods for Molecular Dynamics Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2022, 4, 1583. [Google Scholar] [CrossRef]


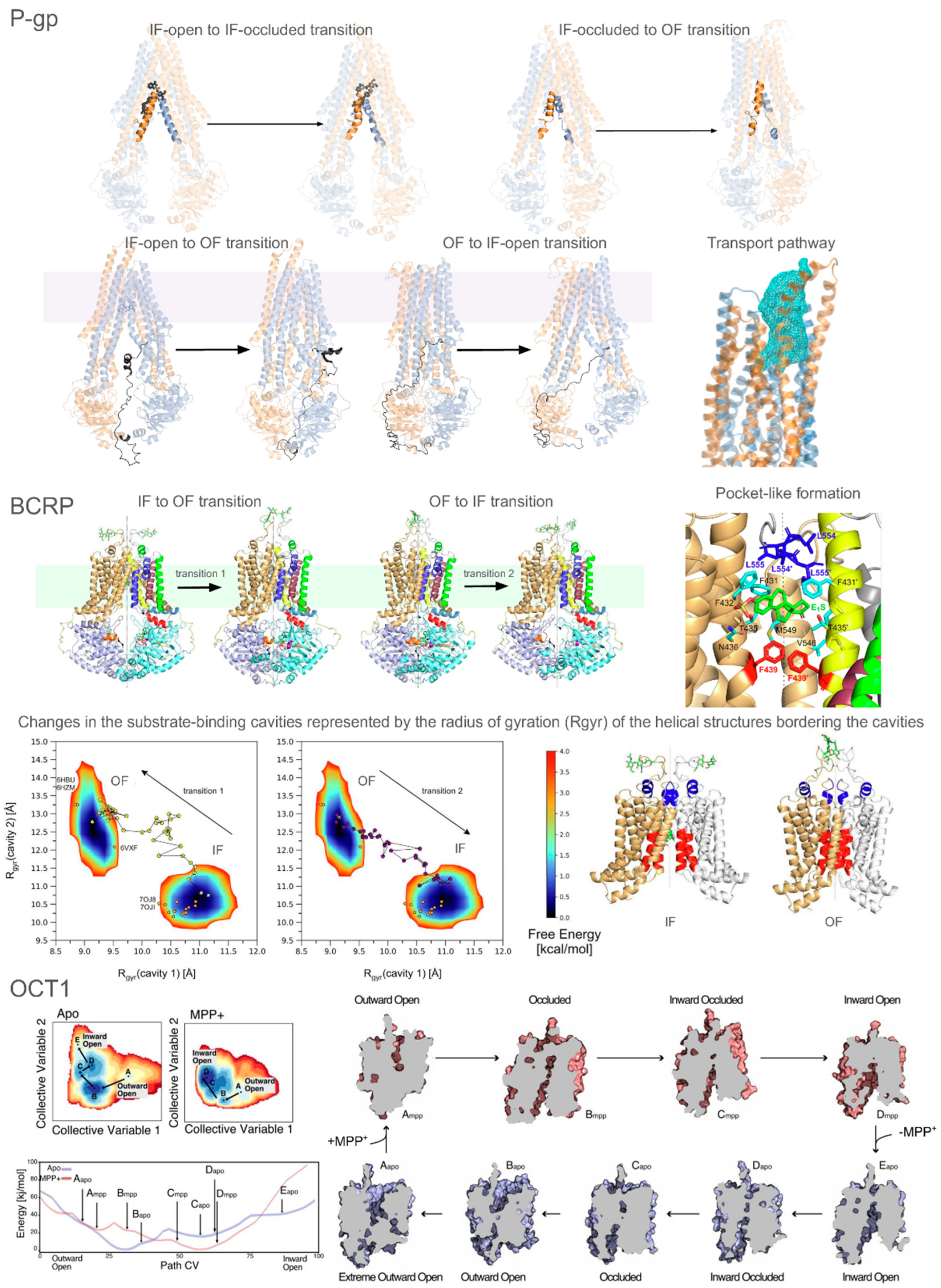
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbahnsi, A.; Dudas, B.; Callebaut, I.; Hinzpeter, A.; Miteva, M.A. ATP-Binding Cassette and Solute Carrier Transporters: Understanding Their Mechanisms and Drug Modulation Through Structural and Modeling Approaches. Pharmaceuticals 2024, 17, 1602. https://doi.org/10.3390/ph17121602
Elbahnsi A, Dudas B, Callebaut I, Hinzpeter A, Miteva MA. ATP-Binding Cassette and Solute Carrier Transporters: Understanding Their Mechanisms and Drug Modulation Through Structural and Modeling Approaches. Pharmaceuticals. 2024; 17(12):1602. https://doi.org/10.3390/ph17121602
Chicago/Turabian StyleElbahnsi, Ahmad, Balint Dudas, Isabelle Callebaut, Alexandre Hinzpeter, and Maria A. Miteva. 2024. "ATP-Binding Cassette and Solute Carrier Transporters: Understanding Their Mechanisms and Drug Modulation Through Structural and Modeling Approaches" Pharmaceuticals 17, no. 12: 1602. https://doi.org/10.3390/ph17121602
APA StyleElbahnsi, A., Dudas, B., Callebaut, I., Hinzpeter, A., & Miteva, M. A. (2024). ATP-Binding Cassette and Solute Carrier Transporters: Understanding Their Mechanisms and Drug Modulation Through Structural and Modeling Approaches. Pharmaceuticals, 17(12), 1602. https://doi.org/10.3390/ph17121602