



Targeting mTOR Kinase with Natural Compounds: Potent ATP-Competitive Inhibition Through Enhanced Binding Mechanisms

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
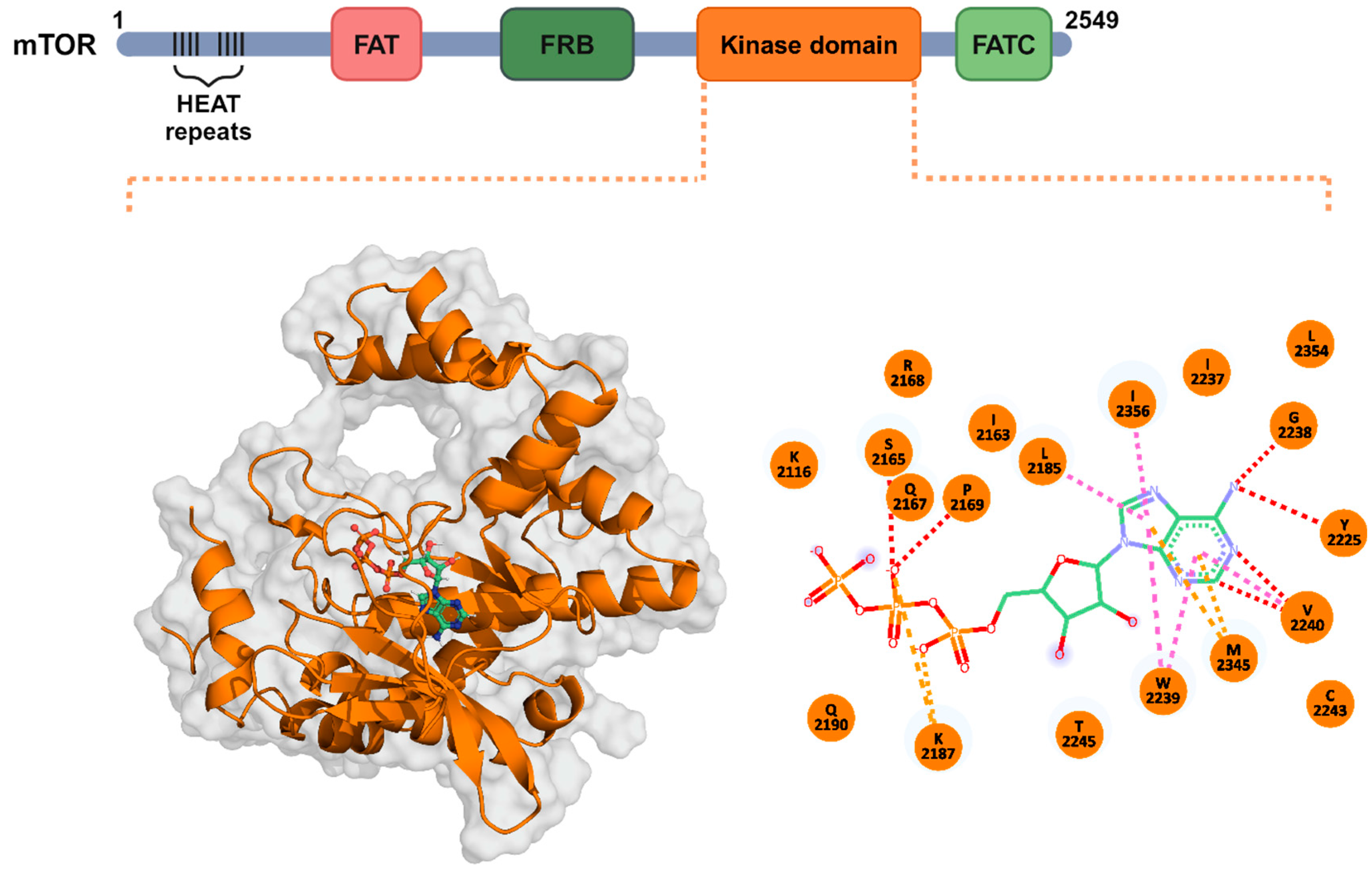
2.1. mTOR Kinase Domain Structural Modeling
2.2. Discovery of the mTOR-KD Inhibitors by Screening the TCM Library
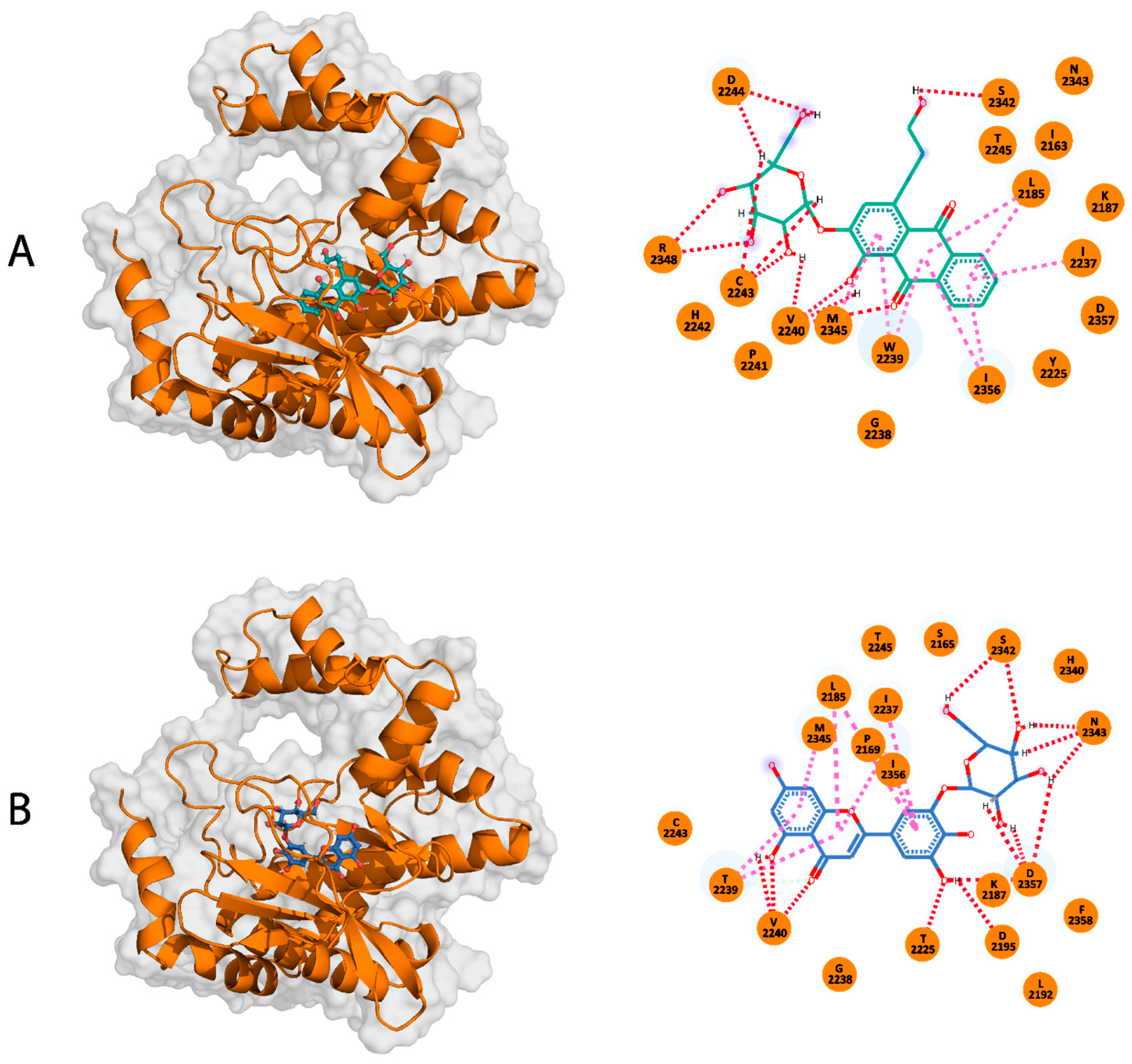
2.3. Binding Modes of the Selected TCM Compounds to the mTOR-KD
2.3.1. Compound A
2.3.2. Compound B
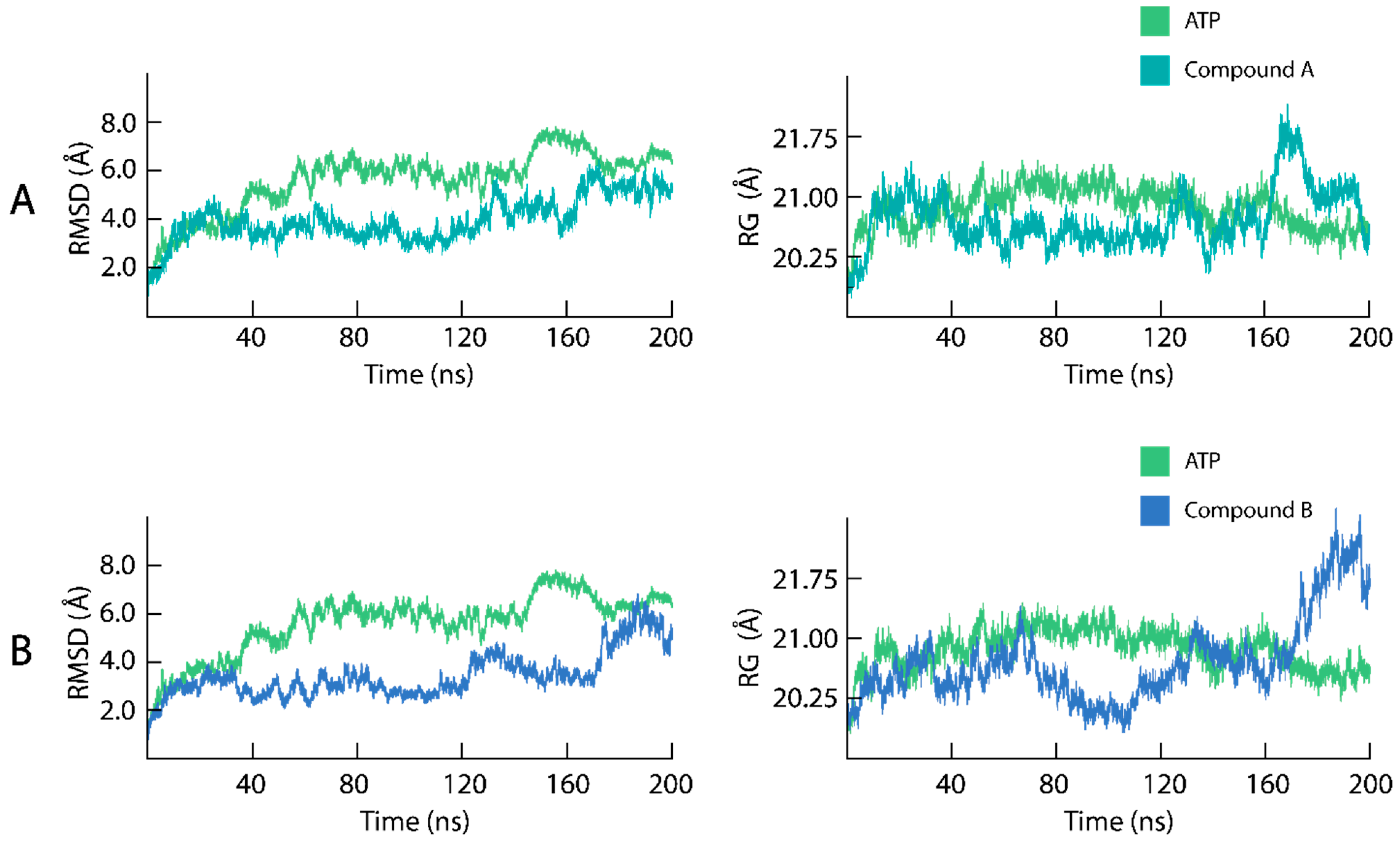
2.4. Dynamic Stability and Compactness Assessment of the mTOR-KD Ligand Binding
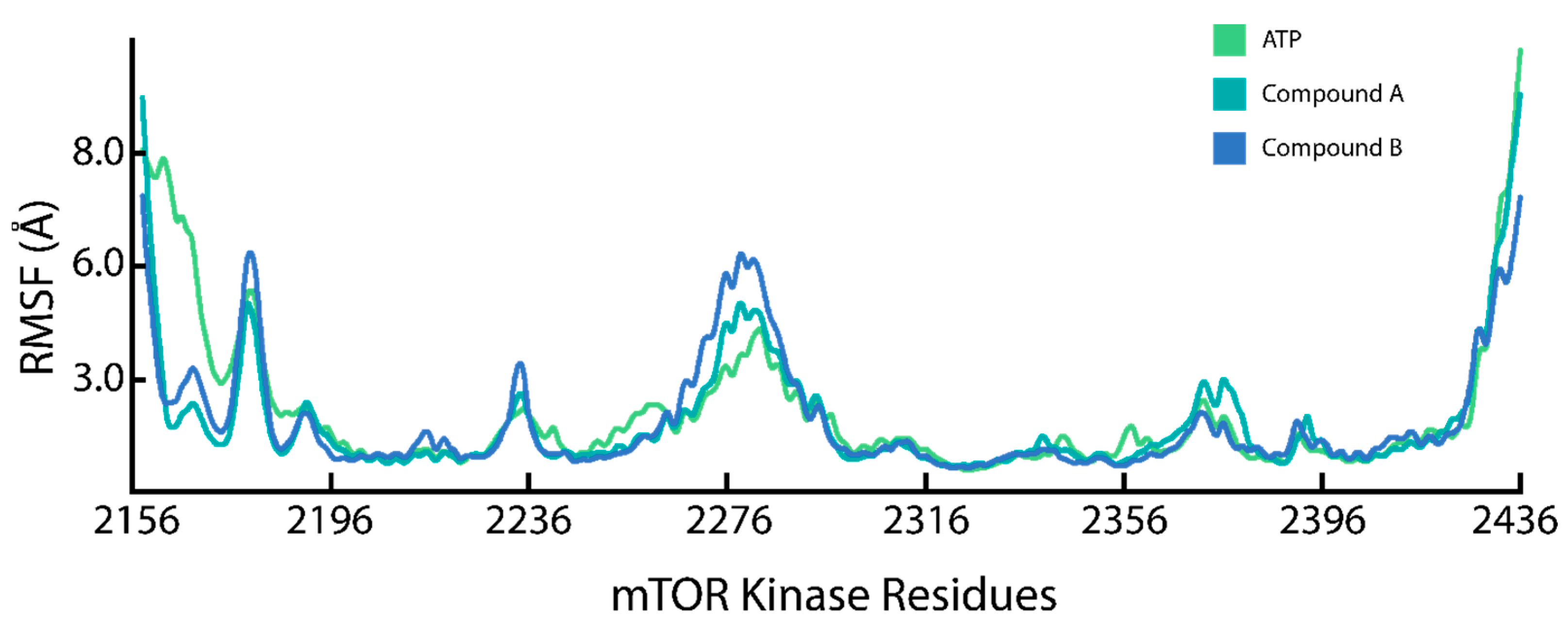
2.5. Residue Flexibility Analysis of the mTOR-KD Ligand Binding
2.6. Binding Free Energy Estimation of the mTOR-KD with Ligands
3. Materials and Methods
3.1. Structures, Sequence Retrieval, and Modeling
3.2. Structures and Validation of Molecular Screening
3.3. Molecular Screening of Natural Product Libraries
3.4. Molecular Simulation of Top Scoring Hits
3.5. Post-Simulation Analysis of the Protein–Ligand Complexes
- ri is the position of the atom at index i;
- mi is the mass of the atom at index i;
- rCM is the center of mass;
- N is the number of atoms being counted;
- r2RG is the square of the radius of gyration.
- B is B-factor, and⟨Δr2⟩ is the mean square deviation (i.e., ⟨Δr2⟩ = RMSD2).
- By rearranging the above equation and accounting for 3 spatial dimensions, we can obtain the RMSF as follows:
3.6. Binding Free Energy Calculation Using the MM/GBSA Approach
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leung, A.; Rangamani, P. Computational modeling of AMPK and mTOR crosstalk in glutamatergic synapse calcium signaling. NPJ Syst. Biol. Appl. 2023, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Blenis, J.; Proud, C.G. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005, 579, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Marques-Ramos, A.; Cervantes, R. Expression of mTOR in normal and pathological conditions. Mol. Cancer 2023, 22, 112. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Z.; Peng, J.; Loor, J.J. Methionine and valine activate the mammalian target of rapamycin complex 1 pathway through heterodimeric amino acid taste receptor (TAS1R1/TAS1R3) and intracellular Ca(2+) in bovine mammary epithelial cells. J. Dairy Sci. 2018, 101, 11354–11363. [Google Scholar] [CrossRef]
- Pende, M.; Kozma, S.C.; Jaquet, M.; Oorschot, V.; Burcelin, R.; Le Marchand-Brustel, Y.; Klumperman, J.; Thorens, B. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000, 408, 994–997. [Google Scholar] [CrossRef]
- Xin-Long, C.; Zhao-Fan, X.; Dao-Feng, B.; Wei, D. mTOR partly mediates insulin resistance by phosphorylation of insulin receptor substrate-1 on serine(307) residues after burn. Burns 2011, 37, 86–93. [Google Scholar] [CrossRef]
- Yu, W.; Li, C.; Zhang, D.; Li, Z.; Xia, P.; Liu, X.; Cai, X.; Yang, P.; Ling, J.; Zhang, J.; et al. Advances in T Cells Based on Inflammation in Metabolic Diseases. Cells 2022, 11, 3554. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-I.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR interacts with Raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.-I.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef]
- Ma, X.J.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Oh, W.; Wu, C.-C.; Kim, S.J.; Facchinetti, V.; Julien, L.-A.; Finlan, M.; Roux, P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef]
- Deng, Y.F.; Wu, S.T.; Peng, H.Y.; Tian, L.; Li, Y.N.; Yang, Y.; Meng, M.; Huang, L.-L.; Xiong, P.-W.; Li, S.-Y.; et al. mTORC2 acts as a gatekeeper for mTORC1 deficiency-mediated impairments in ILC3 development. Acta Pharmacol. Sin. 2023, 44, 2243–2252. [Google Scholar] [CrossRef]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor Phosphorylation Sites Reveals Direct Regulation of mTOR Complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef]
- Tzatsos, A.; Kandror, K.V. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol. Cell. Biol. 2006, 26, 63–76. [Google Scholar] [CrossRef]
- Marafie, S.K.; Al-Shawaf, E.M.; Abubaker, J.; Arefanian, H. Palmitic acid-induced lipotoxicity promotes a novel interplay between Akt-mTOR, IRS-1, and FFAR1 signaling in pancreatic β-cells. Biol. Res. 2019, 52, 44. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell. 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.; Loo, Y.-L.; Patel, J.; Nguyen, J.T.; Kim, J.K.; Krebsbach, P.H. Targeting of lysosomal-bound protein mEAK-7 for cancer therapy. Front. Oncol. 2024, 14, 1375498. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonça, D.B.; Kim, J.K.; Krebsbach, P.H. mEAK-7 Forms an Alternative mTOR Complex with DNA-PKcs in Human Cancer. iScience 2019, 17, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonça, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4, eaao5838. [Google Scholar] [CrossRef]
- Mendonca, D.B.; Nguyen, J.T.; Haidar, F.; Fox, A.L.; Ray, C.; Amatullah, H.; Liu, F.; Kim, J.K.; Krebsbach, P.H. MicroRNA-1911-3p targets mEAK-7 to suppress mTOR signaling in human lung cancer cells. Heliyon 2020, 6, e05734. [Google Scholar] [CrossRef]
- Smithson, L.J.; Gutmann, D.H. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev. 2016, 30, 1383–1388. [Google Scholar] [CrossRef]
- Harwood, F.C.; Geltink, R.I.K.; O’hara, B.P.; Cardone, M.; Janke, L.; Finkelstein, D.; Entin, I.; Paul, L.; Houghton, P.J.; Grosveld, G.C. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci. Adv. 2018, 4, eaar3938. [Google Scholar] [CrossRef]
- Karki, R.; Man, S.M.; Malireddi, R.S.; Kesavardhana, S.; Zhu, Q.; Burton, A.R.; Sharma, B.R.; Qi, X.; Pelletier, S.; Vogel, P.; et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 2016, 540, 583–587. [Google Scholar] [CrossRef]
- Watanabe, R.; Wei, L.; Huang, J. mTOR signaling, function, novel inhibitors, and therapeutic targets. J. Nucl. Med. 2011, 52, 497–500. [Google Scholar] [CrossRef]
- Dancey, J. mTOR signaling and drug development in cancer. Nat. Rev. Clin. Oncol. 2010, 7, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Marafie, S.K.; Al-Mulla, F.; Abubaker, J. mTOR: Its Critical Role in Metabolic Diseases, Cancer, and the Aging Process. Int. J. Mol. Sci. 2024, 25, 6141. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Mitra, K.; Akter, S.; Ramproshad, S.; Mondal, B.; Khan, I.N.; Islam, M.T.; Sharifi-Rad, J.; Calina, D.; Cho, W.C. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 2022, 22, 284. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Zhang, Q.; Ma, L.; Zhao, D.-S.; Zhao, P.; Yan, P. Overview of Research into mTOR Inhibitors. Molecules 2022, 27, 5295. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Zheng, B.; Mao, J.-H.; Qian, L.; Zhu, H.; Gu, D.-H.; Pan, X.-D.; Yi, F.; Ji, D.-M. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef]
- Janes, M.R.; Vu, C.; Mallya, S.; Shieh, M.P.; Limon, J.J.; Li, L.S.; Jessen, K.A.; Martin, M.B.; Ren, P.; Lilly, M.B.; et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia 2013, 27, 586–594. [Google Scholar] [CrossRef]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: Distinct from rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef]
- Korets, S.B.; Musa, F.; Curtin, J.; Blank, S.V.; Schneider, R.J. Dual mTORC1/2 inhibition in a preclinical xenograft tumor model of endometrial cancer. Gynecol. Oncol. 2014, 132, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Y.; Xiong, Z.-Y.; Deng, Z.-Y.; Song, H.-L.; An, Z.-M. Changes of plasma fibroblast growth factor-21 (FGF-21) in oral glucose tolerance test and effects of metformin on FGF-21 levels in type 2 diabetes mellitus. Endokrynol. Pol. 2013, 64, 220–224. [Google Scholar] [PubMed]
- Subbiah, V.; Coleman, N.; Piha-Paul, S.A.; Tsimberidou, A.M.; Janku, F.; Rodon, J.; Pant, S.; Dumbrava, E.E.I.; Fu, S.; Hong, D.S.; et al. Phase I Study of mTORC1/2 Inhibitor Sapanisertib (CB-228/TAK-228) in Combination with Metformin in Patients with mTOR/AKT/PI3K Pathway Alterations and Advanced Solid Malignancies. Cancer Res. Commun. 2024, 4, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K.; Hong, S.-E.; Lee, D.-H.; Kim, J.-Y.; Ye, S.-K.; Hong, J.; Park, I.-C.; Jin, H.-O. Inhibition of mTORC1 through ATF4-induced REDD1 and Sestrin2 expression by Metformin. BMC Cancer 2021, 21, 803. [Google Scholar]
- Melnik, B.C.; Schmitz, G. Metformin: An Inhibitor of mTORC1 Signaling. J. Endocrinol. Diabetes Obes. 2014, 2, 1029. [Google Scholar]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: A tale of two complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef]
- Chen, C.Y.-C. TCM Database@ Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, P.; He, W.; Qin, C.; Chen, S.; Tao, L.; Wang, Y.; Tan, Y.; Gao, D.; Wang, B.; et al. NPASS: Natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018, 46, D1217–D1222. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of open natural products database. J. Cheminformatics 2021, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Shahab, M.; Nasir, F.; Waheed, Y.; Alshammari, A.; Mohammad, A.; Zichen, G.; Li, R.; Wei, D.Q. Exploring the Traditional Chinese Medicine (TCM) database chemical space to target I7L protease from monkeypox virus using molecular screening and simulation approaches. SAR QSAR Environ. Res. 2023, 34, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.-C.; Wu, Y.-C.; Chen, Z.-W.; Yang, W.-C. Naturally Occurring Anthraquinones: Chemistry and Therapeutic Potential in Autoimmune Diabetes. Evid.-Based Complement. Altern. Med. 2015, 2015, 357357. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.S.; Alsantali, R.I.; Jassas, R.S.; Alsimaree, A.A.; Syed, R.; Alsharif, M.A.; Kalpana, K.; Morad, M.; Althagafi, I.I.; Ahmed, S.A. Journey of anthraquinones as anticancer agents—A systematic review of recent literature. RSC Adv. 2021, 11, 35806–35827. [Google Scholar] [CrossRef]
- Qun, T.; Zhou, T.; Hao, J.; Wang, C.; Zhang, K.; Xu, J.; Wang, X.; Zhou, W. Antibacterial activities of anthraquinones: Structure–activity relationships and action mechanisms. RSC Med. Chem. 2023, 14, 1446–1471. [Google Scholar] [CrossRef]
- Jasemi, S.V.; Khazaei, H.; Morovati, M.R.; Joshi, T.; Aneva, I.Y.; Farzaei, M.H.; Echeverría, J. Phytochemicals as treatment for allergic asthma: Therapeutic effects and mechanisms of action. Phytomedicine 2024, 122, 155149. [Google Scholar] [CrossRef]
- Zhao, L.; Zheng, L. A Review on Bioactive Anthraquinone and Derivatives as the Regulators for ROS. Molecules 2023, 28, 8139. [Google Scholar] [CrossRef]
- Soto-Blanco, B. Chapter 12—Herbal glycosides in healthcare. In Herbal Biomolecules in Healthcare Applications; Mandal, S.C., Nayak, A.K., Dhara, A.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 239–282. [Google Scholar]
- Zhou, Y.; He, Y.-J.; Wang, Z.-J.; Hu, B.-Y.; Xie, T.-Z.; Xiao, X.; Zhou, Z.-S.; Sang, X.-Y.; Luo, X.-D. A review of plant characteristics, phytochemistry and bioactivities of the genus Glechoma. J. Ethnopharmacol. 2021, 271, 113830. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef]
- Kerhoas, L.; Aouak, D.; Cingöz, A.; Routaboul, J.-M.; Lepiniec, L.; Einhorn, J.; Birlirakis, N. Structural Characterization of the Major Flavonoid Glycosides from Arabidopsis thaliana Seeds. J. Agric. Food Chem. 2006, 54, 6603–6612. [Google Scholar] [CrossRef] [PubMed]
- Vukics, V.; Guttman, A. Structural characterization of flavonoid glycosides by multi-stage mass spectrometry. Mass Spectrom. Rev. 2010, 29, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed]
- May, A.; Zacharias, M. Accounting for global protein deformability during protein-protein and protein-ligand docking. Biochim. Biophys. Acta 2005, 1754, 225–231. [Google Scholar] [CrossRef]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Functional aspects of protein flexibility. Cell Mol. Life Sci. 2009, 66, 2231–2247. [Google Scholar] [CrossRef]
- Verma, R.; Mitchell-Koch, K. In Silico Studies of Small Molecule Interactions with Enzymes Reveal Aspects of Catalytic Function. Catalysts 2017, 7, 212. [Google Scholar] [CrossRef]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef]
- Virtanen, S.I.; Niinivehmas, S.P.; Pentikainen, O.T. Case-specific performance of MM-PBSA, MM-GBSA, and SIE in virtual screening. J. Mol. Graph Model 2015, 62, 303–318. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLOS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Bell, J.; Cao, Y.; Gunn, J.R.; Day, T.; Gallicchio, E.; Zhou, Z.; Levyb, R.; Farid, R. PrimeX and the Schrödinger Computational Chemistry Suite of Programs. 2012. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1107/97809553602060000864 (accessed on 10 April 2024).
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- Wang, N.; Zhou, K.; Liang, Z.; Sun, R.; Tang, H.; Yang, Z.; Zhao, W.; Peng, Y.; Song, P.; Zheng, S.; et al. RapaLink-1 outperforms rapamycin in alleviating allogeneic graft rejection by inhibiting the mTORC1-4E-BP1 pathway in mice. Int. Immunopharmacol. 2023, 125, 111172. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D Structure | Compound Name | Docking Scores | Identifier |
|---|---|---|---|
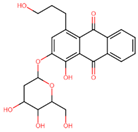 | 1-hydroxy-4-(3-hydroxypropyl)-2-[(2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyanthracene-9,10-dione | −15.455 | A |
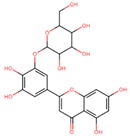 | 2-[3,4-dihydroxy-5-[(2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-5,7-dihydroxychromen-4-one | −14.886 | B |
| Molecular. Formula | Compound | MW. (g/mol) | Source | Molecule Class | Biological Activity | Lipinski Violation | Pfizer Rule * | ** AMES Toxicity | *** IGC50 | HBD | HBA | Rotatable Bonds No. | TPSA (Å2) | Bioactivity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C23H24O10 | Compound A | 460.44 | Rubiaceae, Rhamnaceae, and Polygonaceae families | Anthraquinone | Antimicrobial | 1 | pass | ++ | 4.638 | 6 | 10 | 6 | 174 | 0.45 |
| C21H20O12 | Compound B | 464.38 | Ocimum sanctum, Phyllostachys species | Flavonoid | Anti-inflammatory | 2 | pass | + | 3.878 | 8 | 12 | 4 | 211 | 0.44 |
| Parameters | ATP | Compound A | Compound B |
|---|---|---|---|
| VDWAALS | −30.83 ± 0.20 | −41.15 ± 0.13 | −40.92 ± 0.18 |
| EEL | −81.28 ± 1.25 | −40.45 ± 0.34 | −99.52 ± 0.62 |
| EGB | 104.94 ± 1.12 | 49.28 ± 0.27 | 91.88 ± 0.37 |
| ESURF | −4.39 ± 0.03 | −6.03 ± 0.01 | −7.67 ± 0.01 |
| DELTA G gas | −112.11 ± 1.27 | −81.61 ± 0.36 | −140.45 ± 0.57 |
| DELTA G solv | 100.55 ± 1.12 | 43.25 ± 0.26 | 84.21 ± 0.36 |
| DELTA TOTAL | −11.56 ± 0.46 | −38.36 ± 0.18 | −56.23 ± 0.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marafie, S.K.; Alshawaf, E.; Al-Mulla, F.; Abubaker, J.; Mohammad, A. Targeting mTOR Kinase with Natural Compounds: Potent ATP-Competitive Inhibition Through Enhanced Binding Mechanisms. Pharmaceuticals 2024, 17, 1677. https://doi.org/10.3390/ph17121677
Marafie SK, Alshawaf E, Al-Mulla F, Abubaker J, Mohammad A. Targeting mTOR Kinase with Natural Compounds: Potent ATP-Competitive Inhibition Through Enhanced Binding Mechanisms. Pharmaceuticals. 2024; 17(12):1677. https://doi.org/10.3390/ph17121677
Chicago/Turabian StyleMarafie, Sulaiman K., Eman Alshawaf, Fahd Al-Mulla, Jehad Abubaker, and Anwar Mohammad. 2024. "Targeting mTOR Kinase with Natural Compounds: Potent ATP-Competitive Inhibition Through Enhanced Binding Mechanisms" Pharmaceuticals 17, no. 12: 1677. https://doi.org/10.3390/ph17121677
APA StyleMarafie, S. K., Alshawaf, E., Al-Mulla, F., Abubaker, J., & Mohammad, A. (2024). Targeting mTOR Kinase with Natural Compounds: Potent ATP-Competitive Inhibition Through Enhanced Binding Mechanisms. Pharmaceuticals, 17(12), 1677. https://doi.org/10.3390/ph17121677