
Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

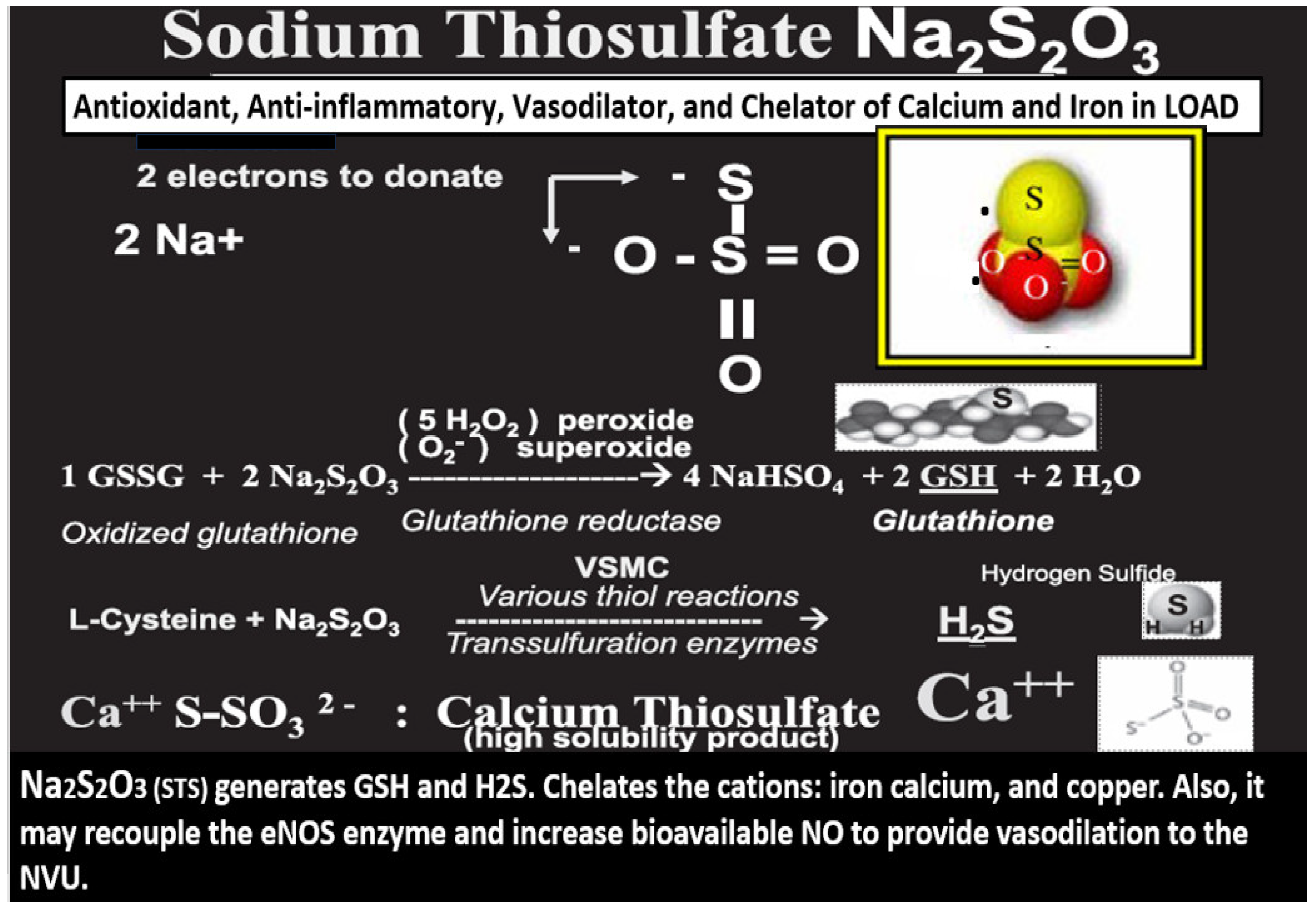
2. Antioxidant Effects of STS
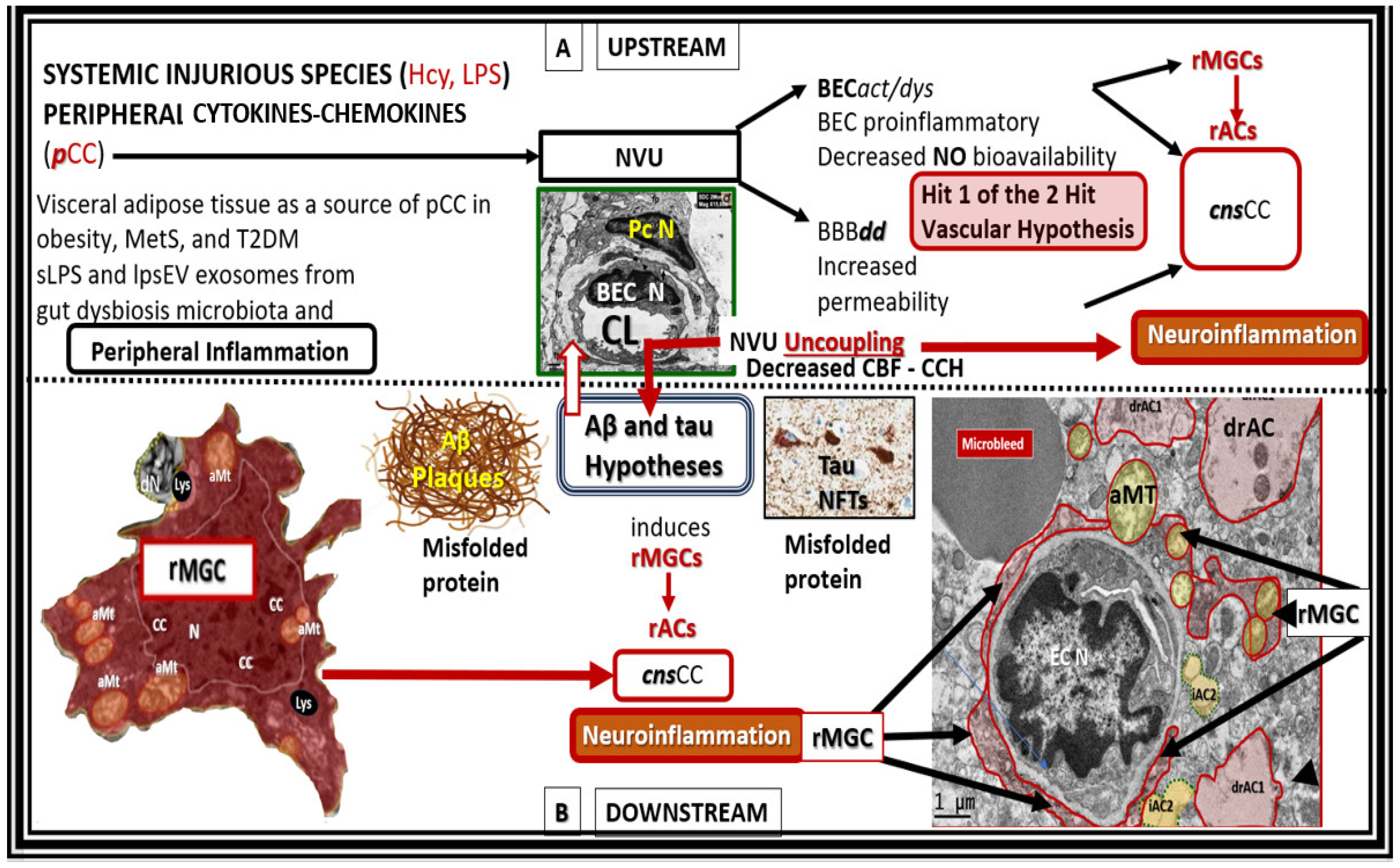
3. Anti-Inflammatory Effects of STS
4. Chelation Effects of STS in Regard to Excess Calcium, Iron, and Copper
4.1. Glutamate Excitotoxicity (GlutET) and Calcium (Ca2+) Excess/Overload

4.2. Iron Overload Due to Microvessel Small Vessel Disease (mvSVD): Cerebral Microbleed(s) (CMBs) in LOAD
4.3. Copper (Cu) Dyshomeostasis and Cu Excess in LOAD
5. Neurovascular Effects of STS: Restoration of the Neurovascular Unit Brain Endothelial Cell Pro-Oxidative, Proinflammatory, and Proconstrictive State (Vascular Hypothesis)
6. Possible Intravenous Sodium Thiosulfate Treatment Paradigms in LOAD
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rabinovici, G.D. Late-onset Alzheimer Disease. Contin. Lifelong Learn. Neurol. 2019, 25, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Type 2 Diabetes Mellitus Increases The Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Medicina 2021, 58, 16. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged ≥65 years. Alzheimer’s Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef]
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J.; Buckley, R.F.; Hohman, T.J. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three Dimensions of the Amyloid Hypothesis: Time, Space, and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Paying Homage to Microvessel Remodeling and Small Vessel Disease in Neurodegeneration: Implications for the Development of Late-Onset Alzheimer’s Disease. J. Vasc. Dis. 2024, 3, 419–452. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Alhazmi, H.A.; Albratty, M. An update on the novel and approved drugs for Alzheimer disease. Saudi Pharm J. 2022, 30, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Medications for Memory, Cognition and Dementia-Related Diseases. FDA-Approved Drugs for Alzheimer’s. Available online: https://www.alz.org/alzheimers-dementia/treatments/medications-for-memory (accessed on 6 September 2024).
- Agis-Torres, A.; Sölhuber, M.; Fernandez, M.; Sanchez-Montero, J.M. Multi-Target-Directed Ligands and other Therapeutic Strategies in the search of a Real Solution for Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Domanowska, A.; Wódkiewicz, E.; Malinowski, B. Neuroprotective Properties of Resveratrol and Its Derivatives—Influence on Potential Mechanisms Leading to the Development of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2749. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer Disease, a Multifactorial Disorder Seeking Multi-therapies. Alzheimer’s Dement. 2010, 6, 420–424. [Google Scholar] [CrossRef]
- Rao, R.V.; Subramaniam, K.G.; Gregory, J.; Bredesen, A.L.; Coward, C.; Okada, S.; Kelly, L.; Bredesen, D.E. Rationale for a Multi-Factorial Approach for the Reversal of Cognitive Decline in Alzheimer’s Disease and MCI: A Review. Int. J. Mol. Sci. 2023, 24, 1659. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Cummings, J.L.; Goldman, D.P.; Simmons-Stern, N.R.; Ponton, E. The costs of developing treatments for Alzheimer’s disease: A retrospective exploration. Alzheimer’s Dement. 2022, 18, 469–477. [Google Scholar] [CrossRef]
- Cicone, J.S.; Petronis, J.B.; Embert, C.D.; Spector, D.A. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am. J. Kidney Dis. 2004, 43, 1104–1108. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C.; Kolb, L.; Sowers, J.R.; Khanna, R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: The emerging role of sodium thiosulfate. Cardiovasc. Diabetol. 2005, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Kolb, L.G.; Khanna, R. Calciphylaxis and the cardiometabolic syndrome. J. Cardiometab. Syndr. 2006, 1, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Yerram, P.; Saab, G.; Karuparthi, P.R.; Hayden, M.R.; Khanna, R. Nephrogenic systemic fibrosis: A mysterious disease in patients with renal failure--role of gadolinium-based contrast media in causation and the beneficial effect of intravenous sodium thiosulfate. Clin. J. Am. Soc. Nephrol. 2007, 2, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Calciphylaxis and the cardiometabolic syndrome: The emerging role of sodium thiosulfate as a novel treatment option. J. Cardiometab. Syndr. 2008, 3, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Goldsmith, D.; Sowers, J.R.; Khanna, R. Calciphylaxis: Calcific uremic arteriolopathy and the emerging role of sodium thiosulfate. Int. Urol. Nephrol. 2008, 40, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Goldsmith, D.J. Sodium thiosulfate: New hope for the treatment of calciphylaxis. Semin. Dial. 2010, 23, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Sowers, K.M.; Hayden, M.R. Calcific uremic arteriolopathy: Pathophysiology, reactive oxygen species and therapeutic approaches. Oxidative Med. Cell. Longev. 2010, 3, 109–121. [Google Scholar] [CrossRef]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Sodium thiosulfate attenuates glial-mediated neuroinflammation in degenerative neurological diseases. J. Neuroinflamm. 2016, 13, 32. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G.; Lee, M. Medical uses of Sodium thiosulfate. J. Neurol. Neuromed. 2016, 1, 28–30. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Dugbartey, G.J.; Juriasingani, S.; Sener, A. Hydrogen Sulfide Metabolite, Sodium Thiosulfate: Clinical Applications and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6452. [Google Scholar] [CrossRef] [PubMed]
- Macabrey, D.; Longchamp, A.; MacArthur, M.R.; Lambelet, M.; Urfer, S.; Deglise, S.; Allagnat, F. Sodium thiosulfate acts as a hydrogen sulfide mimetic to prevent intimal hyperplasia via inhibition of tubulin polymerisation. EBioMedicine 2022, 78, 103954. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Deleon, E.R.; Gao, Y.; Hurley, K.; Sadauskas, V.; Batz, C.; Stoy, G.F. Thiosulfate: A readily accessible source of hydrogen sulfide in oxygen sensing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R592–R603. [Google Scholar] [CrossRef] [PubMed]
- Snijder, P.M.; Frenay, A.R.; de Boer, R.A.; Pasch, A.; Hillebrands, J.L.; Leuvenink, H.G.D.; van Goor, H. Exogenous administration of thiosulfate, a donor of hydrogen sulfide, attenuates angiotensin II-induced hypertensive heart disease in rats. Br. J. Pharmacol. 2015, 172, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Yamada, M.; Ida, T.; Tokuda, K.; Ikeda, K.; Kai, S.; Shirozu, K.; Hayashida, K.; Kosugi, S.; Hanaoka, K.; et al. Thiosulfate mediates cytoprotective effects of hydrogen sulfide against neuronal ischemia. J. Am. Heart Assoc. 2015, 4, e002125. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef]
- Zhang, X.; Bian, J.S. Hydrogen Sulfide: A Neuromodulator and Neuroprotectant in the Central Nervous System. ACS Chem. Neurosci. 2014, 5, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Niu, Y.Y.; Jiang, W.Z.; Tang, H.L.; Zhang, C.; Xia, Q.M.; Tang, X.Q. Neuroprotective effects of hydrogen sulfide and the underlying signaling pathways. Rev. Neurosci. 2015, 26, 129–142. [Google Scholar] [CrossRef]
- White, A.R.; Huang, X.; Jobling, M.F.; Barrow, C.J.; Beyreuther, K.; Masters, C.L.; Bush, A.I.; Cappai, R. Homocysteine potentiates copper- and amyloid beta peptide mediated toxicity in primary neuronal cultures: Possible risk factors in the Alzheimer’s-type neurodegenerative pathways. J. Neurochem. 2001, 76, 1509–1520. [Google Scholar] [CrossRef]
- Ho, P.I.; Collins, S.C.; Dhitavat, S.; Ortiz, D.; Ashline, D.; Rogers, E.; Shea, T.B. Homocysteine potentiates beta-amyloid neurotoxicity: Role of oxidative stress. J. Neurochem. 2001, 78, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.J.; Xu, J.H.; Li, M.H.; Tang, J.P.; Zou, W.; Zhang, P.; Wang, L.; Wag, C.Y.; Tang, X.Q. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 2014, 35, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.Q.; Yang, C.T.; Chen, J.; Yin, W.L.; Tian, S.W.; Hu, B.; Feng, J.; Li, Y. Effect of hydrogen sulphide on beta-amyloid-induced damage in PC12 cells. Clin. Exp. Pharmacol. Physiol. 2008, 35, 180–186. [Google Scholar] [CrossRef]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef]
- Disbrow, E.; Stokes, K.Y.; Ledbetter, C.; Patterson, J.; Kelley, R.; Pardue, S.; Reekes, T.; Larmeu, L.; Batra, V.; Yuan, S.; et al. Plasma hydrogen sulfide: A biomarker of Alzheimer’s disease and related dementias. Alzheimer’s Dement. 2021, 17, 1391–1402. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, J.; Chang, H.; Liu, X.; Zhu, R. Homocysteine and Folic Acid: Risk Factors for Alzheimer’s Disease—An Updated Meta-Analysis. Front. Aging Neurosci. 2021, 13, 665114. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. One-Carbon Metabolism and Alzheimer’s Disease: Focus on Epigenetics. Curr. Genom. 2010, 11, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Pieper, A.A. Protective Roles of Hydrogen Sulfide in Alzheimer’s Disease and Traumatic Brain Injury. Antioxidants 2023, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen sulfide as a neuromodulator. Mol. Neurobiol. 2002, 26, 13–19. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. The Mighty Mitochondria Are Unifying Organelles and Metabolic Hubs in Multiple Organs of Obesity, Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes: An Observational Ultrastructure Study. Int. J. Mol. Sci. 2022, 23, 4820. [Google Scholar] [CrossRef]
- Long, J.M.; Maloney, B.; Rogers, J.T.; Lahiri, D.K. Novel upregulation of amyloid-beta precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2019, 24, 345–363. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Altamura, S.; Muckenthaler, M.U. Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimer’s Dis. 2009, 16, 879–895. [Google Scholar] [CrossRef]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Read, A.D.; Bentley, R.E.T.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron–sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef] [PubMed]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimer’s Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef]
- Isaya, G. Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease. Front. Pharmacol. 2014, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chang, X.; Lang, M. Iron Homeostasis Disorder and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12442. [Google Scholar] [CrossRef] [PubMed]
- Iciek, M.; Bilska-Wilkosz, A.; Kozdrowicki, M.; Górny, M. Reactive sulfur species and their significance in health and disease. Biosci. Rep. 2022, 42, BSR20221006. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc. Diabetol. 2002, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Wong-Guerra, M.; Calfio, C.; Maccioni, R.B.; Rojo, L.E. Revisiting the neuroinflammation hypothesis in Alzheimer’s disease: A focus on the druggability of current targets. Front. Pharmacol. 2023, 14, 1161850. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Sun, Y.; Peng, G. Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease. Clin. Interv. Aging 2022, 17, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Akama, K.T.; Eldik, L.J.V. β-Amyloid Stimulation of Inducible Nitric-oxide Synthase in Astrocytes Is Interleukin-1β- and Tumor Necrosis Factor-α (TNFα)-dependent, and Involves a TNFα Receptor-associated Factor- and NFκB-inducing Kinase-dependent Signaling Mechanism. J. Biol. Chem. 2000, 275, 7918–7924. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of β-Amyloid-Stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.T.; Sheng, J.G.; Roberts, G.W.; Mrak, R.E. Interleukin-1 expression in different plaque types in Alzheimer’s disease: Significance in plaque evolution. J. Neuropathol. Exp. Neurol. 1995, 54, 276–281. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Akiyama, H.; Itagaki, S.; McGeer, E.G. Immune system response in Alzheimer’s disease. Can. J. Neurol. Sci. 1989, 16, 516–527. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S.T. Common inflammatory mechanisms in Lewy Body disease and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2007, 66, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Sheng, J.G.; Griffin, W.S.T. Glial cytokines in Alzheimer’s disease: Review and pathogenic implications. Hum. Pathol. 1995, 26, 816–823. [Google Scholar] [CrossRef]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Boche, D. Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 42. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.; Vugts, D.J.; Funke, U.; Molenaar, G.T.; Kruijer, P.S.; van Berckel, B.N.M. Imaging of neuroinflammation in Alzheimer’s disease, multiple sclerosis and stroke: Recent developments in positron emission tomography. Biochim. Biophys. Acta 2016, 1862, 425–441. [Google Scholar] [CrossRef]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuro-psychopharmacol. Biol. Psychiatry 2018, 80 Pt B, 123–131. [Google Scholar] [CrossRef]
- Lagarde, J.; Sarazin, M.; Bottlaender, M. In vivo PET imaging of neuroinflammation in Alzheimer’s disease. J. Neural. Transm. 2018, 125, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking neuroinflammation in Alzheimer’s disease: The role of positron emission tomography imaging. J. Neuroinflamm. 2014, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Shulyatnikova, T.; Banks, W.A.; Hayden, M.R. Ultrastructural Remodeling of the Blood-Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 1640. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Marutani, E.; Shin, H.S.; Chen, W.; Hanaoka, K.; Xian, M.; Ichinose, F. Sodium thiosulphate attenuates acute lung injury in mice. Anesthesiology 2014, 121, 1248–1257. [Google Scholar] [CrossRef]
- Acero, G.; Catorce, M.N.; González-Mendoza, R.; Meraz-Rodrıguez, M.A.; Hernandez-Zimbron, L.F.; González-Salinas, R.; Gevorkian, G. Sodium thiosulphate attenuates brain inflammation induced by systemic lipopolysaccharide administration in C57BL/6J mice. Inflammopharmacology 2017, 25, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Pollay, M.; Kaplan, R.J. The movement of sulfate and thiosulfate into in vivo choroid plexus. J. Neurobiol. 1971, 2, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Dronamraju, V.; Xie, W.; More, S.S. Sulfur-containing therapeutics in the treatment of Alzheimer’s disease. Med. Chem. Res. 2021, 30, 305–352. [Google Scholar] [CrossRef] [PubMed]
- Solis, E., Jr.; Hascup, K.N.; Hascup, E.R. Alzheimer’s Disease: The Link Between Amyloid-β and Neurovascular Dysfunction. J. Alzheimer’s Dis. 2020, 76, 1179–1198. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Cerebral Microbleeds Associate with Brain Endothelial Cell Activation-Dysfunction and Blood–Brain Barrier Dysfunction/Disruption with Increased Risk of Hemorrhagic and Ischemic Stroke. Biomedicines 2024, 12, 1463. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonclaves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Stanika, R.I.; Pivovarova, N.B.; Watts, C.A.; Winters, C.A.; Andrews, S.B. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9854–9859. [Google Scholar] [CrossRef]
- Ge, M.; Zhang, J.; Chen, S.; Huang, Y.; Chen, W.; He, L.; Zhang, Y. Role of Calcium Homeostasis in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2022, 18, 487–498. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef]
- Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; González-Reyes, R.E.; Nava-Mesa, M.O. Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 9067. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
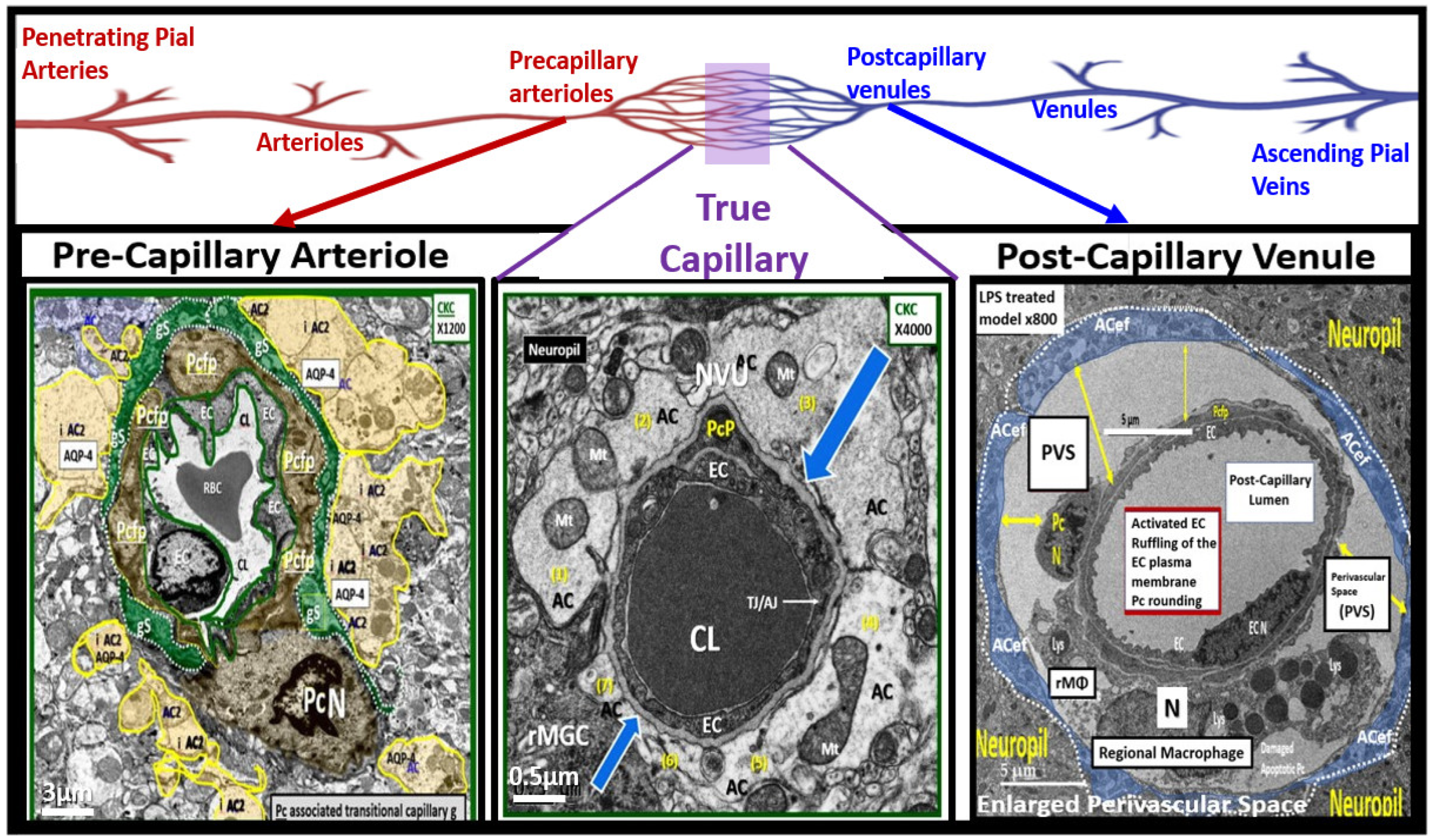
- Hayden, M.R. Brain endothelial cell activation and dysfunction associate with and contribute to the development of enlarged perivascular spaces and cerebral small vessel disease. Histol. Histopathol. 2024, 35, 18792. [Google Scholar] [CrossRef]
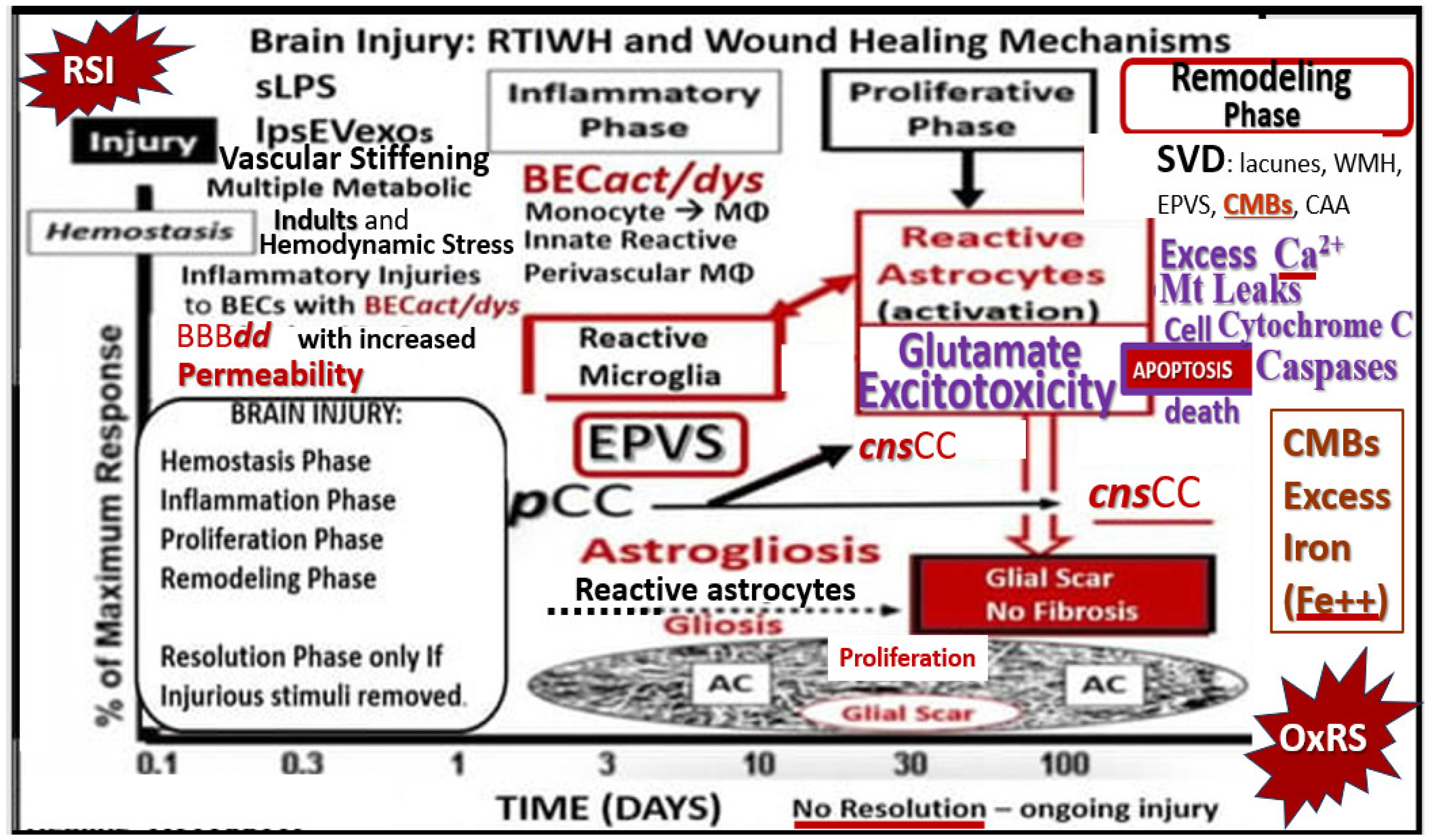
- Hayden, M.R. Brain Injury: Response to Injury Wound-Healing Mechanisms and Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Medicina 2023, 59, 1337. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Peng, T.; Zhuo, L.; Wang, Y.; Jun, M.; Li, G.; Wang, L.; Hong, D. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology 2018, 23, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Canzoniero, L.M.T.; Stefano, L.; Sensi, S.L. A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Front. Mol. Neurosci. 2020, 13, 600089. [Google Scholar] [CrossRef]
- Berdichevsky, E.; Riveros, N.; Sánchez-Armáss, S.; Orrego, F. Kainate N-methylaspartate and other excitatory amino acids increase calcium influx into rat brain cortex cells in vitro. Neurosci. Lett. 1983, 36, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Koh, J.Y.; Peters, S. Pharmacology of glutamate neurotoxicity in cortical cell culture: Attenuation by NMDA antagonists. J. Neurosci. 1988, 8, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, U.; Huang, S.F.; Sridhar, S.; Keller, A. The Interplay Between Brain Vascular Calcification and Microglia. Front. Aging Neurosci. 2022, 14, 848495. [Google Scholar] [CrossRef] [PubMed]
- Subhash, N.; Sriram, R.; Kurian, G.A. Sodium thiosulfate protects brain in rat model of adenine induced vascular calcification. Neurochem. Int. 2015, 90, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Schrank, S.; Barrington, N.; Stutzmann, G.E. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035212. [Google Scholar] [CrossRef] [PubMed]
- Zündorf, G.; Reiser, G. Calcium Dysregulation and Homeostasis of Neural Calcium in the Molecular Mechanisms of Neurodegenerative Diseases Provide Multiple Targets for Neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Neves, P.; Gozzelino, R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals 2019, 12, 126. [Google Scholar] [CrossRef]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S277–S281. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Q.; Yue, D.; Liu, J.; Fu, Y. Cerebral Amyloid Angiopathy: An Undeniable Small Vessel Disease. J. Stroke 2024, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Salami, A.; Papenberg, G.; Sitnikov, R.; Laukka, E.J.; Persson, J.; Kalpouzos, G. Elevated neuroinflammation contributes to the deleterious impact of iron overload on brain function in aging. Neuroimage 2021, 230, 117792. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Gleitze, S.; Paula-lima, A.; Núñez, M.T.; Hidalgo, C. The calcium–iron connection in ferroptosis-mediated neuronal death. Free Radic. Biol. Med. 2021, 175, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, H.W.; Wang, W.; Lang, M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. Int. J. Mol. Sci. 2020, 21, 7660. [Google Scholar] [CrossRef]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 2017, 9, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Exley, C.; House, E.; Polwart, A.; Esiri, M.M. Brain Burdens of Aluminum, Iron, and Copper and their Relationships with Amyloid-β Pathology in 60 Human Brains. J. Alzheimer’s Dis. 2012, 31, 725–730. [Google Scholar] [CrossRef]
- Kaden, D.; Bush, A.I.; Danzeisen, R.; Bayer, T.A.; Multhaup, G. Disturbed Copper Bioavailability in Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Vural, H.; Demirin, H.; Kara, Y.; Eren, I.; Delibas, N. Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. J. Trace Elem. Med. Biol. 2010, 24, 169–173. [Google Scholar] [CrossRef]
- Yu, J.; Luo, X.; Xu, H.; Ma, Q.; Yuan, J.; Li, X.; Chang, R.C.-C.; Qu, Z.; Huang, X.; Zhuang, Z.; et al. Identification of the Key Molecules Involved in Chronic Copper Exposure-Aggravated Memory Impairment in Transgenic Mice of Alzheimer’s Disease Using Proteomic Analysis. J. Alzheimer’s Dis. 2015, 44, 455–469. [Google Scholar] [CrossRef]
- Squitti, R.; Siotto, M.; Polimanti, R. Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol. Aging 2014, 35, S40–S50. [Google Scholar] [CrossRef]
- Brewer, G.J. Alzheimer’s disease causation by copper toxicity and treatment with zinc. Front. Aging Neurosci. 2014, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Ceccom, J.; Coslédan, F.; Halley, H.; Francès, B.; Lassalle, J.M.; Meunier, B. Copper Chelator Induced Efficient Episodic Memory Recovery in a Non-Transgenic Alzheimer’s Mouse Model. PLoS ONE 2012, 7, e43105. [Google Scholar] [CrossRef] [PubMed]
- Eskici, G.; Axelsen, P.H. Copper and Oxidative Stress in the Pathogenesis of Alzheimer’s Disease. Biochemistry 2012, 51, 6289–6311. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Münter, L.; Harmeier, A.; Georgiev, O.; Multhaup, G.; Schaffner, W. Toxicity of Alzheimer’s disease-associated Aβ peptide is ameliorated in a Drosophila model by tight control of zinc and copper availability. Biol. Chem. 2011, 392, 919–926. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Liu, N.; Luo, Y.; Zhao, B. Copper ions influence the toxicity of β-amyloid(1–42) in a concentration-dependent manner in a Caenorhabditis elegans model of Alzheimer’s disease. Sci. China Life Sci. 2011, 54, 527–534. [Google Scholar] [CrossRef]
- Li, Y.; Jiao, Q.; Xu, H.; Du, X.; Shi, L.; Jia, F.; Jiang, H. Biometal Dyshomeostasis and Toxic Metal Accumulations in the Development of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Matlack, K.E.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-beta (Abeta) oligomers to restore endocytosis and ameliorate Abeta toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef]
- Borobia, M.; Villanueva-Saz, S.; de Arcaute, M.R.; Fernández, A.; Verde, M.T.; González, J.M.; Navarro, T.; Benito, A.A.; Arnal, J.L.; Heras, M.D.L.; et al. Copper Poisoning, a Deadly Hazard for Sheep. Animals 2022, 12, 2388. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. [Google Scholar] [CrossRef]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
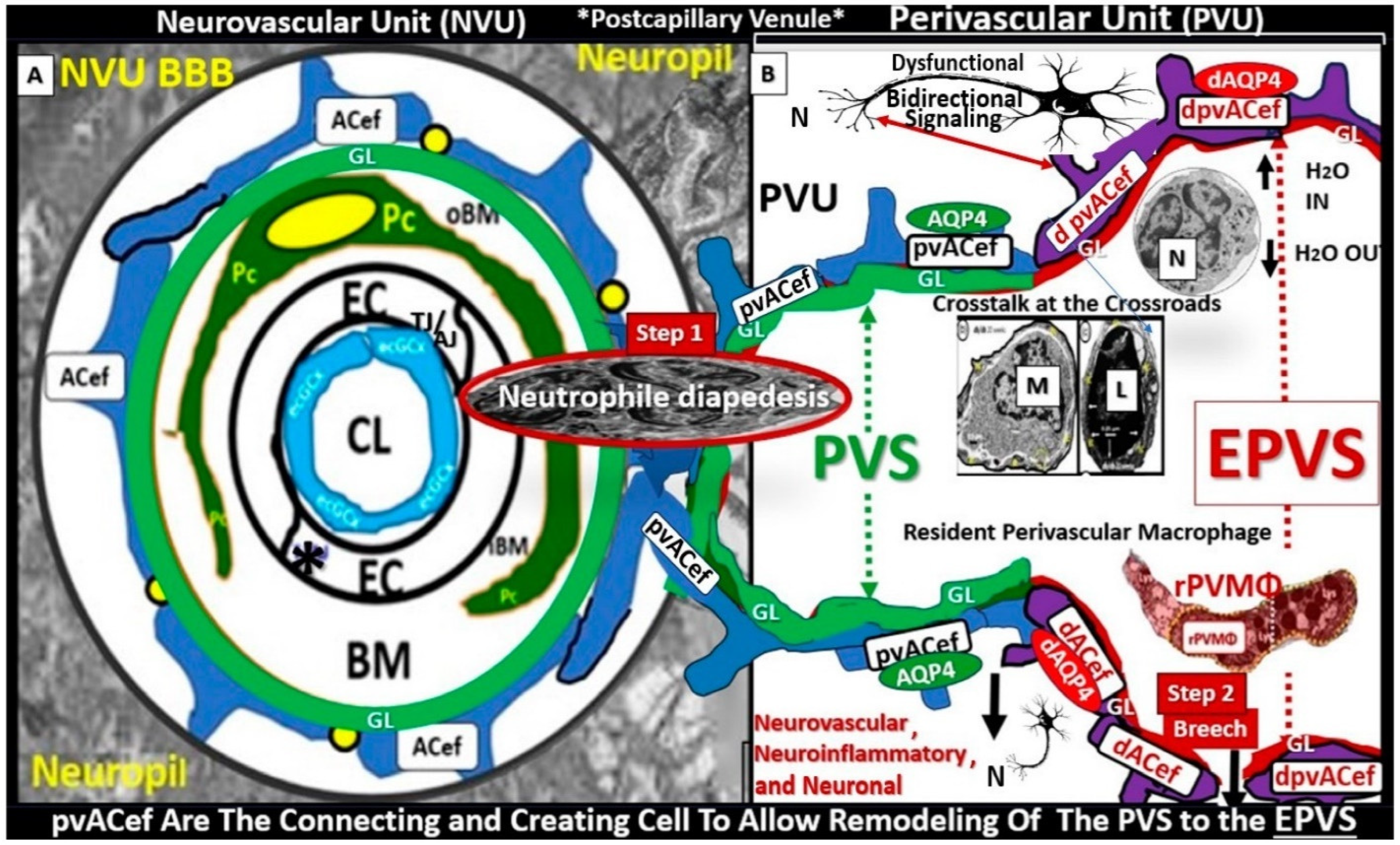
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular Spaces and the Two Steps to Neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. A Closer Look at the Perivascular Unit in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Biomedicines 2024, 12, 96. [Google Scholar] [CrossRef]
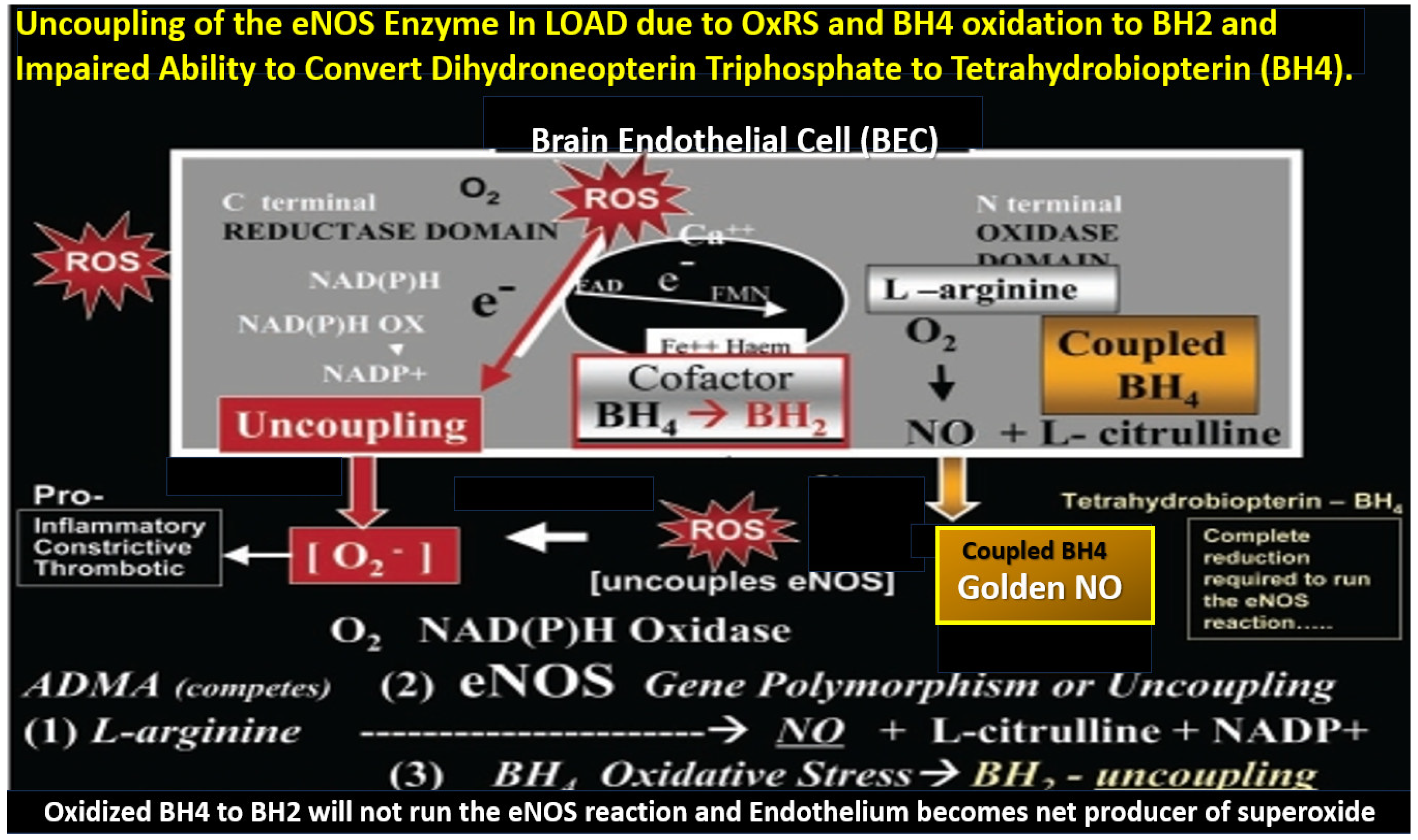
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; d’Uscio, L.V.; He, T. Emerging roles of endothelial nitric oxide synthase in preservation of cognitive health. Stroke 2023, 54, 686–696. [Google Scholar] [CrossRef]
- Barford, P.A.; Blair, J.A.; Eggar, C.; Hamon, C.; Morar, C.; SBWhitburn, S.B. Tetrahydrobiopterin metabolism in the temporal lobe of patients dying with senile dementia of Alzheimer type. J. Neurol. Neurosurg. Psychiatry 1984, 47, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Morar, C.; Whitbum, S.B.; Blair, J.A.; Leeming, R.J.; Wilcock, G.K. Tetrahydrobiopterin metabolism in senile dementia of Alzheimer type. J. Neurol. Neurosurg. Psychiatry 1983, 46, 582. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Pfeifle, C.E.; Howell, S.B.; Felthouse, R.D.; Woliver, T.B.; Andrews, P.A.; Markman, M.; Murphy, M.P. High-dose cisplatin with sodium thiosulfate protection. J. Clin. Oncol. 1985, 3, 237–244.44. [Google Scholar] [CrossRef]
- Brock, P.R.; Maibach, R.; Childs, M.; Rajput, K.; Roebuck, D.; Sullivan, M.J.; Laithier, V.; Ronghe, M.; Dall’Igna, P.; Hiyama, E.; et al. Sodium Thiosulfate for Protection from Cisplatin-Induced Hearing Loss. N. Engl. J. Med. 2018, 378, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Shah, R.C.; Ross, E.A. Rapid resolution of calciphylaxis with intravenous sodium thiosulphate and continuous venovenous haemofiltration using low calcium replacement fluid: Case report. Nephrol. Dial. Transplant. 2005, 20, 1260–1262. [Google Scholar] [CrossRef]
- Brucculeri, M.; Cheigh, J.; Bauer, G.; Serur, D. Long-term intravenous sodium thiosulfate in the treatment of a patient with calciphylaxis. Semin. Dial. 2005, 18, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Bauer, R.; Beier, C.; Betz, C.; Wolter, M.; Kaufmann, R.; Gille, J. Sodium thiosulphate as a promising therapeutic option to treat calciphylaxis. Dermatology 2006, 212, 373–376. [Google Scholar] [CrossRef]
- Mataic, D.; Bastani, B. Intraperitoneal sodium thiosulphate for the treatment of calciphylaxis. Ren. Fail. 2006, 28, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Tokashiki, K.; Ishida, A.; Kouchi, M.; Ishihara, S.; Tomiyama, N.; Kohagura, K.; Iseki, K.; Takishita, S. Successful management of critical limb ischemia with intravenous sodium thiosulphate in a chronic hemodialysis patient. Clin. Nephrol. 2006, 66, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.E.; Fennell, R.S.; Neiberger, R.E.; Dharnidharka, V.R. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin. J. Am. Soc. Nephrol. 2006, 1, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.L.; Fitzgibbons, C.A.; Buescher, L.S. Calciphylaxis responding to sodium thiosulfate therapy. Arch. Dermatol. 2007, 143, 269–270. [Google Scholar] [CrossRef]
- Ackermann, F.; Levy, A.; Daugas, E.; Schartz, N.; Riaux, A.; Derancourt, C.; Urena, P.; Lebbé, C. Sodium thiosulfate as first-line treatment for calciphylaxis. Arch. Dermatol. 2007, 143, 1336–1337. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.; Wallace, H.; Sinniah, R.; Saker, B. Complete resolution of recurrent calciphylaxis with long-term intravenous sodium thiosulfate. Australas. J. Dermatol. 2008, 49, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Leslie, W.D. Bone scan findings in metastatic calcification from calciphylaxis. Clin. Nucl. Med. 2008, 33, 502–504. [Google Scholar] [CrossRef]
- Raymond, C.B.; Wazny, L.D. Sodium thiosulfate, bisphonates and cinacalcet for treatment of calciphylaxis. Am. J. Health Syst. Pharm. 2008, 65, 1419–1429. [Google Scholar] [CrossRef]
- Tindi, A.; Gauray, K.; Panda, M. Non-healing painful ulcers in a patient with chronic kidney disease and role of sodium thiosulfate: A case report. Cases J. 2008, 1, 178. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, I.; Gombou, A.; Griveas, I.; Agroyannis, I.; Retsa, K.; Agroyannis, B. Combination of sodium thiosulfate, cinacalcet, and paricaicitol in the treatment of calciphylaxis with hyperparathyroidism. Int. J. Artif. Organs 2008, 31, 742–744. [Google Scholar] [CrossRef] [PubMed]
- Hackett, B.C.; McAleer, M.A.; Sheehan, G.; Powell, F.C.; O’Donnell, B.F. Calciphylaxis in a patient with normal renal function: Response to treatment with sodium thiosulfate. Clin. Exp. Dermatol. 2009, 34, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.M.; Coates, P.T. Calcific uraemic arteriolopathy: An update. Curr. Opin. Nephrol. Hypertens. 2008, 17, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Schaffner, T.; Huynh-Do, U.; Frey, B.M.; Frey, F.J.; Farese, S. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008, 74, 1444–1453. [Google Scholar] [CrossRef]
- Sen, U.; Vacek, T.P.; Hughes, W.M.; Kumar, M.; Moshal, K.S.; Tyagi, N.; Metreveli, N.; Hayden, M.R.; Tyagi, S.C. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology 2008, 82, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Dmytriv, T.R.; Duve, K.V.; Storey, K.B.; Lushchak, V.I. Vicious cycle of oxidative stress and neuroinflammation in pathophysiology of chronic vascular encephalopathy. Front. Physiol. 2024, 15, 1443604. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Gabr, M.T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K.; Magoola, M.; Mariam, Z. Innovative Therapeutic Strategies in Alzheimer’s Disease: A Synergistic Approach to Neurodegenerative Disorders. Pharmaceuticals 2024, 17, 741. [Google Scholar] [CrossRef]
- Gong, C.X.; Dai, C.L.; Liu, F.; Iqbal, K. Multi-Targets: An Unconventional Drug Development Strategy for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 837649. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R.; Tyagi, N. Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease. Pharmaceuticals 2024, 17, 1741. https://doi.org/10.3390/ph17121741
Hayden MR, Tyagi N. Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease. Pharmaceuticals. 2024; 17(12):1741. https://doi.org/10.3390/ph17121741
Chicago/Turabian StyleHayden, Melvin R., and Neetu Tyagi. 2024. "Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease" Pharmaceuticals 17, no. 12: 1741. https://doi.org/10.3390/ph17121741
APA StyleHayden, M. R., & Tyagi, N. (2024). Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease. Pharmaceuticals, 17(12), 1741. https://doi.org/10.3390/ph17121741