

Implications of NLRP3 Suppression Using Glibenclamide and miR-223 against Colorectal Cancer

 ,
, 


Abstract
1. Introduction
2. Results
2.1. NLRP3 Activation Suppresses Cell Growth through Proinflammatory Cytokine Secretion, While gli Fails to Oppose the Inflammasome’s Effects
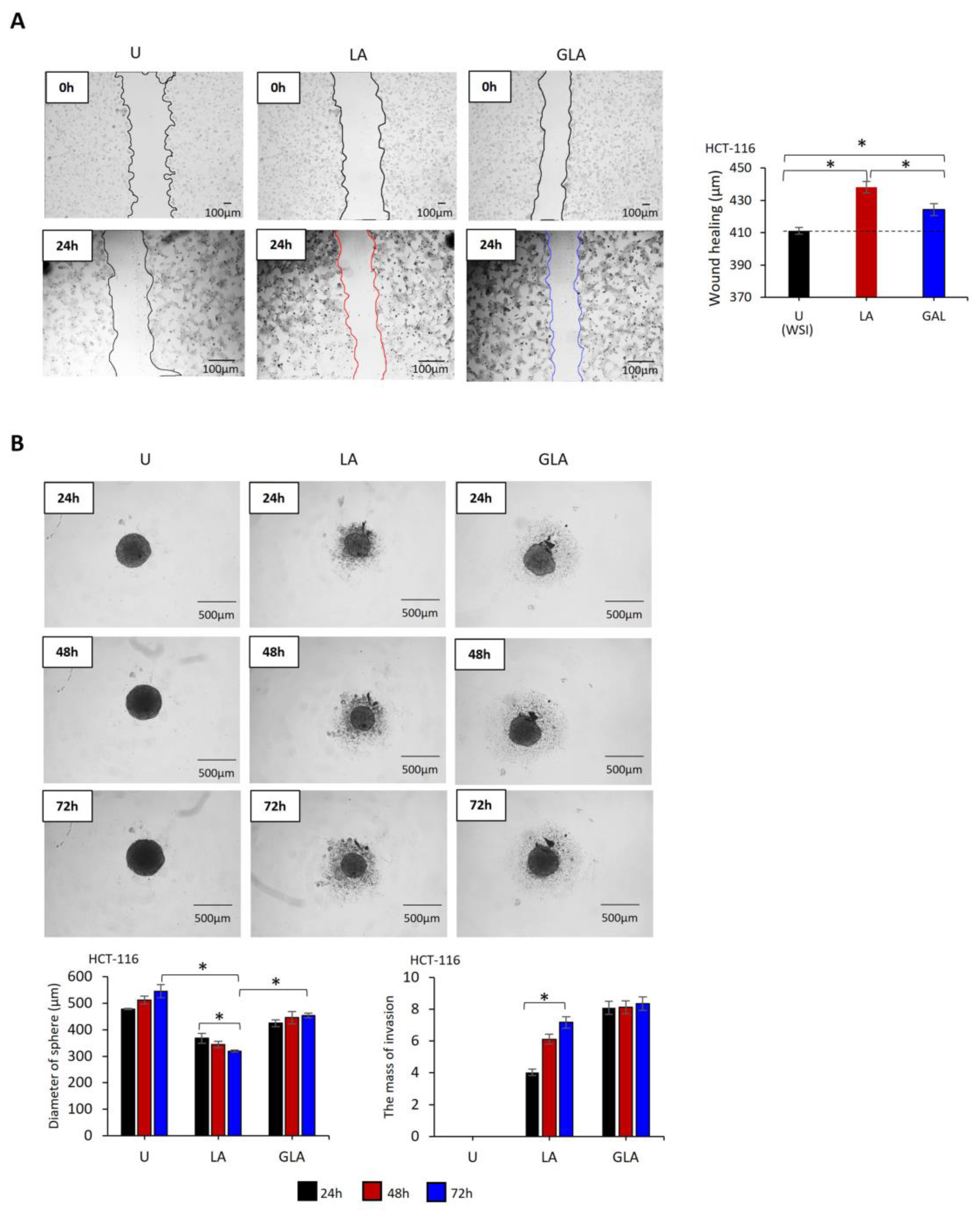
2.2. Gli-Induced CRC Cell Invasion
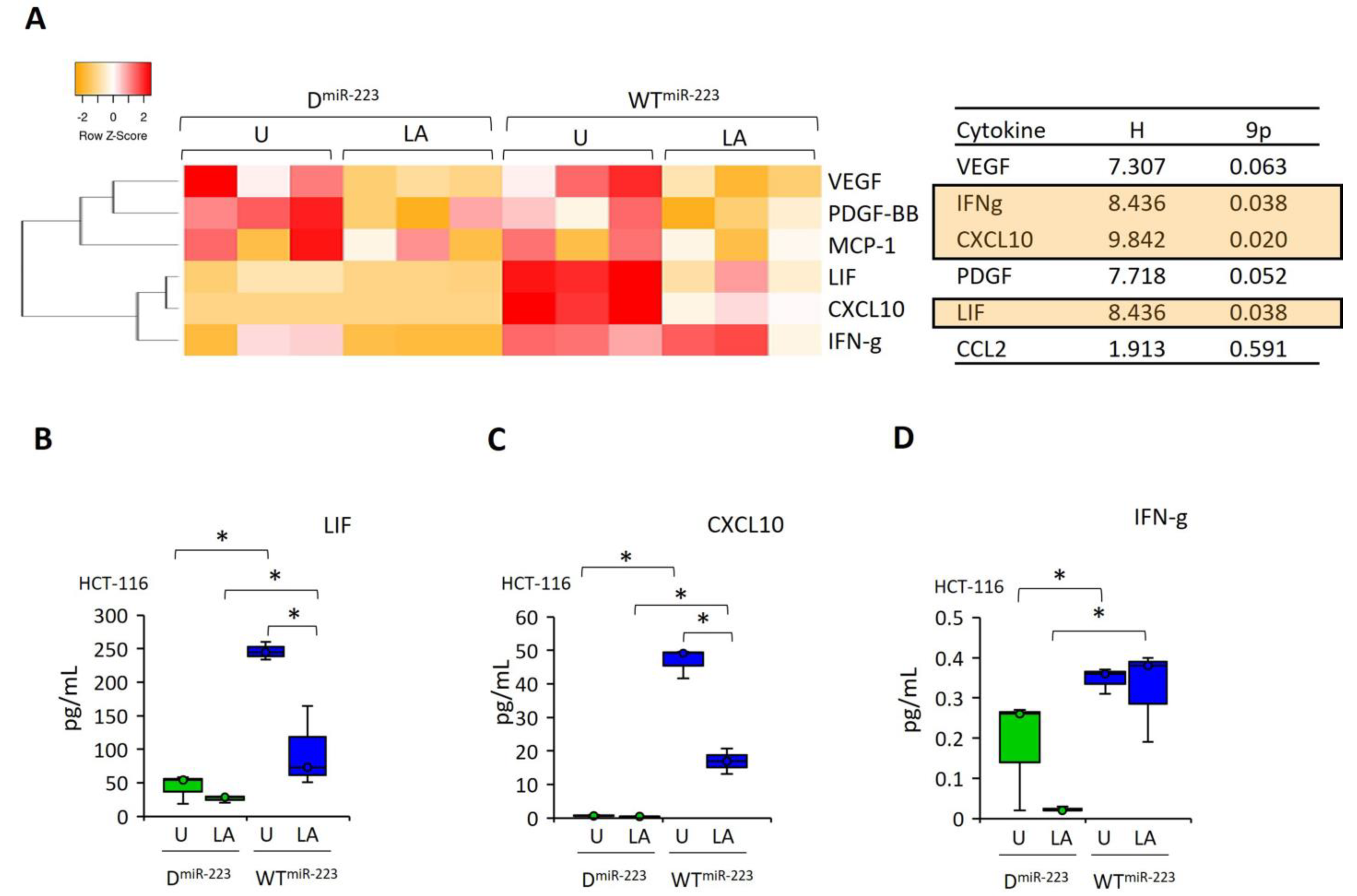
2.3. NLRP3 Has a Dual Effect on Proinflammatory Cytokine Release in HCT-116 Cells, While gli Promotes Angiogenesis
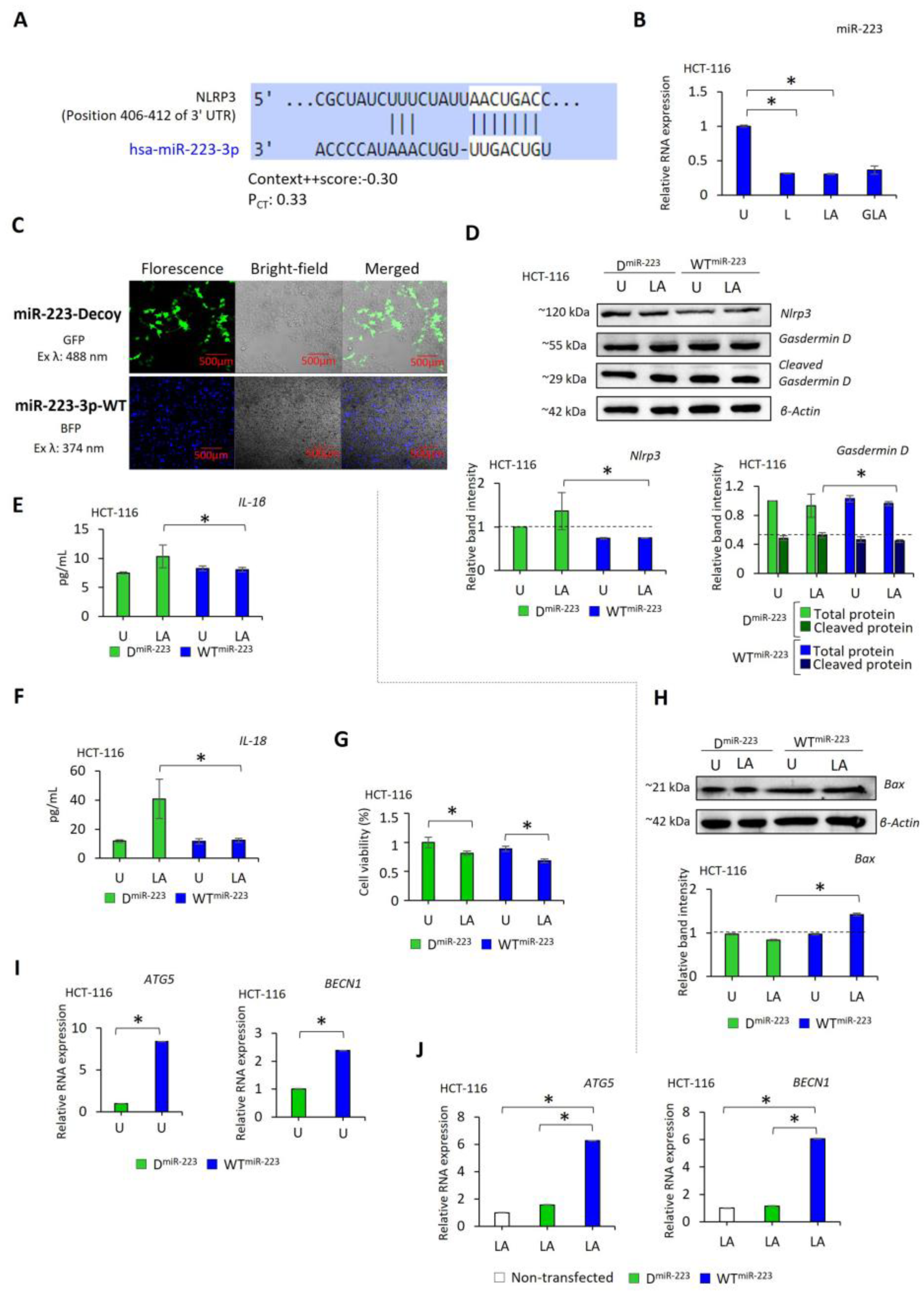
2.4. NLRP3 Activation Suppresses the Expression of miR-223 in CRC Cells, While Adding miR-223 Reduces NLRP3 Activation
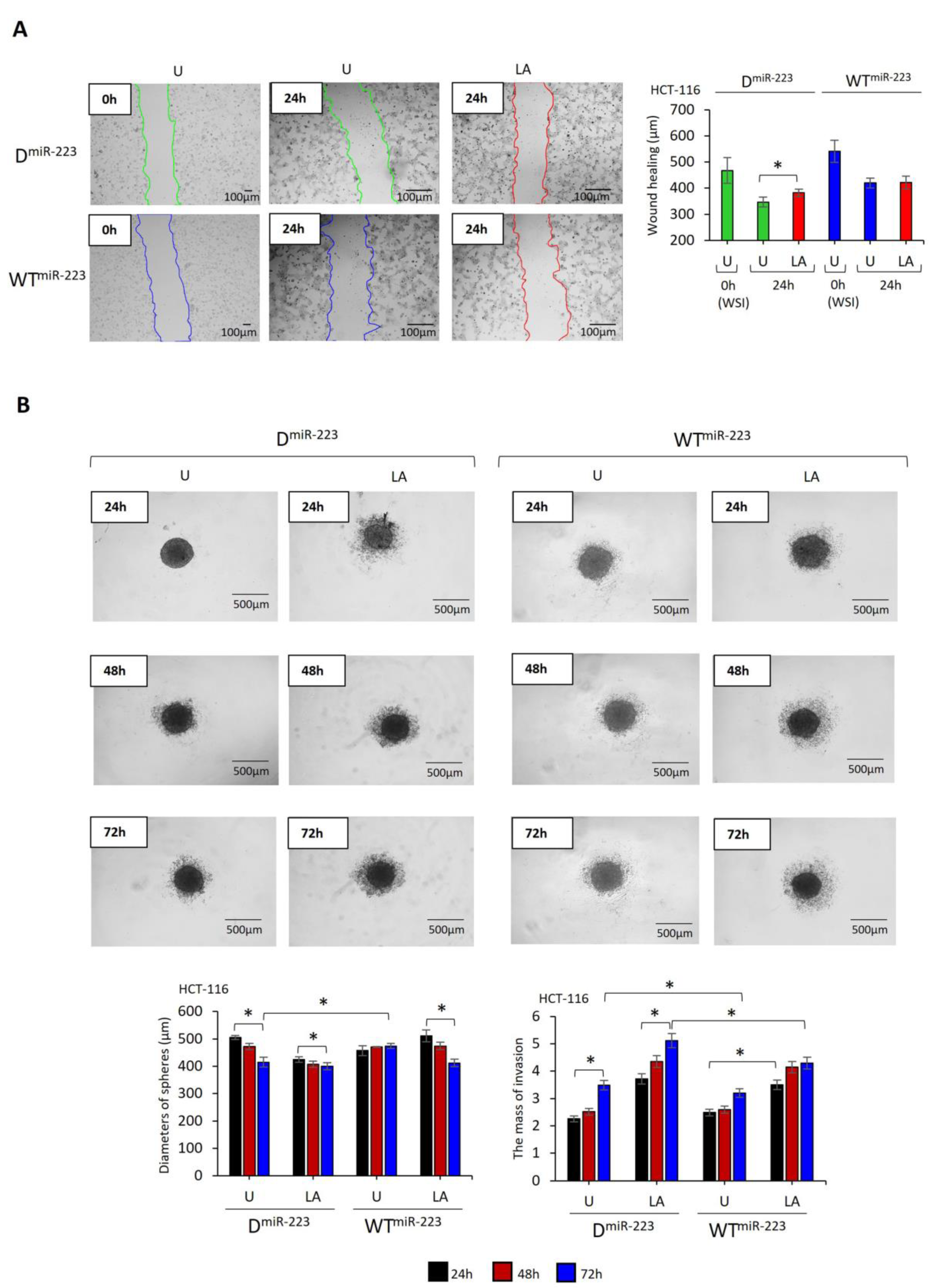
2.5. miR-223 Decreases the Invasion of HCT-116 and HCT-15 Cells
2.6. miR-223 Recovered the Cytokines Production, Which Were Suppressed after NLRP3 Activation by LPS–ATP
3. Discussion
4. Methods
4.1. Cell Lines and Reagents
4.2. Plasmid Transfection
4.3. Western Blot
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Real-Time qPCR
4.6. MTS Assay
4.7. Annexin V assay
4.8. Scratch Wound Healing Assay
4.9. Sphere-Invasion Assay
4.10. Multiplex Analyzes
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. 11 July 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/colorectal-cancer?gad_source=1&gclid=Cj0KCQiAxOauBhCaARIsAEbUSQRmrxLIoxqfZoy-QbAbSd1EIc_udFpS9SDysJiVxdQxohbpuY1DdIwaAjJxEALw_wcB (accessed on 1 December 2023).
- Calle, E.E. Obesity and cancer. BMJ 2007, 335, 1107–1108. [Google Scholar] [CrossRef]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Keshavarz Shahbaz, S.; Koushki, K.; Ayati, S.H.; Bland, A.R.; Bezsonov, E.E.; Sahebkar, A. Inflammasomes and Colorectal Cancer. Cells 2021, 10, 2172. [Google Scholar] [CrossRef]
- Shi, F.; Wei, B.; Lan, T.; Xiao, Y.; Quan, X.; Chen, J.; Zhao, C.; Gao, J. Low NLRP3 expression predicts a better prognosis of colorectal cancer. Biosci. Rep. 2021, 41, BSR20210280. [Google Scholar] [CrossRef]
- Shao, X.; Lei, Z.; Zhou, C. NLRP3 Promotes Colorectal Cancer Cell Proliferation and Metastasis via Regulating Epithelial Mesenchymal Transformation. Anti-Cancer Agents Med. Chem. 2020, 20, 820–827. [Google Scholar] [CrossRef]
- Marandi, Y.; Hashemzade, S.; Tayebinia, H.; Karimi, J.; Zamani, A.; Khodadadi, I. NLRP3-inflammasome activation is associated with epithelial-mesenchymal transition and progression of colorectal cancer. Iran. J. Basic Med. Sci. 2021, 24, 483–492. [Google Scholar]
- Zaharie, R.; Valean, D.; Popa, C.; Fetti, A.; Zdrehus, C.; Puia, A.; Usatiuc, L.; Schlanger, D.; Zaharie, F. The Multifaceted Role and Regulation of Nlrp3 Inflammasome in Colitis-Associated Colo-Rectal Cancer: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3472. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Liu, Y.; Xia, Y.; Chang, R.; Zhang, C. Inflammasome inhibitors: Promising therapeutic approaches against cancer. J. Hematol. Oncol. 2019, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Harnack, U.; Johnen, H.; Pecher, G. IL-1 receptor antagonist anakinra enhances tumour growth inhibition in mice receiving peptide vaccination and beta-(1-3),(1-6)-D-glucan. Anticancer. Res. 2010, 30, 3959–3965. [Google Scholar] [PubMed]
- Xie, J.; Zhang, Y.; Jiang, L. Role of Interleukin-1 in the pathogenesis of colorectal cancer: A brief look at anakinra therapy. Int. Immunopharmacol. 2022, 105, 108577. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Tezcan, G.; Garanina, E.E.; Alsaadi, M.; Gilazieva, Z.E.; Martinova, E.V.; Markelova, M.I.; Arkhipova, S.S.; Hamza, S.; McIntyre, A.; Rizvanov, A.A.; et al. Therapeutic Potential of Pharmacological Targeting NLRP3 Inflammasome Complex in Cancer. Front. Immunol. 2021, 11, 607881. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- Kim, J.A.; Kang, Y.S.; Lee, S.H.; Lee, E.H.; Yoo, B.H.; Lee, Y.S. Glibenclamide induces apoptosis through inhibition of cystic fibrosis transmembrane conductance regulator (CFTR) Cl(-) channels and intracellular Ca(2+) release in HepG2 human hepatoblastoma cells. Biochem. Biophys. Res. Commun. 1999, 261, 682–688. [Google Scholar] [CrossRef]
- Qian, X.; Li, J.; Ding, J.; Wang, Z.; Duan, L.; Hu, G. Glibenclamide exerts an antitumor activity through reactive oxygen species-c-jun NH2-terminal kinase pathway in human gastric cancer cell line MGC-803. Biochem. Pharmacol. 2008, 76, 1705–1715. [Google Scholar] [CrossRef]
- Núñez, M.; Medina, V.; Cricco, G.; Croci, M.; Cocca, C.; Rivera, E.; Bergoc, R.; Martín, G. Glibenclamide inhibits cell growth by inducing G0/G1 arrest in the human breast cancer cell line MDA-MB-231. BMC Pharmacol. Toxicol. 2013, 14, 6. [Google Scholar] [CrossRef]
- Tuccori, M.; Wu, J.W.; Yin, H.; Majdan, A.; Azoulay, L. The Use of Glyburide Compared With Other Sulfonylureas and the Risk of Cancer in Patients With Type 2 Diabetes. Diabetes Care 2015, 38, 2083–2089. [Google Scholar] [CrossRef]
- Olatunde, A.; Nigam, M.; Singh, R.K.; Panwar, A.S.; Lasisi, A.; Alhumaydhi, F.A.; Jyoti Kumar, V.; Mishra, A.P.; Sharifi-Rad, J. Cancer and diabetes: The interlinking metabolic pathways and repurposing actions of antidiabetic drugs. Cancer Cell Int. 2021, 21, 499. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef]
- Chai, B.; Guo, Y.; Cui, X.; Liu, J.; Suo, Y.; Dou, Z.; Li, N. MiR-223-3p promotes the proliferation, invasion and migration of colon cancer cells by negative regulating PRDM1. Am. J. Transl. Res. 2019, 11, 4516–4523. [Google Scholar]
- Mahmoud, H.A.; El Amin, H.A.; Ahmed, E.S.M.; Kenawy, A.G.; El-Ebidi, A.M.; ElNakeeb, I.; Kholef, E.F.M.; Elsewify, W.A.E. Role of MicroRNA-223 and MicroRNA-182 as Novel Biomarkers in Early Detection of Colorectal Cancer. Int. J. Gen. Med. 2022, 15, 3281–3291. [Google Scholar] [CrossRef]
- Aziz, F.; Chakraborty, A.; Khan, I.; Monts, J. Relevance of miR-223 as Potential Diagnostic and Prognostic Markers in Cancer. Biology 2022, 11, 249. [Google Scholar] [CrossRef]
- Seeneevassen, L.; Giraud, J.; Molina-Castro, S.; Sifré, E.; Tiffon, C.; Beauvoit, C.; Staedel, C.; Mégraud, F.; Lehours, P.; Martin, O.C.B.; et al. Leukaemia Inhibitory Factor (LIF) Inhibits Cancer Stem Cells Tumorigenic Properties through Hippo Kinases Activation in Gastric Cancer. Cancers 2020, 12, 2011. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Tezcan, G.; Martynova, E.V.; Gilazieva, Z.E.; McIntyre, A.; Rizvanov, A.A.; Khaiboullina, S.F. MicroRNA Post-transcriptional Regulation of the NLRP3 Inflammasome in Immunopathologies. Front. Pharmacol. 2019, 10, 451. [Google Scholar] [CrossRef]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; Di Paola, R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef]
- Broz, P.; It, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Shadab, A.; Mahjoor, M.; Abbasi-Kolli, M.; Afkhami, H.; Moeinian, P.; Safdarian, A.R. Divergent functions of NLRP3 inflammasomes in cancer: A review. Cell Commun. Signal. 2023, 21, 232. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef]
- Si, Y.; Liu, L.; Fan, Z. Mechanisms and effects of NLRP3 in digestive cancers. Cell Death Discov. 2024, 10, 10. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Liu, X.; Zhang, Y. NLRP3 Inflammasome Activation in MΦs-CRC Crosstalk Promotes Colorectal Cancer Metastasis. Ann. Clin. Lab. Sci. 2022, 52, 571–579. [Google Scholar]
- Xie, W.; Peng, M.; Liu, Y.; Zhang, B.; Yi, L.; Long, Y. Simvastatin induces pyroptosis via ROS/caspase-1/GSDMD pathway in colon cancer. Cell Commun. Signal. 2023, 21, 329. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, S.; Pi, D.; Wu, Y.; Zuo, Q.; Li, C.; Ouyang, M. Luteolin induces pyroptosis in HT-29 cells by activating the Caspase1/Gasdermin D signalling pathway. Front. Pharmacol. 2022, 13, 952587. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Tarassishin, L.; Lim, J.; Weatherly, D.B.; Angeletti, R.H.; Lee, S.C. Interleukin-1-induced changes in the glioblastoma secretome suggest its role in tumor progression. J. Proteom. 2014, 99, 152–168. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Oh, J.M.; Venters, C.C.; Di, C.; Pinto, A.M.; Wan, L.; Younis, I.; Cai, Z.; Arai, C.; So, B.R.; Duan, J.; et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef]
- Reuten, R.; Zendehroud, S.; Nicolau, M.; Fleischhauer, L.; Laitala, A.; Kiderlen, S.; Nikodemus, D.; Wullkopf, L.; Nielsen, S.R.; McNeilly, S.; et al. Basement membrane stiffness determines metastases formation. Nat. Mater. 2021, 20, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.Y.; Kim, S.Y.; Jung, K.; Cho, M.L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 10133. [Google Scholar] [CrossRef]
- Yang, D.; Liang, Y.; Zhao, S.; Ding, Y.; Zhuang, Q.; Shi, Q.; Ai, T.; Wu, S.Q.; Han, J. ZBP1 mediates interferon-induced necroptosis. Cell. Mol. Immunol. 2020, 17, 356–368. [Google Scholar] [CrossRef]
- Ding, M.; He, S.J.; Yang, J. MCP-1/CCL2 Mediated by Autocrine Loop of PDGF-BB Promotes Invasion of Lung Cancer Cell by Recruitment of Macrophages Via CCL2-CCR2 Axis. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2019, 39, 224–232. [Google Scholar] [CrossRef]
- Zhang, Y.; Manouchehri Doulabi, E.; Herre, M.; Cedervall, J.; Qiao, Q.; Miao, Z.; Hamidi, A.; Hellman, L.; Kamali-Moghaddam, M.; Olsson, A.K. Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment. Cancers 2022, 14, 1947. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Zheng, Y.; Yu, Q.; Zeng, M.; Bai, L.; Yang, L.; Guo, M.; Jiang, X.; Gan, J. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases (Review). Int. J. Mol. Med. 2023, 51, 35. [Google Scholar] [CrossRef]
- Giannuzzo, A.; Pedersen, S.F.; Novak, I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol. Cancer 2015, 14, 203. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.Y.; Lee, S.W.; Shin, Y.H.; Lee, J.H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef]
- Yasukagawa, T.; Niwa, Y.; Simizu, S.; Umezawa, K. Suppression of cellular invasion by glybenclamide through inhibited secretion of platelet-derived growth factor in ovarian clear cell carcinoma ES-2 cells. FEBS Lett. 2012, 586, 1504–1509. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, X.; Luo, Y.; Yuan, Y.; Wu, Y.; Wang, M.; Liu, D. Glibenclamide-Induced Autophagy Inhibits Its Insulin Secretion-Improving Function in β Cells. Int. J. Endocrinol. 2019, 2019, 1265175. [Google Scholar] [CrossRef]
- Saresella, M.; Zoia, C.P.; La Rosa, F.; Bazzini, C.; Sala, G.; Grassenis, E.; Marventano, I.; Hernis, A.; Piancone, F.; Conti, E.; et al. Glibenclamide-Loaded Engineered Nanovectors (GNVs) Modulate Autophagy and NLRP3-Inflammasome Activation. Pharmaceuticals 2023, 16, 1725. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.R.; Jia, W.; Johnson, R.S.; Dent, P.; Mitchell, C.; Loria, R.M. Autophagy and the functional roles of Atg5 and beclin-1 in the anti-tumor effects of 3beta androstene 17alpha diol neuro-steroid on malignant glioma cells. J. Steroid Biochem. Mol. Biol. 2009, 115, 137–145. [Google Scholar] [CrossRef]
- Liu, B.; Wen, X.; Cheng, Y. Survival or death: Disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013, 4, e892. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Alan Diehl, J.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013, 32, 4932–4940. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-Formation Assay: Three-Dimensional in vitro Culturing of Prostate Cancer Stem/Progenitor Sphere-Forming Cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef]
- Yan, B.; Peng, Z.; Xing, X.; Du, C. Glibenclamide induces apoptosis by activating reactive oxygen species dependent JNK pathway in hepatocellular carcinoma cells. Biosci. Rep. 2017, 37, BSR20170685. [Google Scholar] [CrossRef]
- Subramaniyam, N.; Arumugam, S.; Ezthupurakkal, P.B.; Ariraman, S.; Biswas, I.; Muthuvel, S.K.; Balakrishnan, A.; Alshammari, G.M.; Chinnasamy, T. Unveiling anticancer potential of glibenclamide: Its synergistic cytotoxicity with doxorubicin on cancer cells. J. Pharm. Biomed. Anal. 2018, 154, 294–301. [Google Scholar] [CrossRef]
- Monami, M.; Balzi, D.; Lamanna, C.; Barchielli, A.; Masotti, G.; Buiatti, E.; Marchionni, N.; Mannucci, E. Are sulphonylureas all the same? A cohort study on cardiovascular and cancer-related mortality. Diabetes Metab. Res. Rev. 2007, 23, 479–484. [Google Scholar] [CrossRef]
- Gao, R.; Yang, T.; Xu, W. Enemies or weapons in hands: Investigational anti-diabetic drug glibenclamide and cancer risk. Expert Opin. Investig. Drugs 2017, 26, 853–864. [Google Scholar] [CrossRef]
- Hendriks, A.M.; Schrijnders, D.; Kleefstra, N.; de Vries, E.G.E.; Bilo, H.J.G.; Jalving, M.; Landman, G.W.D. Sulfonylurea derivatives and cancer, friend or foe? Eur. J. Pharmacol. 2019, 861, 172598. [Google Scholar] [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Wu, L.; Li, H.; Jia, C.Y.; Cheng, W.; Yu, M.; Peng, M.; Zhu, Y.; Zhao, Q.; Dong, Y.W.; Shao, K.; et al. MicroRNA-223 regulates FOXO1 expression and cell proliferation. FEBS Lett. 2012, 586, 1038–1043. [Google Scholar] [CrossRef]
- Wan, L.; Yuan, X.; Liu, M.; Xue, B. miRNA-223-3p regulates NLRP3 to promote apoptosis and inhibit proliferation of hep3B cells. Exp. Ther. Med. 2018, 15, 2429–2435. [Google Scholar] [CrossRef]
- Ding, Q.; Shen, L.; Nie, X.; Lu, B.; Pan, X.; Su, Z.; Yan, A.; Yan, R.; Zhou, Y.; Li, L.; et al. MiR-223-3p overexpression inhibits cell proliferation and migration by regulating inflammation-associated cytokines in glioblastomas. Pathol. Res. Pract. 2018, 214, 1330–1339. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Zang, Y.; Wang, F. NLRP3 inflammasome inactivation driven by miR-223-3p reduces tumor growth and increases anticancer immunity in breast cancer. Mol. Med. Rep. 2019, 19, 2180–2188. [Google Scholar] [CrossRef]
- Manikandan, M.; Deva Magendhra Rao, A.K.; Arunkumar, G.; Manickavasagam, M.; Rajkumar, K.S.; Rajaraman, R.; Munirajan, A.K. Oral squamous cell carcinoma: microRNA expression profiling and integrative analyses for elucidation of tumourigenesis mechanism. Mol. Cancer 2016, 15, 28. [Google Scholar] [CrossRef]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009, 16, 1135–1145. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, L.; Francalanci, M.; Annunziato, F.; Lazzeri, E.; Giannini, S.; Cosmi, L.; Sagrinati, C.; Mazzinghi, B.; Orlando, C.; Maggi, E.; et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J. Exp. Med. 2003, 197, 1537–1549. [Google Scholar] [CrossRef]
- Bodnar, R.J.; Yates, C.C.; Wells, A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ. Res. 2006, 98, 617–625. [Google Scholar] [CrossRef]
- Hao, Q.; Tang, H. Interferon-γ and Smac mimetics synergize to induce apoptosis of lung cancer cells in a TNFα-independent manner. Cancer Cell Int. 2018, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Ping, Y.; Zhang, K.; Yang, L.; Li, F.; Zhang, C.; Cheng, S.; Yue, D.; Maimela, N.R.; Qu, J.; et al. Low-Dose IFNγ Induces Tumor Cell Stemness in Tumor Microenvironment of Non-Small Cell Lung Cancer. Cancer Res. 2019, 79, 3737–3748. [Google Scholar] [CrossRef]
- Lo, U.G.; Pong, R.C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFNγ-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019, 79, 1098–1112. [Google Scholar] [CrossRef]
- Chen, H.C.; Chou, A.S.; Liu, Y.C.; Hsieh, C.H.; Kang, C.C.; Pang, S.T.; Yeh, C.T.; Liu, H.P.; Liao, S.K. Induction of metastatic cancer stem cells from the NK/LAK-resistant floating, but not adherent, subset of the UP-LN1 carcinoma cell line by IFN-γ. Lab. Investig. J. Tech. Methods Pathol. 2011, 91, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, S.G.; Aksoy, M.O.; Yang, Y.; Shahabuddin, S.; Litvin, J.; Safadi, F.; Rogers, T.J. The chemokine receptor CXCR3 and its splice variant are expressed in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L584–L591. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Viswanadhapalli, S.; Dileep, K.V.; Zhang, K.Y.J.; Nair, H.B.; Vadlamudi, R.K. Targeting LIF/LIFR signaling in cancer. Genes Dis. 2021, 9, 973–980. [Google Scholar] [CrossRef]
- Gam, J.J.; Babb, J.; Weiss, R. A mixed antagonistic/synergistic miRNA repression model enables accurate predictions of multi-input miRNA sensor activity. Nat. Commun. 2018, 9, 2430. [Google Scholar] [CrossRef]
- Mullokandov, G.; Baccarini, A.; Ruzo, A.; Jayaprakash, A.D.; Tung, N.; Israelow, B.; Evans, M.J.; Sachidanandam, R.; Brown, B.D. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods 2012, 9, 840–846. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5439. [Google Scholar] [CrossRef]
- Tezcan, G.; Garanina, E.E.; Zhuravleva, M.N.; Hamza, S.; Rizvanov, A.A.; Khaiboullina, S.F. Rab GTPase Mediating Regulation of NALP3 in Colorectal Cancer. Molecules 2020, 25, 4834. [Google Scholar] [CrossRef]
- Lagarkova, M.A.; Shutova, M.V.; Bogomazova, A.N.; Vassina, E.M.; Glazov, E.A.; Zhang, P.; Rizvanov, A.A.; Chestkov, I.V.; Kiselev, S.L. Induction of pluripotency in human endothelial cells resets epigenetic profile on genome scale. Cell Cycle 2010, 9, 937–946. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Fuel, M.; Mesas, C.; Martínez, R.; Ortiz, R.; Quiñonero, F.; Bermúdez, F.; Gutiérrez, N.; Torres, A.M.; Kapravelou, G.; Lozano, A.; et al. Antioxidant and Chemopreventive Activity of Protein Hydrolysates from Raw and Germinated Flour of Legumes with Commercial Interest in Colorectal Cancer. Antioxidants 2022, 11, 2421. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACTB | F | 5′-GAC AGG ATG CAG AAG GAG ATT ACT-3 | [30] |
| R | 5′-TGA TCC ACA TCT GCT GGA AGG T-3′ | ||
| NLRP3 | F | 5′-ATG AGT GCT GCT TCG ACA TC-3′ | [30] |
| R | 5′-TTG TCA CTC AGG TCC AGC TC-3 | ||
| BECN1 | F | 5′-AGA CCC AGG AGG AAG AGA CT-3′ | |
| R | 5′-AGC TGT TGG CAC TTT CTG TG-3′ | ||
| ATG5 | F | 5′-CTG GGC TGG TCT TAC TTT GC-3′ | |
| R | 5′-GGC CAA AGG TTT CAG CTT CA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamza, S.; Garanina, E.E.; Shkair, L.; Alsaadi, M.; Khaiboullina, S.F.; Tezcan, G. Implications of NLRP3 Suppression Using Glibenclamide and miR-223 against Colorectal Cancer. Pharmaceuticals 2024, 17, 299. https://doi.org/10.3390/ph17030299
Hamza S, Garanina EE, Shkair L, Alsaadi M, Khaiboullina SF, Tezcan G. Implications of NLRP3 Suppression Using Glibenclamide and miR-223 against Colorectal Cancer. Pharmaceuticals. 2024; 17(3):299. https://doi.org/10.3390/ph17030299
Chicago/Turabian StyleHamza, Shaimaa, Ekaterina E. Garanina, Layaly Shkair, Mohammad Alsaadi, Svetlana F. Khaiboullina, and Gulcin Tezcan. 2024. "Implications of NLRP3 Suppression Using Glibenclamide and miR-223 against Colorectal Cancer" Pharmaceuticals 17, no. 3: 299. https://doi.org/10.3390/ph17030299
APA StyleHamza, S., Garanina, E. E., Shkair, L., Alsaadi, M., Khaiboullina, S. F., & Tezcan, G. (2024). Implications of NLRP3 Suppression Using Glibenclamide and miR-223 against Colorectal Cancer. Pharmaceuticals, 17(3), 299. https://doi.org/10.3390/ph17030299