
Cytotoxic and Antiproliferative Activity of LASSBio-2208 and the Attempts to Determine Its Drug Metabolism and Pharmacokinetics In Vitro Profile

 ,
,  ,
,  ,
,  , and
, and 
Abstract
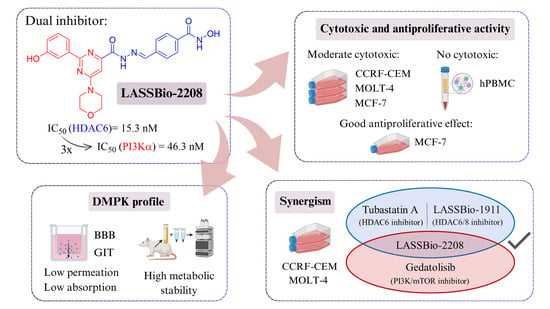
1. Introduction

2. Results and Discussion
2.1. Pharmacological Experiments
2.1.1. Cell Viability by MTT and CC50 Determination
2.1.2. Determination of Selectivity Index (SI) by MTT Assay
2.1.3. Cell Viability and Antiproliferative Profile by SRB Assay
2.1.4. Synergism Study by MTT on CCRF-CEM Cell Line
2.1.5. Synergism Study by MTT on MOLT-4 Cell Line
2.2. In Vitro DMPK Studies
2.2.1. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.2.2. Metabolic Stability
3. Materials and Methods
3.1. Cell Culture
3.2. Compounds and LC/UV Analysis
3.3. Cell Viability Assay by MTT
3.4. PBMC Viability Assay by MTT
3.5. Cell Viability Assay by SRB
3.6. Synergism Assay by MTT
3.7. Parallel Artificial Membrane Permeability Assay (PAMPA)
3.8. Plasma Stability
3.9. Microsomal Stability
3.10. Non-Specific Microsome Protein Binding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ranganna, K.; Selvam, C.; Shivachar, A.; Yousefipour, Z. Histone deacetylase inhibitors as multitarget-directed epi-drugs in blocking pi3k oncogenic signaling: A polypharmacology approach. Int. J. Mol. Sci. 2020, 21, 8198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Lai, C.J.; Bao, R.; Wang, D.G.; Wang, J.; Xu, G.X.; Atoyan, R.; Qu, H.; Yin, L.; Samson, M.; et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin. Cancer Res. 2012, 18, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.A.; Guerra, F.S.; Sagrillo, F.S.; De Sena, M.; Pinheiro, P.; Alves, M.A.; Thota, S.; Chaves, L.S.; Sant’Anna, C.M.R.; Fernandes, P.D.; et al. Design, Synthesis, and Pharmacological Evaluation of First-in-Class Multitarget N-Acylhydrazone Derivatives as Selective HDAC6/8 and PI3Kα Inhibitors. ChemMedChem 2020, 15, 539–551. [Google Scholar] [CrossRef]
- Guerra, F.S.; Rodrigues, D.A.; Fraga, C.A.M.; Fernandes, P.D. Novel single inhibitor of hdac6/8 and dual inhibitor of pi3k/hdac6 as potential alternative treatments for prostate cancer. Pharmaceuticals 2021, 14, 387. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Ferreira-Silva, G.À.; Ferreira, A.C.; Fernandes, R.A.; Kwee, J.K.; Sant’Anna, C.M.; Ionta, M.; Fraga, C.A. Design, Synthesis, and Pharmacological Evaluation of Novel N-Acylhydrazone Derivatives as Potent Histone Deacetylase 6/8 Dual Inhibitors. J. Med. Chem. 2016, 59, 655–670. [Google Scholar] [CrossRef]
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.; Chen, Z.; Dos Santos, O.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis(morpholino-1,3,5-triazine) Derivatives: Potent Adenosine 5′-Triphosphate Competitive Phosphatidylinositol-3-kinase/Mammalian Target of Rapamycin Inhibitors: Discovery of Compound 26 (PKI-587), a Highly Efficacious Dual Inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin, A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.M.X.; Aparecida-Silva, C.; Gamba, L.E.R.; de Melo, T.N.; Barbosa, G.; Moraes Junior, M.O.d.; de Oliveira, V.R.T.; de Amorim, C.S.; Moraes, J.A.; Barreiro, E.J.; et al. Design, Synthesis and Phenotypic Profiling of Simplified Gedatolisib Analogues. Pharmaceuticals 2023, 16, 209. [Google Scholar] [CrossRef]
- Vlietstra, R.J.; van Alewijk, D.C.; Hermans, K.G.; van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar] [PubMed]
- Beaver, J.A.; Gustin, J.P.; Yi, K.H.; Rajpurohit, A.; Thomas, M.; Gilbert, S.F.; Rosen, D.M.; Ho Park, B.; Lauring, J. PIK3CA and AKT1 mutations have distinct effects on sensitivity to targeted pathway inhibitors in an isogenic luminal breast cancer model system. Clin. Cancer Res. 2013, 19, 5413–5422. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.J.; Liu, Y.P.; Dai, H.Y.; Shiue, Y.L.; Tsai, C.J.; Huang, M.S.; Yeh, Y.T. Nuclear HDAC6 inhibits invasion by suppressing NF-kB/MMP2 and is inversely correlated with metastasis of non-small cell lung cancer. Oncotarget 2015, 6, 30263–30276. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.; Yao, T. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Verdel, A.; Curtet, S.; Brocard, M.P.; Rousseaux, S.; Lemercier, C.; Yoshida, M.; Khochbin, S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr. Biol. 2000, 10, 747–749. [Google Scholar] [CrossRef]
- Citalingam, K.; Abas, F.; Lajis, N.H.; Othman, I.; Naidu, R. Anti-proliferative effect and induction of apoptosis in androgen-independent human prostate cancer cells by 1,5-bis(2-hydroxyphenyl)-1,4-pentadiene-3-one. Molecules 2015, 20, 3406–3430. [Google Scholar] [CrossRef]
- Ghasemi, M.; Turnbull, T.; Sebastian, S.; Kempson, I. The MTT Assay: Utility, Limitations, Pitfalls, and Interpretation in Bulk and Single-Cell Analysis. Int. J. Mol. Sci. 2021, 22, 12827. [Google Scholar] [CrossRef]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.-C.; Martin, N.; CombuSyn. CompuSyn for Drug Combinations: User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug CompuSyn for Drug Combinations, and the Determination of IC50 and ED50 Values. 2024. Available online: https://www.combosyn.com/register.html (accessed on 11 January 2024).
- Zhi, L.; Can, Z.; Ge, H.; Yujie, W.; Yang, W.; Xiaodong, M. Identification of PI3K/HDAC Dual-targeted inhibitors with subtype selectivity as potential therapeutic agents against solid Tumors: Building HDAC6 potency in a Quinazolinone-based PI3Kδ-selective template. Bioorganic Med. Chem. 2022, 73, 117028. [Google Scholar] [CrossRef]
- Yamada, T.; Horinaka, M.; Shinnoh, M.; Yoshioka, T.; Miki, T.; Sakai, T. A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma. Int. J. Oncol. 2013, 43, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, A.; Alves, G.; Soares-da-Silva, P.; Falcão, A. Optimization of a parallel artificial membrane permeability assay for the fast and simultaneous prediction of human intestinal absorption and plasma protein binding of drug candidates: Application to dibenz[b,f]azepine-5-carboxamide derivatives. J. Pharm. Sci. 2012, 101, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Pérez, D.I.; Pistolozzi, M.; Palomo, V.; Redondo, M.; Fortugno, C.; Gil, C.; Felix, G.; Martinez, A.; Bertucci, C. 5-Imino-1,2-4-thiadiazoles and quinazolines derivatives as glycogen synthase kinase 3β (GSK-3β) and phosphodiesterase 7 (PDE7) inhibitors: Determination of blood–brainbarrier penetration and binding to human serum albumin. Eur. J. Pharm. Sci. 2012, 45, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.X.; Strojnowski, M.J.; Hu, J.K.; Smith, B.J.; Eichhold, T.H.; Wehmeyer, K.R.; Pikul, S.; Almstead, N.G. Gas chromatographic-mass spectrometric analysis of hydroxylamine for monitoring the metabolic hydrolysis of metalloprotease inhibitors in rat and human liver microsomes. J. Chromatogr. B Biomed. Sci. Appl. 1999, 724, 181–187. [Google Scholar] [CrossRef]
- Singh, A.; Gao, M.; Beck, M.W. Human carboxylesterases and fluorescent probes to image their activity in live cells. RSC Med. Chem. 2021, 12, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Bosc, D.; Piveteau, C.; Gealageas, R.; Lam, B.; Ronco, C.; Roignant, M.; Tolojanahary, H.; Jean, L.; Renard, P.Y.; et al. Controlling Plasma Stability of Hydroxamic Acids: A MedChem Toolbox. J. Med. Chem. 2017, 60, 9067–9089. [Google Scholar] [CrossRef] [PubMed]
- Halouska, S.; Fenton, R.J.; Zinniel, D.K.; Marshall, D.D.; Barletta, R.G.; Powers, R. Metabolomics analysis identifies d-Alanine-d-Alanine ligase as the primary lethal target of d-Cycloserine in mycobacteria. J. Proteome Res. 2014, 13, 1065–1076. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef]
- McLure, J.A.; Miners, J.O.; Birkett, D.J. Nonspecific binding of drugs to human liver microsomes. Br. J. Clin. Pharmacol. 2000, 49, 453–461. [Google Scholar] [CrossRef]
- Koyanagi, M.; Kawakabe, S.; Arimura, Y. A comparative study of colorimetric cell proliferation assays in immune cells. Cytotechnology 2015, 68, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. lmmunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Verma, S.C.; Murakami, M.; Bajaj, B.; Robertson, E.S. Isolation of Human Peripheral Blood Mononuclear Cells (PBMCs). Curr. Protoc. Microbiol. 2007, A4C.1-A4C.9. [Google Scholar] [CrossRef] [PubMed]
- Riedhammer, C.; Halbritter, D.; Weissert, R. Peripheral blood mononuclear cells: Isolation, freezing, thawing, and culture. Methods Mol. Biol. 2015, 1304, 53–61. [Google Scholar] [CrossRef]
- Kleiveland, C. Peripheral blood mononuclear cells. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 161–167. [Google Scholar] [CrossRef]
- Orellana, E.; Kasinski, A. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-Protocol 2016, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; Mcmahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- de Queiroz, A.C.; Barbosa, G.; de Oliveira, V.R.T.; de Mattos Alves, H.; Alves, M.A.; Carregaro, V.; da Silva, J.S.; Barreiro, E.J.; Alezandre-Moreira, M.S.; Lima, L.M. Pre-clinical evaluation of LASSBio-1491: From in vitro pharmacokinetic study to in vivo leishmanicidal activity. PLoS ONE 2022, 17, e0269447. [Google Scholar] [CrossRef]
- Alves, M.A.; de Queiroz, A.C.; Leite, A.B.; Martins, F.T.; Doriguetto, A.C.; Barreiro, E.J.; Alexandre, M.S.; Lima, L.M. Carbamoyl-N-aryl-imine-urea: A new framework to obtain a putative leishmanicidal drug-candidate. RSC Adv. 2020, 10, 12384–12394. [Google Scholar] [CrossRef]
- Cabrera, M.; Lavaggi, M.L.; Hernández, P.; Merlino, A.; Gerpe, A.; Porcal, W.; Boiani, M.; Ferreira, A.; Monge, A.; de Cerain, A.L.; et al. Cytotoxic, mutagenic and genotoxic effects of new anti-T. cruzi 5-phenylethenylbenzofuroxans. Contribution of phase I metabolites on the mutagenicity induction. Toxicol. Lett. 2009, 190, 140–149. [Google Scholar] [CrossRef]
- Gardner, I.; Xu, M.; Han, C.; Wang, Y.; Jiao, X.; Jamei, M.; Khalidi, H.; Kilford, P.; Neuhoff, S.; Southall, R.; et al. Non-specific binding of compounds in in vitro metabolism assays: A comparison of microsomal and hepatocyte binding in different species and an assessment of the accuracy of prediction models. Xenobiotica 2022, 52, 943–956. [Google Scholar] [CrossRef]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.S.; Jones, B.C. The Prediction of Human Pharmacokinetic Parameters from Preclinical and In Vitro Metabolism Data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar]

{kind=link}
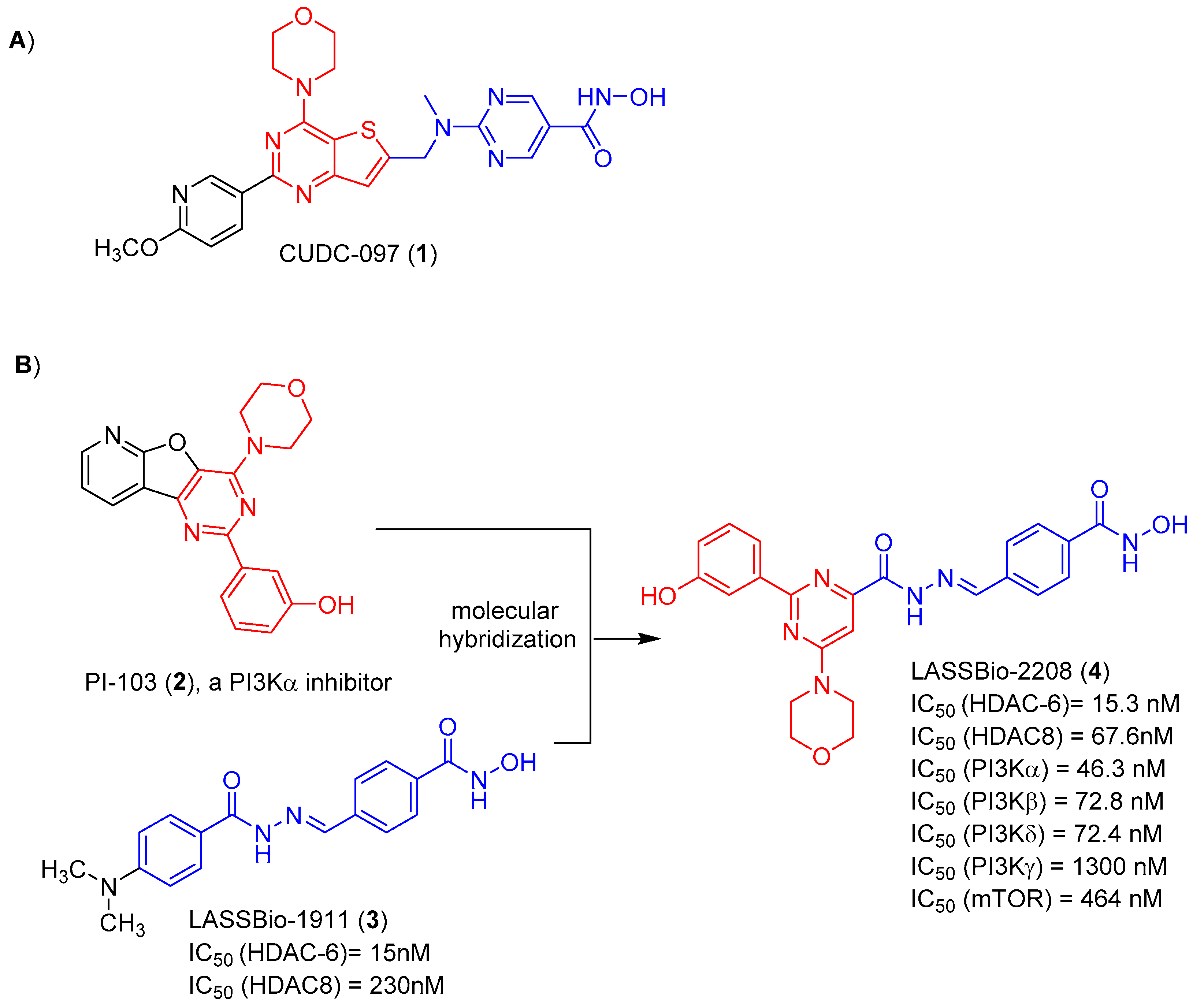{kind=link}
| MTT Assay | Cell Line | Gedatolisib CC50 (µM) | Tubastatin A CC50 (µM) | LASSBio-1911 CC50 (µM) | LASSBio-2208 CC50 (µM) |
|---|---|---|---|---|---|
| 24 h | PC-3 | >100 a Emax = 48% | >100 a Emax = 41.8% | >100 a Emax = 26.4% | >100 a Emax = 18.4% |
| MCF-7 | 5.04 (2.86–8.88) Emax = 79.9% | >100 a Emax = 40.1% | >100 a Emax = 21.6% | >100 a Emax = 29.5% | |
| CCRF-CEM | 0.31 (0.24–0.41) Emax = 98.6% | 12.51 (5.95–26.31) Emax = 95.8% | 9.69 (8.07–11.64) Emax = 98% | 27.75 (21.70–35.50) Emax = 69.4% | |
| MOLT-4 | 0.35 (0.25–0.47) Emax = 97.2% | 5.67 (3.95–8.12) Emax = 94.6% | 13.75 (2.55–74.05) Emax = 96.5% | 7.14 (3.51–14.54) Emax = 84.1% | |
| 48 h | PC-3 | 5.05 (2.59–9.82) Emax = 60.5% | 65.94 (57.29–75.88) Emax = 67.8% | >100 a Emax = 42.6% | >100 a Emax = 39.3% |
| MCF-7 | 0.21 (0.13–0.34) Emax = 96.8% | >100 a Emax = 48.2% | 42.62 (30.76–59.05) Emax = 65.4% | 31.71 (19.45–51.69) Emax = 57.3% | |
| CCRF-CEM | 0.085 (0.07–0.11) Emax = 99.5% | 9.38 (5.18–17.03) Emax = 99.1% | 6.98 (4.28–11.37) Emax = 98.8% | 8.68 (6.99–10.30) Emax = 91.7% | |
| MOLT-4 | 0.08 (0.07–0.11) Emax = 99.3% | 7.50 (5.79–9.70) Emax = 98.2% | 6.15 (4.24–8.90) Emax = 98.9% | 8.26 (5.82–11.71) Emax = 83.2% | |
| 72 h | PC-3 | 0.86 (0.59–1.25) Emax = 81.1% | 34.89 (30.83–39.47) Emáx = 83.6% | 77.10 (63.96–92.95) Emax = 59.2% | >100 a Emax = 42.1% |
| MCF-7 | 0.06 (0.04–0.08) Emax = 99.3% | 83.56 (70.22–99.42) Emax = 56.8% | 38.43 (31.37–47.06) Emax = 75.4% | 23.0 (15.50–34.14) Emax = 60.7% | |
| CCRF-CEM | 0.09 (0.08–0.11) Emax = 100% | 7.11 (3.32–15.19) Emax = 99.3% | 6.92 (2.97–16.08) Emax = 97.1% | 8.54 (6.30–11.57) Emax = 90.4% | |
| MOLT-4 | 0.07 (0.06–0.08) Emax = 99.8% | 5.65 (3.51–9.09) Emax = 98.7% | 5.27 (3.45–8.03) Emax = 99.2% | 7.15 (5.20–9.81) Emax = 91.2% |
| Cell Line | LASSBio-1911 | Tubastatin A | LASSBio-2208 | |||
|---|---|---|---|---|---|---|
| CC50 (µM) 48 h | SI | CC50 (µM) 48 h | SI | CC50 (µM) 48 h | SI | |
| Human PBMC | >100 a Emax = 31.1% | N.A. | 85.66 (39.7–184.8) Emax = 58.2% | N.A. | >100 a Emax = 29.9% | N.A. |
| PC-3 | >100 Emax = 42.6% | N.D. | 65.94 (57.29–75.88) Emax = 67.8% | 1.29 | >100 a Emax = 39.3% | N.D. |
| MCF-7 | 42.62 (30.76–59.05) Emax = 65.4% | N.D. | >100 a Emax = 48.2% | N.D. | 31.71 (19.45–51.69) Emax = 57.3% | N.D. |
| MOLT-4 | 6.15 (4.24–8.90) Emax = 98.9% | N.D. | 7.50 (5.79–9.70) Emax = 98.2% | 11.42 | 8.26 (5.82–11.71) Emax = 83.2% | N.D. |
| CCRF-CEM | 6.98 (4.28–11.37) Emax = 98.8% | N.D. | 9.38 (5.18–17.03) Emax = 99.1% | 9.13 | 8.68 (6.99–10.30) Emax = 91.7% | N.D. |
| Cell Line | Gedatolisib CC50 (µM) | LASSBio-1911 CC50 (µM) | Tubastatin A CC50 (µM) | LASSBio-2208 CC50 (µM) |
|---|---|---|---|---|
| PC-3 | 6.16 (1.42–26.7) Emax = 56.2% | >100 Emax = 34.04% | >100 Emax = 39.3% | 98.93 (85.02–115.1) Emax = 51.4% |
| MCF-7 | 0.03 (0.01–0.06) Emax = 95.7% | 39.14 (29.36–52.17) Emax = 86.9% | 66.01 (57.45–75.95) Emax = 71.2% | 5.44 (3.15–9.41) Emax = 67.4% |
| MOLT-4 | 3.68 (2.04–6.62) Emax = 69.2% | 53.09 (34.03–82.84) Emax = 61% | 10.45 (1.43–76.18) Emax = 54.4% | >100 |
| CCRF-CEM | 0.78 (0.25–2.37) Emax = 66.2% | 25.18 (16.61–38.18) Emax = 69.2% | 9.69 (4.39–21.39) Emax = 69.5% | >100 |
| Combinations: X×CC50 (A) + X×CC50 (B) | Gedatolisib (A) + LASSBio-1911 (B) | Gedatolisib (A) + Tubastatin A (B) | Gedatolisib (A) + LASSBio-2208 (B) | LASSBio-2208 (A) + LASSBio-1911 (B) | Tubastatin A (A) + LASSBio-2208 (B) |
|---|---|---|---|---|---|
| X = 0.25 | Moderate antagonism (CI = 1.25) | Nearly additive (CI = 1.05) | Moderate antagonism (CI = 1.24) | Moderate antagonism (CI = 1.23) | Slight synergism (CI = 0.86) |
| X = 0.5 | Moderate antagonism (CI =1.27) | Moderate antagonism (CI =1.32) | Slight synergism (CI = 0.89) | Moderate synergism (CI = 0.81) | Antagonism (CI =1.53) |
| X = 1 | Nearly additive (CI = 0.96) | Synergism (CI = 0.48) | Synergism (CI = 0.63) | Synergism (CI = 0.56) | Synergism (CI = 0.38) |
| X = 2 | Synergism (CI = 0.53) | Synergism (CI = 0.51) | Synergism (CI = 0.33) | Synergism (CI = 0.61) | Synergism (CI = 0.48) |
| X = 4 | Moderate synergism (CI = 0.77) | Nearly additive (CI = 1.01) | Strong synergism (CI = 0.15) | Nearly additive (CI = 0.96) | Nearly additive (CI = 0.97) |
| Combinations: X×CC50 (A) + X×CC50 (B) | Gedatolisib (A) + LASSBio-1911 (B) | Gedatolisib (A) + Tubastatin A (B) | Gedatolisib (A) + LASSBio-2208 (B) | LASSBio-2208 (A) + LASSBio-1911 (B) | Tubastatin A (A) + LASSBio-2208 (B) |
|---|---|---|---|---|---|
| X = 0.25 | Moderate antagonism (CI = 1.21) | Moderate synergism (CI = 0.72) | Moderate synergism (CI = 0.77) | Moderate antagonism (CI = 1.28) | Strong antagonism (CI = 3.69) |
| X = 0.5 | Nearly additive (CI = 0.91) | Synergism (CI = 0.69) | Slight synergism (CI = 0.65) | Slight antagonism (CI = 1.18) | Nearly additive (CI =1.02) |
| X = 1 | Synergism (CI = 0.49) | Synergism (CI = 0.41) | Synergism (CI = 0.44) | Synergism (CI = 0.54) | Synergism (CI = 0.53) |
| X = 2 | Synergism (CI = 0.54) | Moderate synergism (CI = 0.80) | Synergism (CI = 0.34) | Synergism (CI = 0.64) | Slight synergism (CI = 0.87) |
| X = 4 | Nearly additive (CI = 1.03) | Antagonism (CI = 1.59) | Synergism (CI = 0.47) | Moderate antagonism (CI = 1.31) | Antagonism (CI = 1.86) |
| Compounds | Pe. Exp. BBB (10−6 cm/s) | Classification BBB | Pe. Exp. GIT (10−6 cm/s) | Fraction Absorbed (%) | Classification GIT |
|---|---|---|---|---|---|
| LASSBio-2208 | 0.69 | CNS− | 0.39 | 13.78 | Low |
| LASSBio-1911 | 0.29 | CNS− | 0.592 | 20.15 | Low |
| Gedatolisib | Insoluble | 1.74 | 48.40 | Medium | |
| Tubastatin A | 5.54 | CNS+ | 1.982 | 52.93 | Medium |
| Compounds | Metabolization Rate (%) | Elimination Rate Constant (k) | t1/2 (min) | Recovery (%) |
|---|---|---|---|---|
| LASSBio-2208 | 20.61 | 0.0008 | 866.25 | 75.47 |
| LASSBio-1911 | 36.14 | 0.0018 | 385.00 | 73.36 |
| Compounds | Percentage of ƒu(mic) |
|---|---|
| Tolbutamide * | 1.113 |
| LASSBio-2208 | 0.937 |
| LASSBio-1911 | 0.004 |
| Gedatolisib | 0.503 |
| Tubastatin A | 0.131 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pillpe-Meza, R.M.; Gouveia, W.L.; Barbosa, G.; Fraga, C.A.M.; Barreiro, E.J.; Lima, L.M. Cytotoxic and Antiproliferative Activity of LASSBio-2208 and the Attempts to Determine Its Drug Metabolism and Pharmacokinetics In Vitro Profile. Pharmaceuticals 2024, 17, 389. https://doi.org/10.3390/ph17030389
Pillpe-Meza RM, Gouveia WL, Barbosa G, Fraga CAM, Barreiro EJ, Lima LM. Cytotoxic and Antiproliferative Activity of LASSBio-2208 and the Attempts to Determine Its Drug Metabolism and Pharmacokinetics In Vitro Profile. Pharmaceuticals. 2024; 17(3):389. https://doi.org/10.3390/ph17030389
Chicago/Turabian StylePillpe-Meza, Raysa Magali, Wesley Leandro Gouveia, Gisele Barbosa, Carlos A. M. Fraga, Eliezer J. Barreiro, and Lidia Moreira Lima. 2024. "Cytotoxic and Antiproliferative Activity of LASSBio-2208 and the Attempts to Determine Its Drug Metabolism and Pharmacokinetics In Vitro Profile" Pharmaceuticals 17, no. 3: 389. https://doi.org/10.3390/ph17030389
APA StylePillpe-Meza, R. M., Gouveia, W. L., Barbosa, G., Fraga, C. A. M., Barreiro, E. J., & Lima, L. M. (2024). Cytotoxic and Antiproliferative Activity of LASSBio-2208 and the Attempts to Determine Its Drug Metabolism and Pharmacokinetics In Vitro Profile. Pharmaceuticals, 17(3), 389. https://doi.org/10.3390/ph17030389