
Current Perspectives on Biological Screening of Newly Synthetised Sulfanilamide Schiff Bases as Promising Antibacterial and Antibiofilm Agents


 ,
,  , ,
, ,  ,
, 
 and
and 
Abstract
1. Introduction
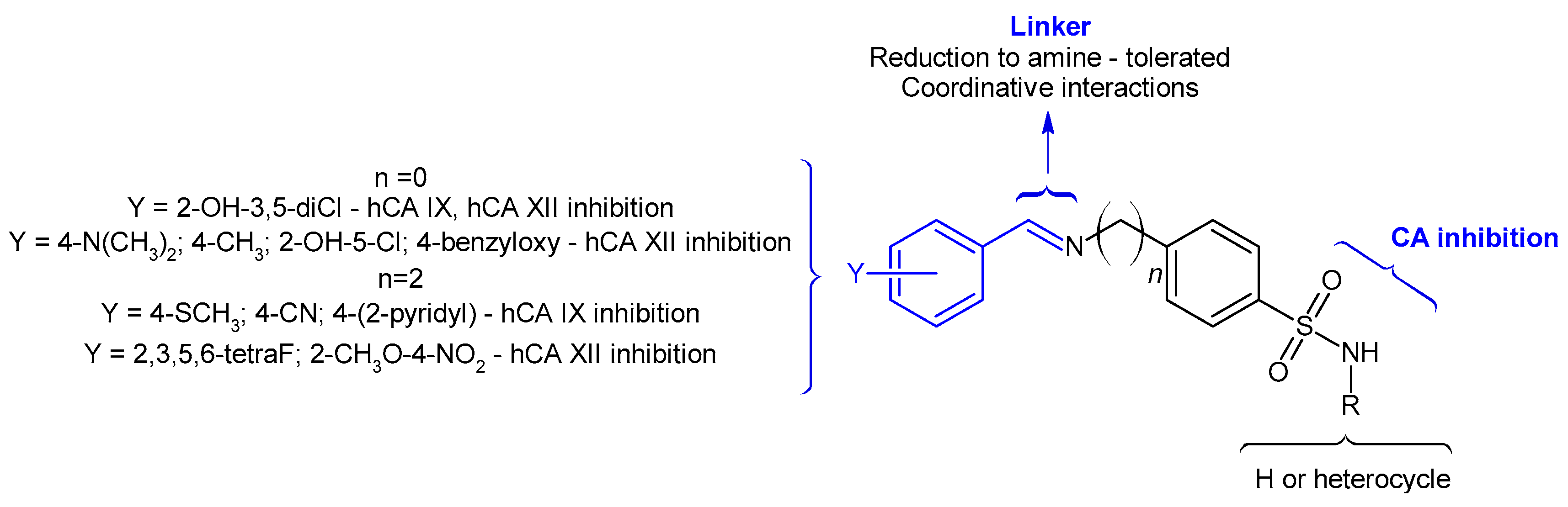
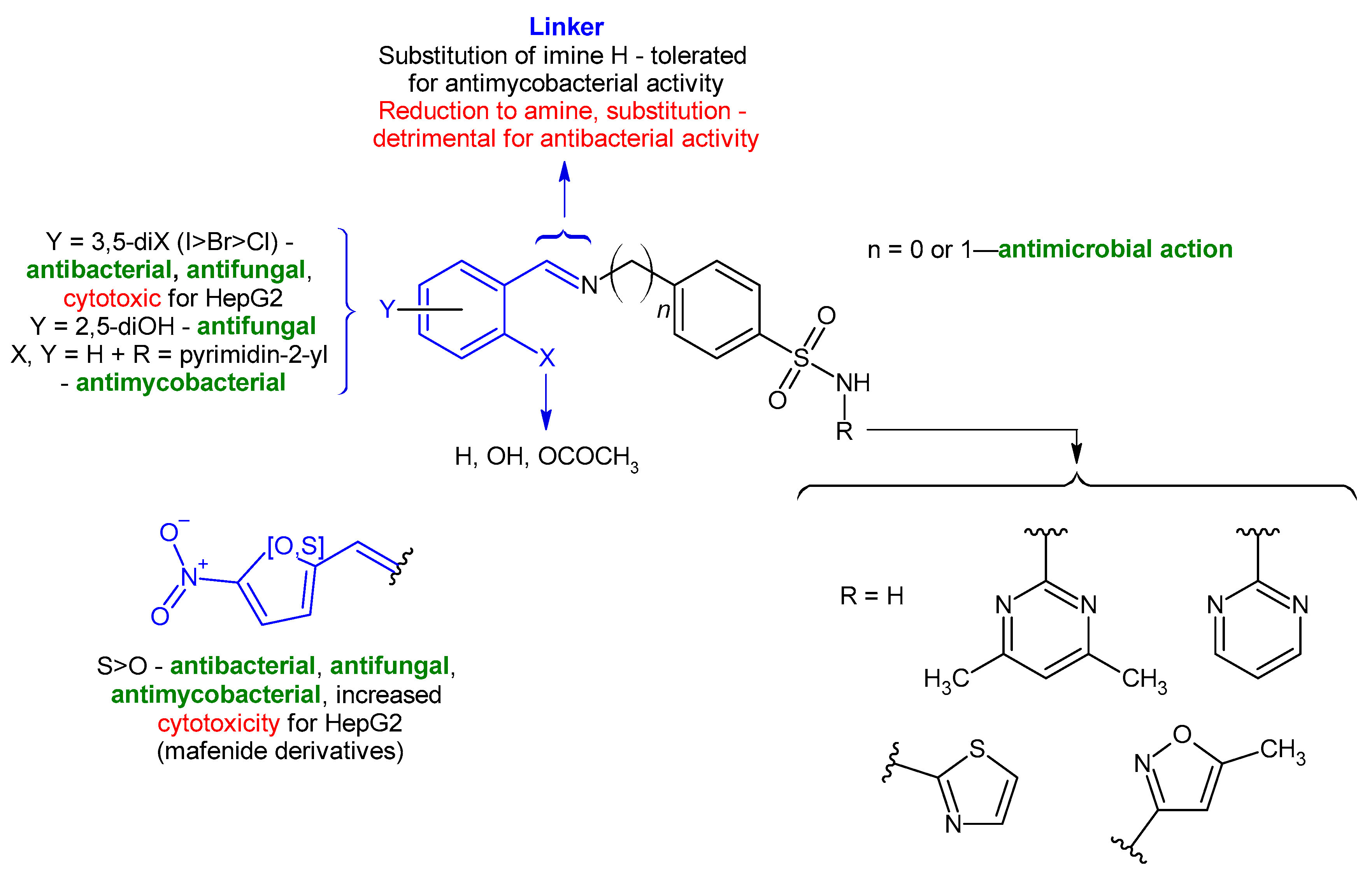
2. Results
2.1. Chemistry and Spectral Data
2.2. In Silico Studies
2.2.1. Assessment of the Compounds’ Drug- and Lead-Likeness Features
2.2.2. Pharmacokinetics and Pharmacogenomics Profiles of Molecules 1a–e
2.2.3. Computational Pharmacodynamic Profiles of Molecules 1a–e
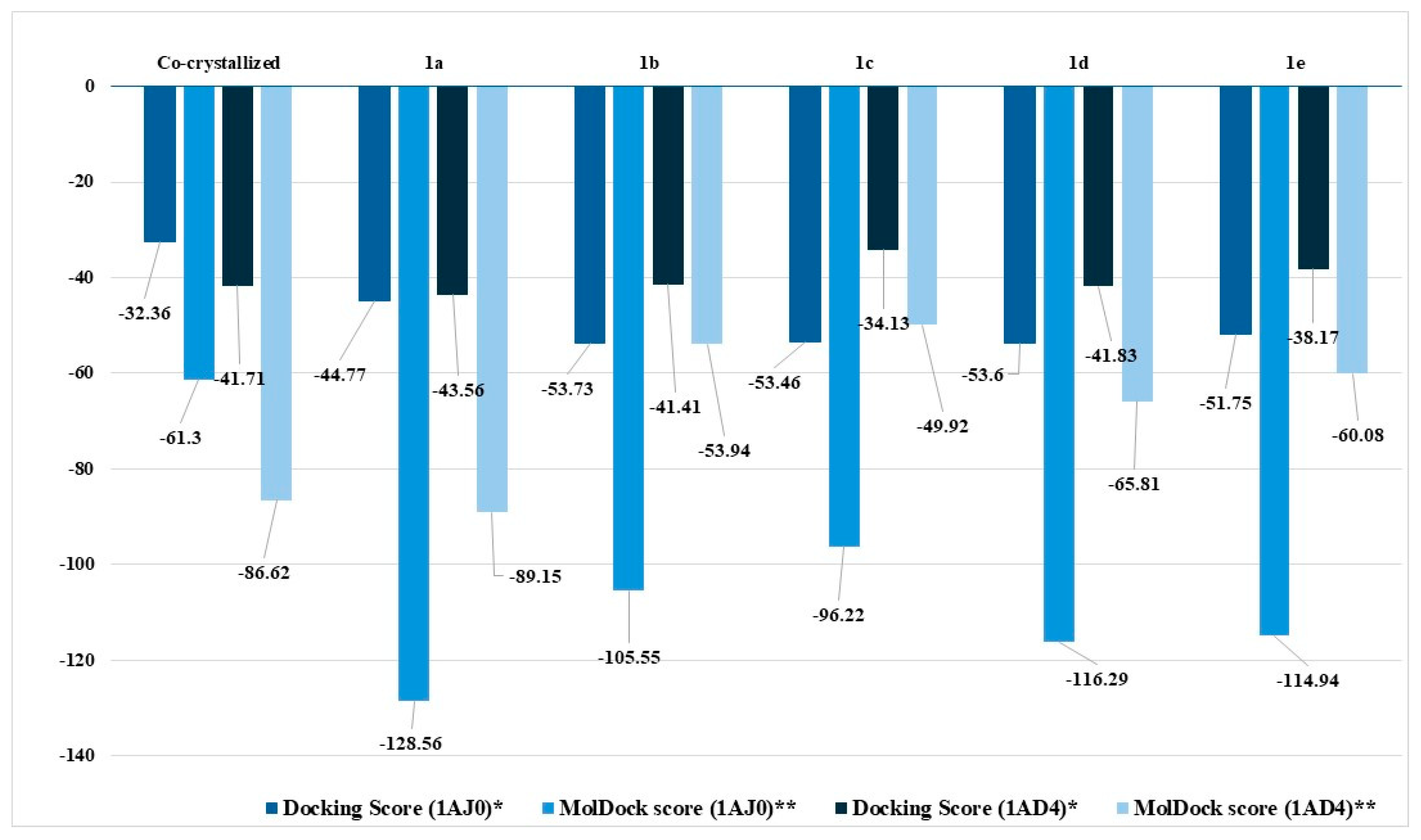
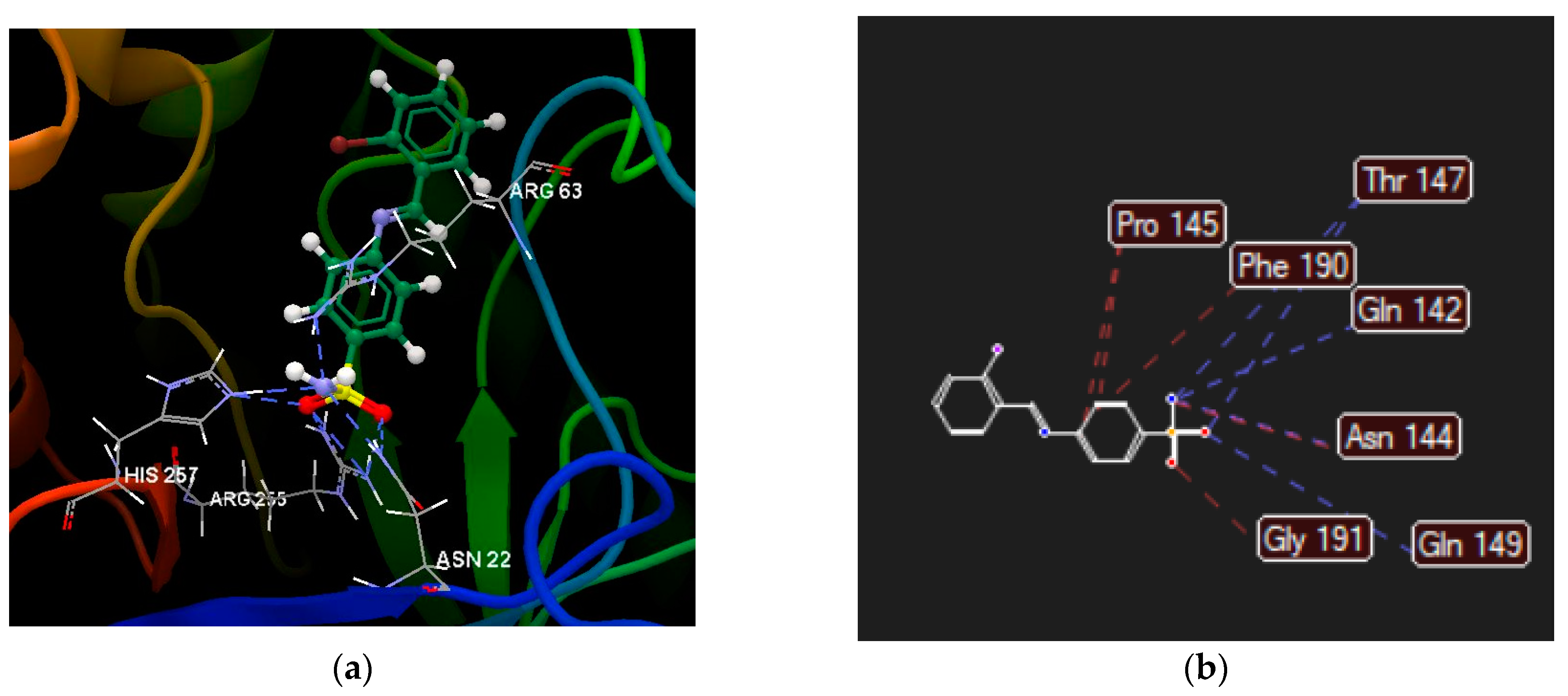
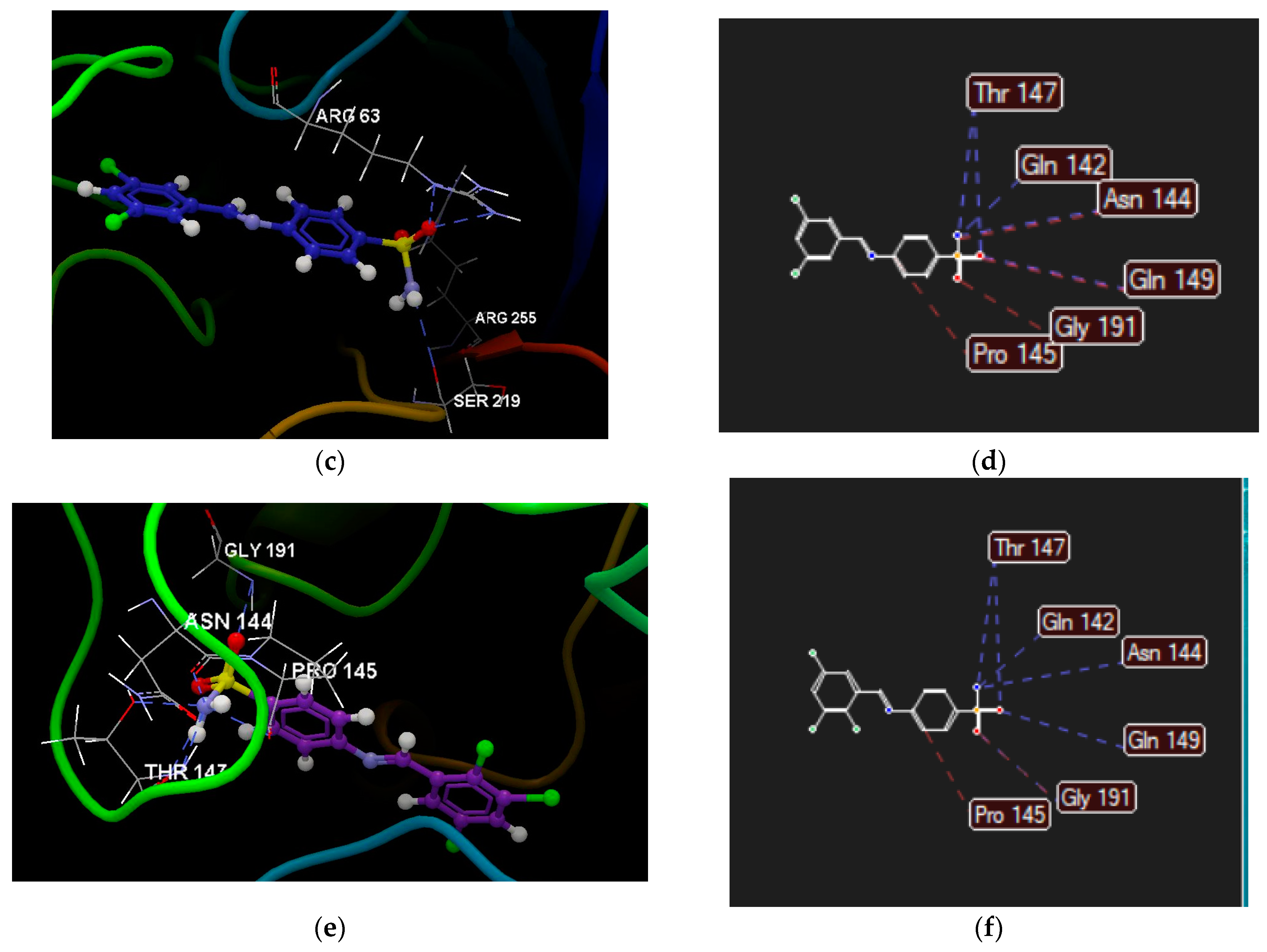
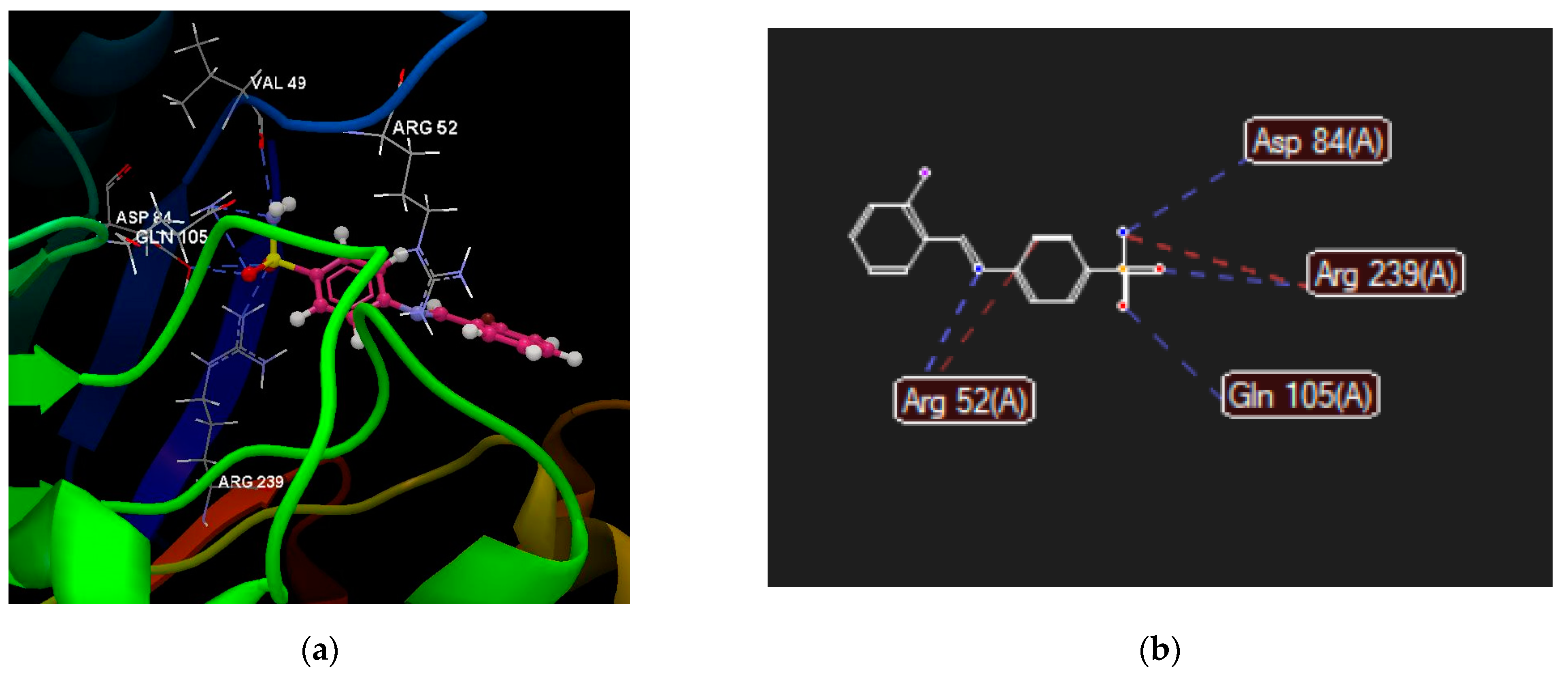
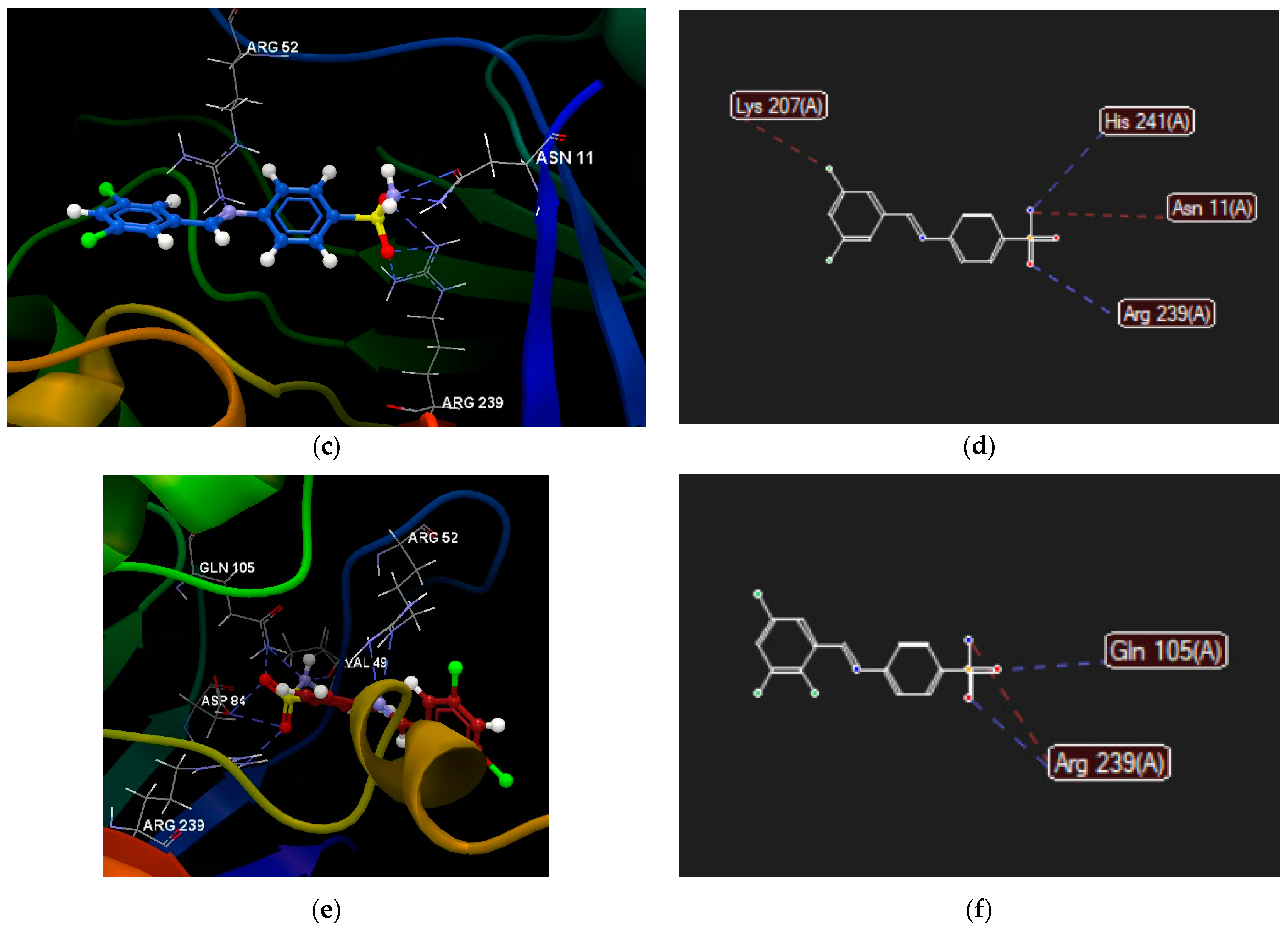
2.2.4. Molecular Docking
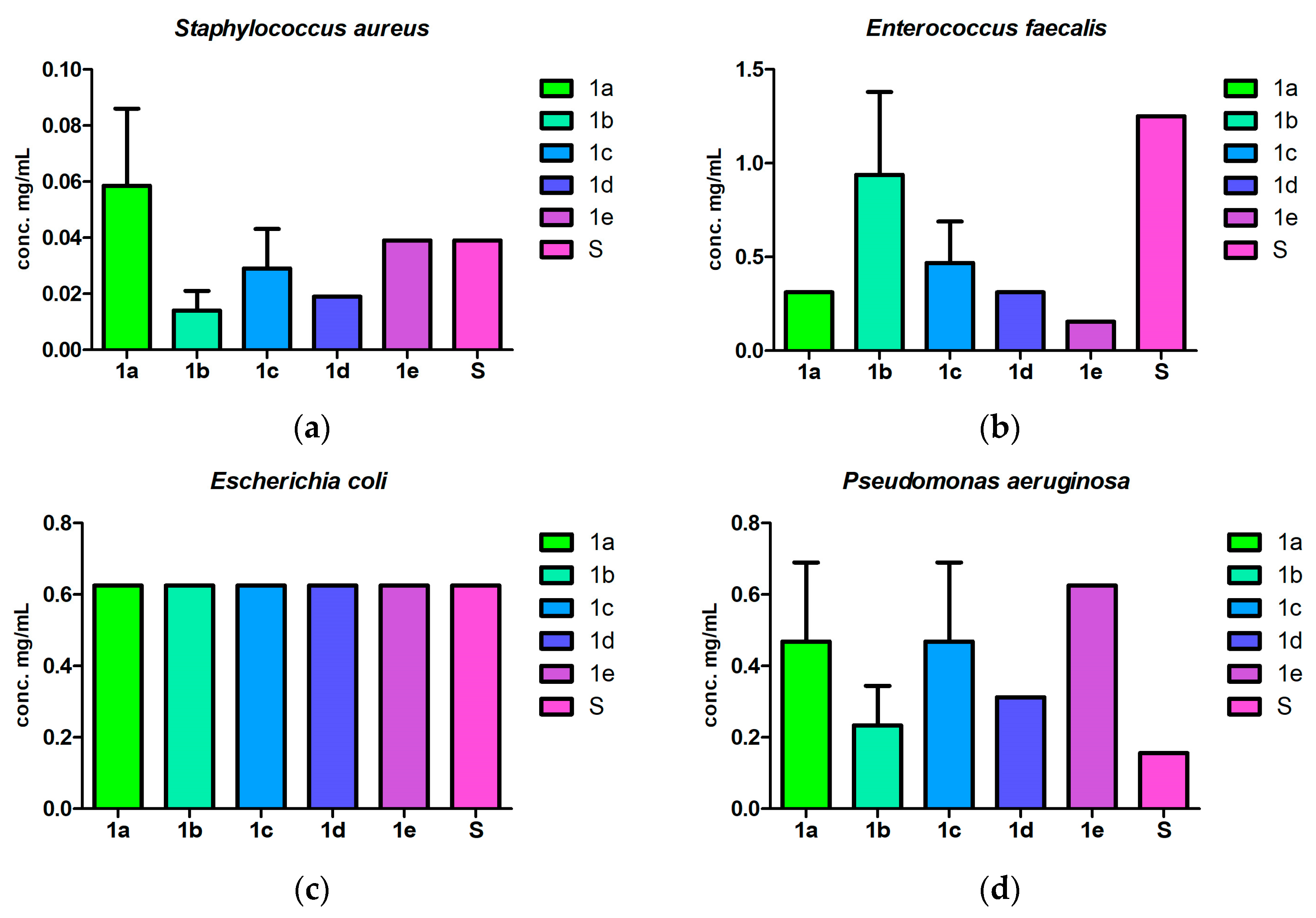
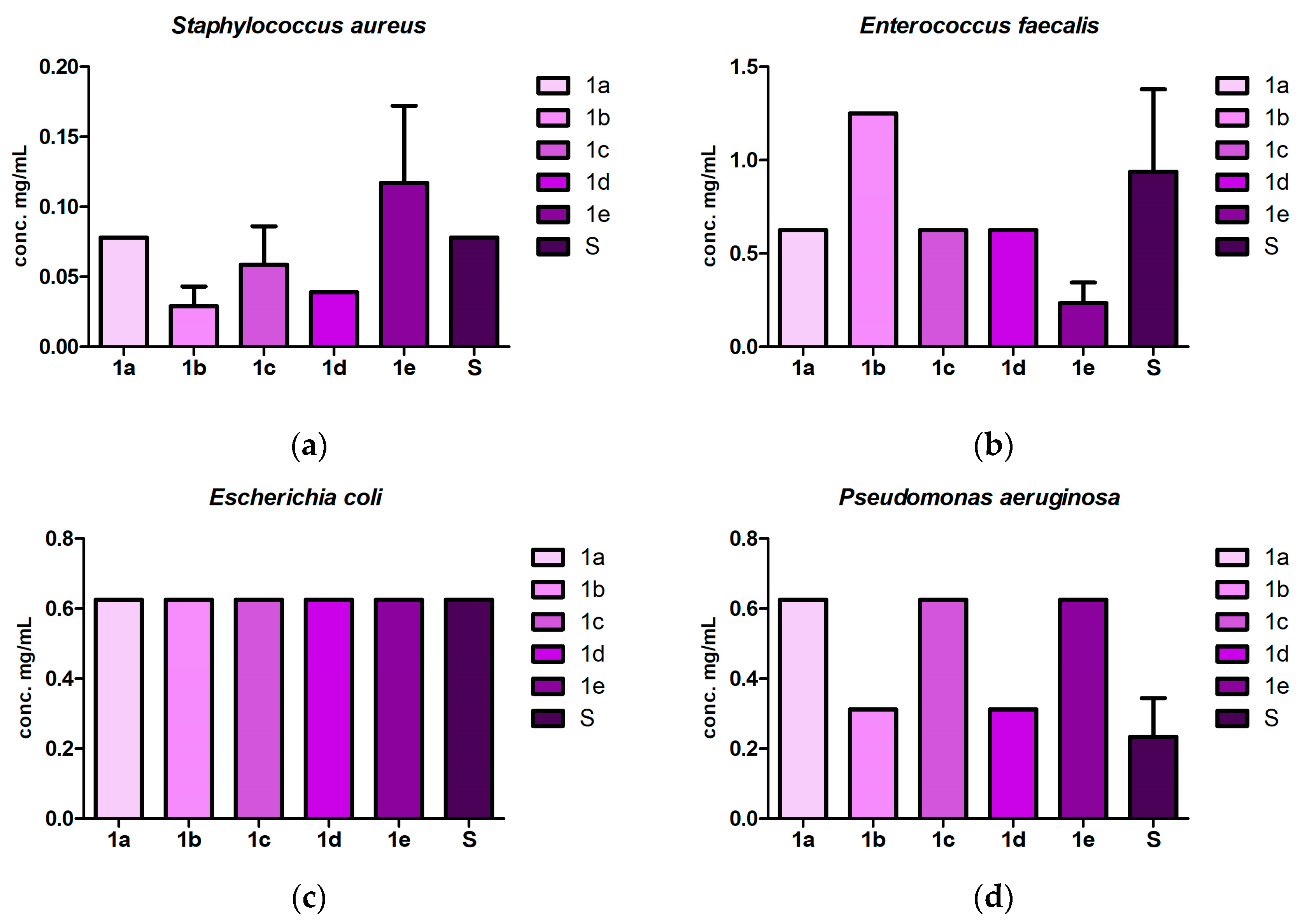
2.3. Antibacterial and Antibiofilm Screening
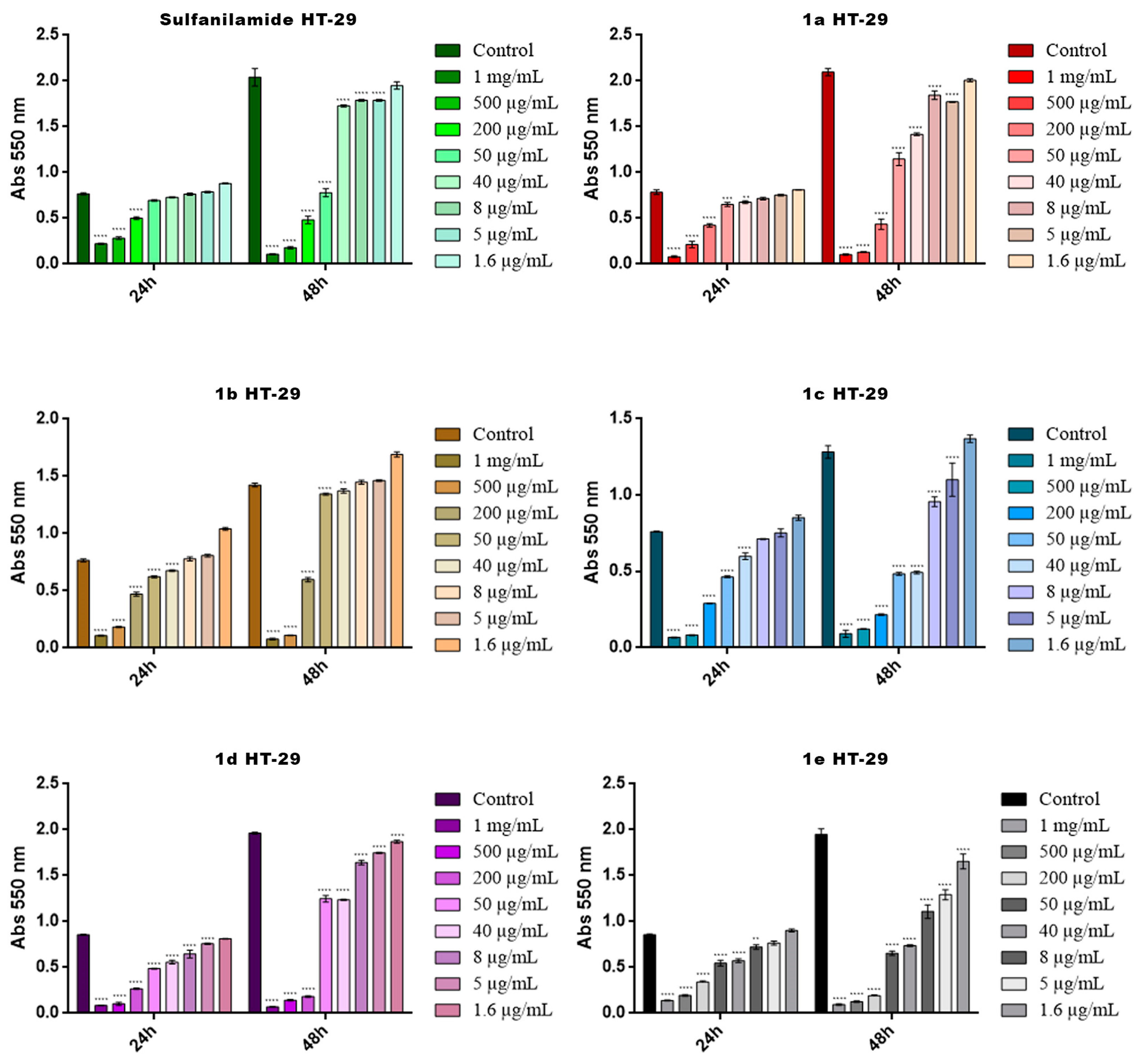
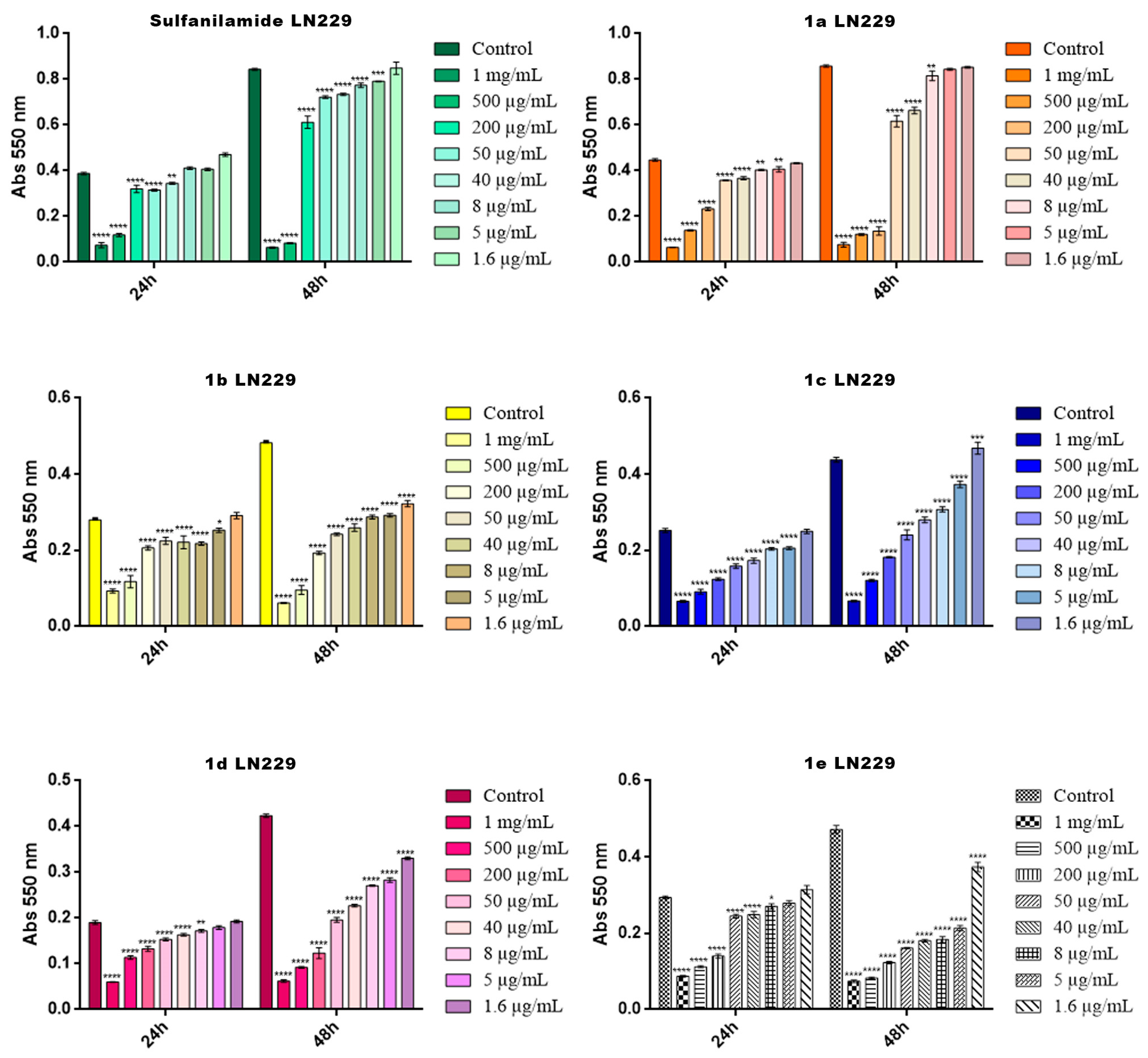
2.4. Cytotoxicity Screening
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Information
4.1.2. Synthesis and Characterization of the Compounds
4.2. Computational Strategy
4.2.1. Molecular Modeling of Chemical Structures 1a–e
4.2.2. Assessment of Compounds’ Drug- and Lead-Likeness Features
4.2.3. Computational Pharmacokinetics, Pharmacogenomics and Toxicological Profiles of Sulfanilamide Schiff Bases 1a–e
4.2.4. Computational Pharmacodynamic Profiles of Molecules 1a–e
4.2.5. Molecular Docking
4.3. Antibacterial and Antibiofilm Bioactivity Screening
4.3.1. General Information
4.3.2. Methods
4.4. In Vitro Cytotoxicity on Cancer Cells
4.4.1. General Information
4.4.2. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- New WHO Report Highlights Progress, but Also Remaining Gaps, in Ensuring a Robust Pipeline of Antibiotic Treatments to Combat Antimicrobial Resistance (AMR). Available online: https://www.who.int/news/item/15-05-2023-new-who-report-highlights-progress-but-also-remaining-gaps-in-ensuring-a-robust-pipeline-of-antibiotic-treatments-to-combat-antimicrobial-resistance-(amr) (accessed on 23 May 2023).
- Høiby, N.; Bjarnsholt, T.; Moser, C.; Bassi, G.L.; Coenye, T.; Donelli, G.; Hall-Stoodley, L.; Holá, V.; Imbert, C.; Kirketerp-Møller, K.; et al. ESCMID* Guideline for the Diagnosis and Treatment of Biofilm Infections 2014. Clin. Microbiol. Infect. 2015, 21, S1–S25. [Google Scholar] [CrossRef] [PubMed]
- Jorge, P.; Magalhães, A.P.; Grainha, T.; Alves, D.; Sousa, A.M.; Lopes, S.P.; Pereira, M.O. Antimicrobial Resistance Three Ways: Healthcare Crisis, Major Concepts and the Relevance of Biofilms. FEMS Microbiol. Ecol. 2019, 95, fiz115. [Google Scholar] [CrossRef] [PubMed]
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular Mechanisms of Antibiotic Resistance Revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar] [CrossRef]
- Ceri, H.; Gorman, S.P.; Gilmore, B.F. Microbial Biofilms: Consequences for Health. In Hugo and Russell’s Pharmaceutical Microbiology; Denyer, S.P., Hodges, N., Gorman, S.P., Gilmore, B.F., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2011; pp. 121–130. ISBN 9781444330632. [Google Scholar]
- Patrick, G.L. Antibacterial Agents. In An Introduction to Medicinal Chemistry; Oxford University Press: Oxford, UK, 2017; pp. 425–432. ISBN 9780198749691. [Google Scholar]
- Werth, B.J. Sulfonamides. Available online: https://www.msdmanuals.com/professional/infectious-diseases/bacteria-and-antibacterial-drugs/sulfonamides?query=sulfonamideantibiotics (accessed on 23 February 2023).
- Mercer, M.A. Sulfonamides and Sulfonamide Combinations Use in Animals. Available online: https://www.msdvetmanual.com/pharmacology/antibacterial-agents/sulfonamides-and-sulfonamide-combinations-use-in-animals (accessed on 23 February 2023).
- Kaye, K.S.; Gales, A.C.; Dubourg, G. Old Antibiotics for Multidrug-Resistant Pathogens: From in Vitro Activity to Clinical Outcomes. Int. J. Antimicrob. Agents 2017, 49, 542–548. [Google Scholar] [CrossRef]
- Younus, H.A.; Saleem, F.; Hameed, A.; Al-Rashida, M.; Al-Qawasmeh, R.A.; El-Naggar, M.; Rana, S.; Saeed, M.; Khan, K.M. Part-II: An Update of Schiff Bases Synthesis and Applications in Medicinal Chemistry-a Patent Review (2016–2023). Expert Opin. Ther. Pat. 2023, 33, 841–864. [Google Scholar] [CrossRef]
- Ceramella, J.; Iacopetta, D.; Catalano, A.; Cirillo, F.; Lappano, R.; Sinicropi, M.S. A Review on the Antimicrobial Activity of Schiff Bases: Data Collection and Recent Studies. Antibiotics 2022, 11, 191. [Google Scholar] [CrossRef]
- Coandă, M.; Limban, C.; Nuță, D.C. Small Schiff Base Molecules—A Possible Strategy to Combat Biofilm-Related Infections. Antibiotics 2024, 13, 75. [Google Scholar] [CrossRef]
- Hameed, A.; al-Rashida, M.; Uroos, M.; Abid Ali, S.; Khan, K.M. Schiff Bases in Medicinal Chemistry: A Patent Review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 63–79. [Google Scholar] [CrossRef]
- Zafar, W.; Sumrra, S.H.; Hassan, A.U.; Chohan, Z.H. A Review on ‘Sulfonamides’: Their Chemistry and Pharmacological Potentials for Designing Therapeutic Drugs in Medical Science. J. Coord. Chem. 2023, 76, 546–580. [Google Scholar] [CrossRef]
- Krátký, M.; Konečná, K.; Janoušek, J.; Janďourek, O.; Maixnerová, J.; Kalivodová, S.; Trejtnar, F.; Vinšová, J. Sulfonamide-Salicylaldehyde Imines Active against Methicillin- and Trimethoprim/Sulfonamide-Resistant Staphylococci. Future Med. Chem. 2021, 13, 1945–1962. [Google Scholar] [CrossRef]
- Krátký, M.; Dzurková, M.; Janoušek, J.; Konečná, K.; Trejtnar, F.; Stolaříková, J.; VinŠová, J. Sulfadiazine Salicylaldehyde-Based Schiff Bases: Synthesis, Antimicrobial Activity and Cytotoxicity. Molecules 2017, 22, 1573. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Volková, M.; Buchta, V.; Trejtnar, F.; Stolaříková, J. Antimicrobial Activity of Sulfonamides Containing 5-Chloro-2- Hydroxybenzaldehyde and 5-Chloro-2-Hydroxybenzoic Acid Scaffold. Eur. J. Med. Chem. 2012, 50, 433–440. [Google Scholar] [CrossRef]
- Mondal, S.; Mandal, S.M.; Mondal, T.K.; Sinha, C. Structural Characterization of New Schiff Bases of Sulfamethoxazole and Sulfathiazole, Their Antibacterial Activity and Docking Computation with DHPS Protein Structure. Spectrochim. Acta—Part A Mol. Biomol. Spectrosc. 2015, 150, 268–279. [Google Scholar] [CrossRef]
- Krátký, M.; Konečná, K.; Šimková, A.; Janďourek, O.; Maixnerová, J.; Stolaříková, J.; Vejsová, M.; Voxová, B.; Trejtnar, F.; Vinšová, J. Improving the Antimicrobial Activity of Old Antibacterial Drug Mafenide: Schiff Bases and Their Bioactivity Targeting Resistant Pathogens. Future Med. Chem. 2023, 15, 255–274. [Google Scholar] [CrossRef]
- Sarikaya, B.; Ceruso, M.; Carta, F.; Supuran, C.T. Inhibition of Carbonic Anhydrase Isoforms I, II, IX and XII with Novel Schiff Bases: Identification of Selective Inhibitors for the Tumor-Associated Isoforms over the Cytosolic Ones. Bioorganic Med. Chem. 2014, 22, 5883–5890. [Google Scholar] [CrossRef] [PubMed]
- Durgun, M.; Turkmen, H.; Ceruso, M.; Supuran, C.T. Synthesis of 4-Sulfamoylphenyl-Benzylamine Derivatives with Inhibitory Activity against Human Carbonic Anhydrase Isoforms I, II, IX and XII. Bioorg. Med. Chem. 2016, 24, 982–988. [Google Scholar] [CrossRef]
- Koyuncu, I.; Tülüce, Y.; Slahaddin Qadir, H.; Durgun, M.; Supuran, C.T. Evaluation of the Anticancer Potential of a Sulphonamide Carbonic Anhydrase IX Inhibitor on Cervical Cancer Cells. J. Enzyme Inhib. Med. Chem. 2019, 34, 703–711. [Google Scholar] [CrossRef]
- Taslimi, P.; Işık, M.; Türkan, F.; Durgun, M.; Türkeş, C.; Gülçin, İ.; Beydemir, Ş. Benzenesulfonamide Derivatives as Potent Acetylcholinesterase, α-Glycosidase, and Glutathione S-Transferase Inhibitors: Biological Evaluation and Molecular Docking Studies. J. Biomol. Struct. Dyn. 2020, 39, 5449–5460. [Google Scholar] [CrossRef]
- Işık, M.; Demir, Y.; Durgun, M.; Türkeş, C.; Necip, A.; Beydemir, Ş. Molecular Docking and Investigation of 4-(Benzylideneamino)- and 4-(Benzylamino)-Benzenesulfonamide Derivatives as Potent AChE Inhibitors. Chem. Pap. 2020, 74, 1395–1405. [Google Scholar] [CrossRef]
- Hamad, A.; Khan, M.A.; Rahman, K.M.; Ahmad, I.; Ul-Haq, Z.; Khan, S.; Shafiq, Z. Development of Sulfonamide-Based Schiff Bases Targeting Urease Inhibition: Synthesis, Characterization, Inhibitory Activity Assessment, Molecular Docking and ADME Studies. Bioorg. Chem. 2020, 102, 104057. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The Role of Carbonic Anhydrase IX Overexpression in Kidney Cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.; Sly, W.S. Carbonic Anhydrase XII Functions in Health and Disease. Gene 2017, 623, 33–40. [Google Scholar] [CrossRef]
- Krungkrai, J.; Supuran, C.T. The Alpha-Carbonic Anhydrase from the Malaria Parasite and Its Inhibition. Curr. Pharm. Des. 2008, 14, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Maji, M.; Acharya, S.; Bhattacharya, I.; Gupta, A.; Mukherjee, A. Effect of an Imidazole-Containing Schiff Base of an Aromatic Sulfonamide on the Cytotoxic Efficacy of N,N-Coordinated Half-Sandwich Ruthenium(II) p-Cymene Complexes. Inorg. Chem. 2021, 60, 4744–4754. [Google Scholar] [CrossRef]
- Arshad, J.Z.; Tabassum, S.; Kiani, M.S.; Arshad, S.; Hashmi, M.A.; Majeed, I.; Ali, H.; Shah, S.S.A. Anticancer Properties of Ru and Os Half-Sandwich Complexes of N,S Bidentate Schiff Base Ligands Derived from Phenylthiocarbamide. Chem. Asian J. 2023, 18, e202300804. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, I.; Temiz, E.; Durgun, M.; Kocyigit, A.; Yuksekdag, O.; Supuran, C.T. Intracellular PH-Mediated Induction of Apoptosis in HeLa Cells by a Sulfonamide Carbonic Anhydrase Inhibitor. Int. J. Biol. Macromol. 2022, 201, 37–46. [Google Scholar] [CrossRef]
- Elie, J.; Vercouillie, J.; Arlicot, N.; Lemaire, L.; Bidault, R.; Bodard, S.; Hosselet, C.; Deloye, J.B.; Chalon, S.; Emond, P.; et al. Design of Selective COX-2 Inhibitors in the (Aza)Indazole Series. Chemistry, In Vitro Studies, Radiochemistry and Evaluations in Rats of a [18F] PET Tracer. J. Enzyme Inhib. Med. Chem. 2019, 34, 1–7. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Achari, A.; Somers, D.O.; Champness, J.N.; Bryant, P.K.; Rosemond, J.; Stammers, D.K. Crystal Structure of the Anti-Bacterial Sulfonamide Drug Target Dihydropteroate Synthase. Nat. Struct. Biol. 1997, 4, 490–497. [Google Scholar] [CrossRef]
- Hampele, I.C.; D’Arcy, A.; Dale, G.E.; Kostrewa, D.; Nielsen, J.; Oefner, C.; Page, M.G.P.; Schönfeld, H.-J.; Stüber, D.; Then, R.L. Structure and Function of the Dihydropteroate Synthase from Staphylococcus Aureus11Edited by R. Huber. J. Mol. Biol. 1997, 268, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Sköld, O. Sulfonamide Resistance: Mechanisms and Trends. Drug Resist. Updat. 2000, 3, 155–160. [Google Scholar] [CrossRef]
- Poey, M.E.; Azpiroz, M.F.; Laviña, M. On Sulfonamide Resistance, Sul Genes, Class 1 Integrons and Their Horizontal Transfer in Escherichia Coli. Microb. Pathog. 2019, 135, 103611. [Google Scholar] [CrossRef]
- Razavi, M.; Marathe, N.P.; Gillings, M.R.; Flach, C.F.; Kristiansson, E.; Joakim Larsson, D.G. Discovery of the Fourth Mobile Sulfonamide Resistance Gene. Microbiome 2017, 5, 160. [Google Scholar] [CrossRef]
- Poole, C. Thin-Layer Chromatography. In Clarke’s Analysis of Drugs and Poisons; Moffat, A.C., Osselton, M.D., Widdop, B., Watts, J., Eds.; Pharmaceutical Press: London, UK, 2011; pp. 611–634. ISBN 978 0 85369 711 4. [Google Scholar]
- Avram, S.; Udrea, A.M.; Nuta, D.C.; Limban, C.; Balea, A.C.; Caproiu, M.T.; Dumitrascu, F.; Buiu, C.; Bordei, A.T. Synthesis and Bioinformatic Characterization of New Schiff Bases with Possible Applicability in Brain Disorders. Molecules 2021, 26, 4160. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Movileanu, L.; Mihailescu, D.; Flonta, M.-L. Comparative Study of Some Energetic and Steric Parameters of the Wild Type and Mutants HIV-1 Protease: A Way to Explain the Viral Resistance. J. Cell. Mol. Med. 2002, 6, 251–260. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on Drug Classification and Target Prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- QIAGEN Aarhus A/S. CLC Drug Discovery Workbench, Version 2.4; Silkeborgvej 2 Prismet; QIAGEN Aarhus A/S: Aarhus, Denmark, 2015. [Google Scholar]
- Molegro Virtual Docker, Version 2019; Molexus IVS, Rørt: Odder, Denmark. 2019. Available online: http://www.molexus.io (accessed on 3 January 2024).
- SPARTAN’20, Version 2022; Wavefunction, Inc.: Irvine, CA, USA, 2022; Available online: www.wavefun.com (accessed on 3 January 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ν (NH2) (cm−1) | ν (NH2-SO2) (cm−1) | ν (C=N) (cm−1) | ν (Aromatic Ring) (cm−1) | ν (-SO2-N) (cm−1) |
|---|---|---|---|---|---|
| Sulfanilamide | 3476 3373 | 3265 | - | 1627 1594 1504 | 1311 1144 |
| 1a | - | 3357 3263 | 1626 | 1570 1481 | 1335 1153 |
| 1b | - | 3293 | 1614 | 1580 1486 | 1338 1156 |
| 1c | - | 3313 | 1644 | 1587 1561 1488 | 1339 1152 |
| 1d | - | 3312 | 1621 | 1588 1564 1511 | 1336 1153 |
| 1e | - | 3325 3245 | 1619 | 1551 | 1330 1155 |
| Compound | 1a | 1b | 1c | 1d | 1e | |
|---|---|---|---|---|---|---|
| Parameter | ||||||
| Lipinski #violations | 0 | 0 | 0 | 0 | 0 | |
| Ghose #violations | 0 | 0 | 0 | 0 | 0 | |
| Veber #violations | 0 | 0 | 0 | 0 | 0 | |
| Egan #violations | 0 | 0 | 0 | 0 | 0 | |
| Muegge #violations | 0 | 0 | 0 | 0 | 0 | |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | |
| Lead-likeness #violations | 1 Violations: MW > 350 (1a, 1e) | 0 | 0 | 0 | 2 Violations: MW > 350, XLOGP3 > 3.5 (1e) | |
| Lipophilicity (XLOGP3) | 3.32 | 2.48 | 3.04 | 3.04 | 3.67 | |
| Compound | 1a | 1b | 1c | 1d | 1e | |
|---|---|---|---|---|---|---|
| Parameter | ||||||
| Human intestinal absorption | Yes | Yes | Yes | Yes | Yes | |
| Plasma protein binding | 0.83 | 1.04 | 0.95 | 0.92 | 1.09 | |
| BBB permeability (logBBB) | −0.66 | −0.55 | −0.72 | −0.72 | −0.90 | |
| CNS permeability (logPS) | −2.16 | −2.16 | −2.06 | −2.06 | −1.94 | |
| OCT2 inhibitor | No | No | No | No | No | |
| OCT1 inhibitor | No | No | No | No | No | |
| OATP1B1 inhibitor | Yes | Yes | Yes | Yes | Yes | |
| OATP1B3 inhibitor | Yes | Yes | Yes | Yes | Yes | |
| CYP1A2 inhibitor | Yes | Yes | Yes | Yes | Yes | |
| CYP2C19 inhibitor | Yes | No | No | No | No | |
| CYP2C9 inhibitor/substrate | Yes/No | No/No | No/No | No/No | No/No | |
| CYP2D6 inhibitor/substrate | No/No | No/No | No/No | No/No | No/No | |
| CYP3A4 inhibitor/substrate | Yes/No | Yes/No | Yes/No | Yes/No | Yes/No | |
| Compound | 1a | 1b | 1c | 1d | 1e | |
|---|---|---|---|---|---|---|
| Property | ||||||
| Human Toxicity | ||||||
| Ames mutagenesis | Yes | No | No | No | No | |
| Androgen receptor binding | Yes | No | No | No | No | |
| Aromatase binding | Yes | Yes | Yes | Yes | Yes | |
| Estrogen receptor binding | Yes | Yes | Yes | Yes | Yes | |
| Glucocorticoid receptor binding | Yes | Yes | Yes | No | Yes | |
| Human ether-a-go-go-related gene inhibition | Yes | No | No | No | No | |
| Carcinogenicity (binary) | Yes | No | No | No | No | |
| Mitochondrial toxicity | Yes | Yes | Yes | Yes | Yes | |
| Hepatotoxicity | Yes | Yes | Yes | Yes | Yes | |
| Nephrotoxicity | Yes | No | Yes | No | Yes | |
| Reproductive toxicity | No | No | No | No | No | |
| Respiratory toxicity | No | Yes | Yes | Yes | Yes | |
| Eye corrosion | No | No | No | No | No | |
| Eye irritation | No | No | No | No | No | |
| Other species toxicity | ||||||
| Honey bee toxicity | No | No | No | No | No | |
| Crustacea aquatic toxicity | Yes | No | Yes | Yes | Yes | |
| Fish aquatic toxicity | Yes | Yes | Yes | Yes | Yes | |
| Compounds | Human Targets | Probability (%) | Model Accuracy |
|---|---|---|---|
| 1a | Endoplasmic reticulum-associated amyloid beta-peptide-binding protein | 100 | 70 |
| Transcription intermediary factor 1-alpha | 98 | 96 | |
| Cyclooxygenase-2 | 98 | 90 | |
| 1b | Endoplasmic reticulum-associated amyloid beta-peptide-binding protein | 99.6 | 70 |
| Cyclooxygenase-2 | 98 | 90 | |
| Transcription intermediary factor 1-alpha | 98 | 96 | |
| 1c | Endoplasmic reticulum-associated amyloid beta-peptide-binding protein | 99 | 70 |
| Carbonic anhydrase XII | 99 | 97 | |
| Carbonic anhydrase IX | 99 | 99 | |
| Cyclin-dependent kinase 1 | 98 | 92 | |
| Cyclooxygenase-2 | 98 | 90 | |
| 1d | Endoplasmic reticulum-associated amyloid beta-peptide-binding protein | 99.1 | 70 |
| Carbonic anhydrase XII | 99 | 97 | |
| Carbonic anhydrase IX | 98 | 99 | |
| 1e | Carbonic anhydrase XII | 99 | 97 |
| Cyclooxygenase-2 | 99 | 90 | |
| Carbonic anhydrase IX | 99 | 99 | |
| Dual specificity protein kinase CLK4 | 98 | 94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coanda, M.; Limban, C.; Draghici, C.; Ciobanu, A.-M.; Grigore, G.A.; Popa, M.; Stan, M.; Larion, C.; Avram, S.; Mares, C.; et al. Current Perspectives on Biological Screening of Newly Synthetised Sulfanilamide Schiff Bases as Promising Antibacterial and Antibiofilm Agents. Pharmaceuticals 2024, 17, 405. https://doi.org/10.3390/ph17040405
Coanda M, Limban C, Draghici C, Ciobanu A-M, Grigore GA, Popa M, Stan M, Larion C, Avram S, Mares C, et al. Current Perspectives on Biological Screening of Newly Synthetised Sulfanilamide Schiff Bases as Promising Antibacterial and Antibiofilm Agents. Pharmaceuticals. 2024; 17(4):405. https://doi.org/10.3390/ph17040405
Chicago/Turabian StyleCoanda, Maria, Carmen Limban, Constantin Draghici, Anne-Marie Ciobanu, Georgiana Alexandra Grigore, Marcela Popa, Miruna Stan, Cristina Larion, Speranta Avram, Catalina Mares, and et al. 2024. "Current Perspectives on Biological Screening of Newly Synthetised Sulfanilamide Schiff Bases as Promising Antibacterial and Antibiofilm Agents" Pharmaceuticals 17, no. 4: 405. https://doi.org/10.3390/ph17040405
APA StyleCoanda, M., Limban, C., Draghici, C., Ciobanu, A.-M., Grigore, G. A., Popa, M., Stan, M., Larion, C., Avram, S., Mares, C., Ciornei, M.-C., Dabu, A., Hudita, A., Galateanu, B., Pintilie, L., & Nuta, D. C. (2024). Current Perspectives on Biological Screening of Newly Synthetised Sulfanilamide Schiff Bases as Promising Antibacterial and Antibiofilm Agents. Pharmaceuticals, 17(4), 405. https://doi.org/10.3390/ph17040405