
Optimizing Paclitaxel Oral Absorption and Bioavailability: TPGS Co-Coating via Supercritical Anti-Solvent Fluidized Bed Technology

 and
and
Abstract
1. Introduction
2. Results and Discussion
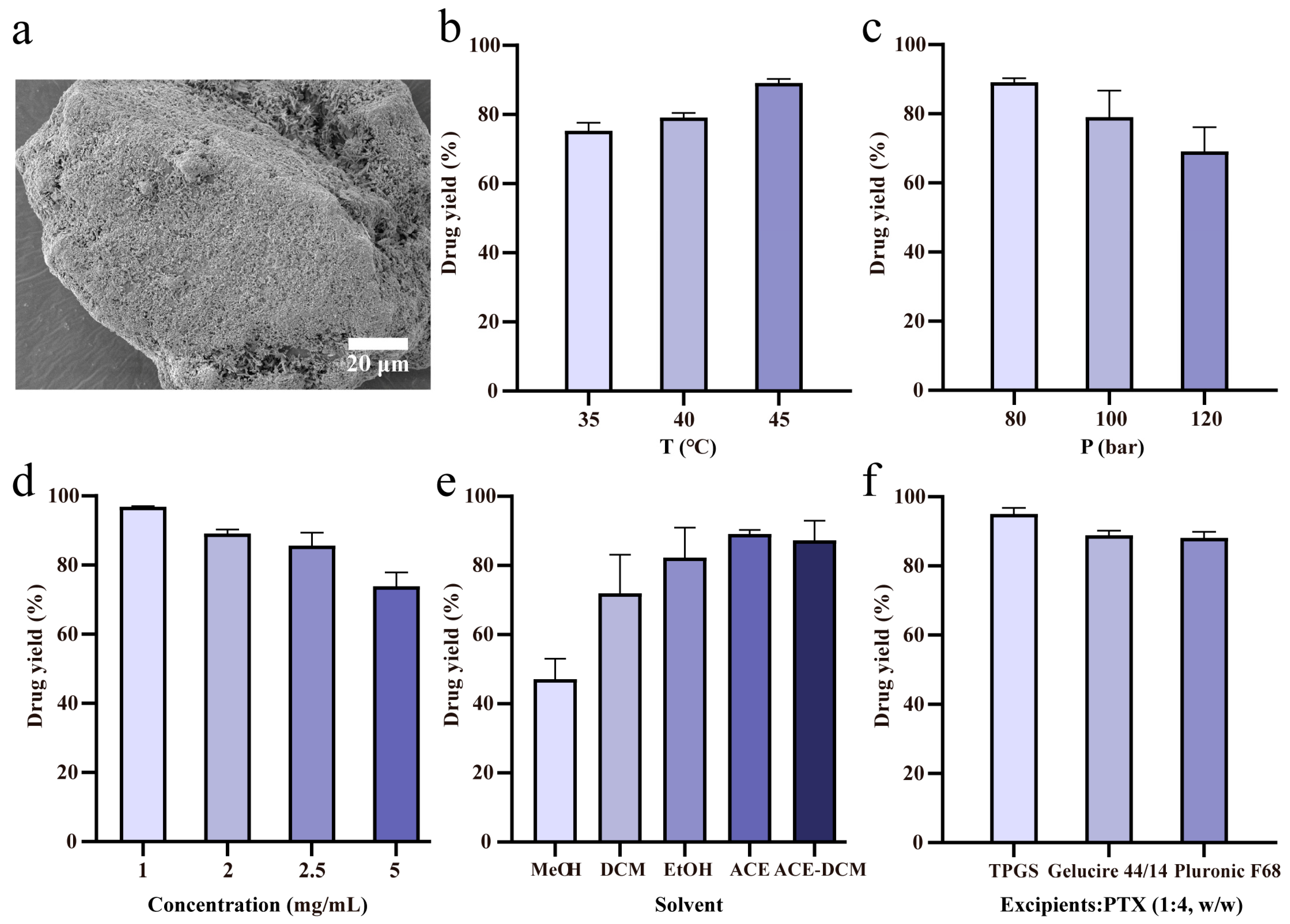
2.1. Drug Loading of SAS-FB Products
2.2. In Vitro Dissolution
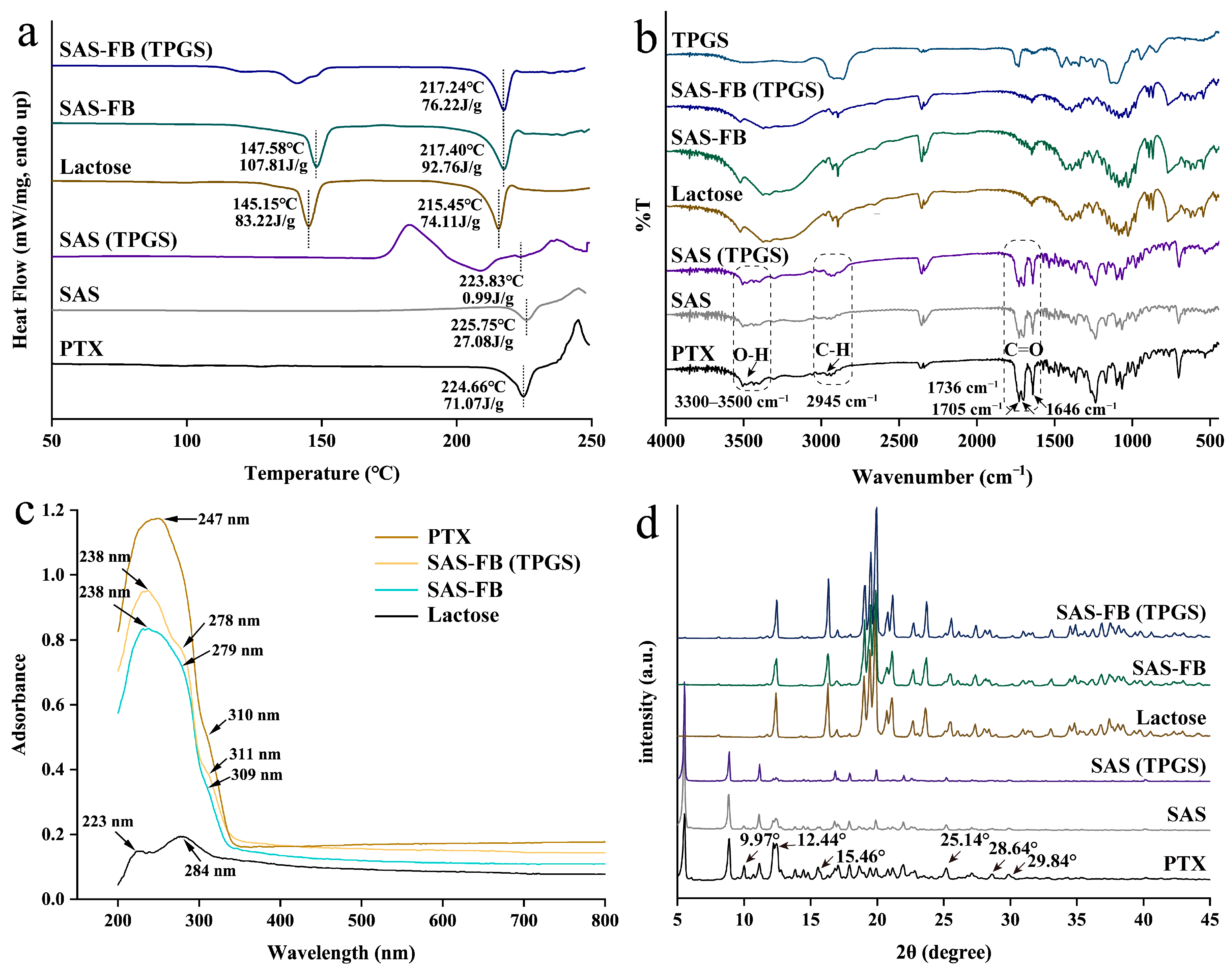
2.3. Particle Characterization
2.4. Cytotoxicity Studies
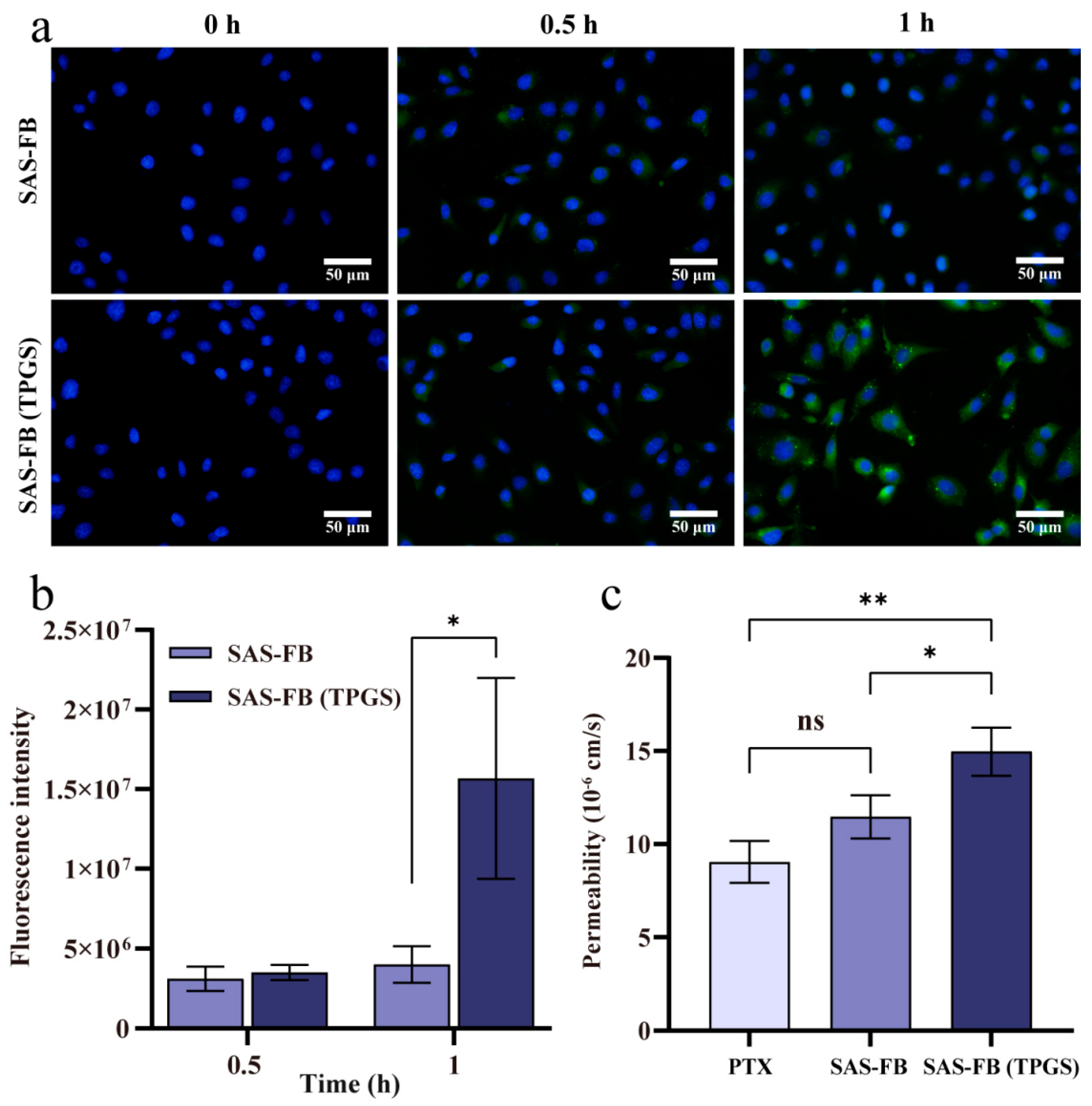
2.5. Cellular Uptake Studies
2.6. Transport Assay
2.7. In Vivo Oral Bioavailability
3. Materials and Methods
3.1. Materials
3.2. SAS-FB Coating Procedure and Optimization
3.3. Quantitative Determination of PTX Yield
3.4. In Vitro Dissolution
3.5. Characterizing the Physicochemical Properties of the Samples
3.5.1. Scanning Electron Microscopy (SEM)
3.5.2. Fluorescence Microscopy (FM)
3.5.3. Differential Scanning Calorimeter (DSC)
3.5.4. Fourier Transform Infrared Spectroscopy (FT-IR)
3.5.5. Diffuse Reflectance Spectroscopy (DRS)
3.5.6. X-ray Powder Diffraction (XRPD)
3.5.7. Stability Test
3.6. Cytotoxicity
3.7. Cellular Uptake of PTX from SAS-FB and SAS-FB (TPGS)
3.8. In Vitro Transport Studies
3.9. In Vivo Oral Bioavailability and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Abu, S.T.; Samec, M.; Liskova, A.; Kubatka, P.; Busselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar]
- Khan, K.; Quispe, C.; Javed, Z.; Iqbal, M.J.; Sadia, H.; Raza, S.; Irshad, A.; Salehi, B.; Reiner, Z.; Sharifi-Rad, J. Resveratrol, curcumin, paclitaxel and miRNAs mediated regulation of PI3K/Akt/mTOR pathway: Go four better to treat bladder cancer. Cancer Cell Int. 2020, 20, 560. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, L.; Yao, X.; Lou, X.; Wan, J.; Duan, X.; Pan, L.; Li, A.; Gu, Z.; Wang, M.; et al. Paclitaxel treatment enhances lymphatic metastasis of B16F10 melanoma cells via CCL21/CCR7 axis. Int. J. Biol. Sci. 2022, 18, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Ozguroglu, M.; Bang, Y.J.; Di Bartolomeo, M.; Mandala, M.; Ryu, M.H.; Fornaro, L.; Olesinski, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Jang, S.K.; Kim, Y.J.; Jin, H.O.; Bae, S.; Hong, J.; Park, I.C.; Lee, J.H. Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 8761. [Google Scholar] [CrossRef]
- Villaruz, L.C.; Socinski, M.A. Is there a role of nab-paclitaxel in the treatment of advanced non-small cell lung cancer? The data suggest yes. Eur. J. Cancer 2016, 56, 162–171. [Google Scholar] [CrossRef]
- Choi, J.S.; Cho, N.H.; Kim, D.H.; Park, J.S. Comparison of paclitaxel solid dispersion and polymeric micelles for improved oral bioavailability and in vitro anti-cancer effects. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 100, 247–259. [Google Scholar] [CrossRef]
- Thanki, K.; Gangwal, R.P.; Sangamwar, A.T.; Jain, S. Oral delivery of anticancer drugs: Challenges and opportunities. J. Control. Release 2013, 170, 15–40. [Google Scholar] [CrossRef]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Wang, F.; Wang, H.; Sun, J. Paclitaxel nanosuspension coated with P-gp inhibitory surfactants: II. Ability to reverse the drug-resistance of H460 human lung cancer cells. Colloids Surf. B Biointerfaces 2014, 117, 122–127. [Google Scholar] [CrossRef]
- Singh, S.; Dash, A.K. Paclitaxel in cancer treatment: Perspectives and prospects of its delivery challenges. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 333–372. [Google Scholar] [CrossRef]
- Mo, R.; Jin, X.; Li, N.; Ju, C.; Sun, M.; Zhang, C.; Ping, Q. The mechanism of enhancement on oral absorption of paclitaxel by N-octyl-O-sulfate chitosan micelles. Biomaterials 2011, 32, 4609–4620. [Google Scholar] [CrossRef]
- Choi, J.S.; Shin, S.C. Enhanced paclitaxel bioavailability after oral coadministration of paclitaxel prodrug with naringin to rats. Int. J. Pharm. 2005, 292, 149–156. [Google Scholar] [CrossRef]
- Ahadian, S.; Finbloom, J.A.; Mofidfar, M.; Diltemiz, S.E.; Nasrollahi, F.; Davoodi, E.; Hosseini, V.; Mylonaki, I.; Sangabathuni, S.; Montazerian, H.; et al. Micro and nanoscale technologies in oral drug delivery. Adv. Drug Deliv. Rev. 2020, 157, 37–62. [Google Scholar] [CrossRef]
- Zhang, T.; Luo, J.; Fu, Y.; Li, H.; Ding, R.; Gong, T.; Zhang, Z. Novel oral administrated paclitaxel micelles with enhanced bioavailability and antitumor efficacy for resistant breast cancer. Colloids Surf. B Biointerfaces 2017, 150, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Tu, L.; Wang, G.; Qi, N.; Wu, W.; Zhang, W.; Feng, J. Multi-functional chitosan polymeric micelles as oral paclitaxel delivery systems for enhanced bioavailability and anti-tumor efficacy. Int. J. Pharm. 2020, 578, 119105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, T.; Wei, S.; Zhou, C.; Lan, Y.; Cao, A.; Yang, J.; Wang, W. Mucus adhesion- and penetration-enhanced liposomes for paclitaxel oral delivery. Int. J. Pharm. 2018, 537, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Godara, S.; Lather, V.; Kirthanashri, S.V.; Awasthi, R.; Pandita, D. Lipid-PLGA hybrid nanoparticles of paclitaxel: Preparation, characterization, in vitro and in vivo evaluation. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 109, 110576. [Google Scholar] [CrossRef]
- Du, X.; Yin, S.; Xu, L.; Ma, J.; Yu, H.; Wang, G.; Li, J. Polylysine and cysteine functionalized chitosan nanoparticle as an efficient platform for oral delivery of paclitaxel. Carbohydr. Polym. 2020, 229, 115484. [Google Scholar] [CrossRef]
- Ding, D.; Sun, B.; Cui, W.; Chen, Q.; Zhang, X.; Zhang, H.; He, Z.; Sun, J.; Luo, C. Integration of phospholipid-drug complex into self-nanoemulsifying drug delivery system to facilitate oral delivery of paclitaxel. Asian J. Pharm. Sci. 2019, 14, 552–558. [Google Scholar] [CrossRef]
- Djerafi, R.; Swanepoel, A.; Crampon, C.; Kalombo, L.; Labuschagne, P.; Badens, E.; Masmoudi, Y. Supercritical antisolvent co-precipitation of rifampicin and ethyl cellulose. Eur. J. Pharm. Sci. 2017, 102, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.T.Y.; Ng, W.K.; Tan, R.B.H. Amorphization of pharmaceutical compound by co-precipitation using supercritical anti-solvent (SAS) process (Part I). J. Supercrit. Fluids 2010, 53, 179–184. [Google Scholar] [CrossRef]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Saad, S.; Ahmad, I.; Kawish, S.M.; Khan, U.A.; Ahmad, F.J.; Ali, A.; Jain, G.K. Improved cardioprotective effects of hesperidin solid lipid nanoparticles prepared by supercritical antisolvent technology. Colloids Surf. B Biointerfaces 2020, 187, 110628. [Google Scholar] [CrossRef]
- Duarte, A.R.; Gordillo, M.D.; Cardoso, M.M.; Simplicio, A.L.; Duarte, C.M. Preparation of ethyl cellulose/methyl cellulose blends by supercritical antisolvent precipitation. Int. J. Pharm. 2006, 311, 50–54. [Google Scholar] [CrossRef]
- Chen, T.; Liu, L.; Zhang, L.; Lu, T.; Matos, R.L.; Jiang, C.; Lin, Y.; Yuan, T.; Ma, Z.; He, H.; et al. Optimization of the supercritical fluidized bed process for sirolimus coating and drug release. Int. J. Pharm. 2020, 589, 119809. [Google Scholar] [CrossRef]
- He, H.; Huang, Y.; Zhang, X.; Ouyang, Y.; Pan, P.; Lan, Y.; Zhong, Z.; Ping, L.; Lu, T.; Chen, Z.; et al. Supercritical fluid coating of flavonoids on excipients enhances drug release and antioxidant activity. Int. J. Pharm. 2023, 632, 122593. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, X.; Ping, L.; Zhong, Z.; Zhang, X.; Zhuang, X.; Wang, G.; Guo, Q.; Zhan, S.; Qiu, Z.; et al. Supercritical antisolvent-fluidized bed for the preparation of dry powder inhaler for pulmonary delivery of nanomedicine. Int. J. Pharm. 2023, 648, 123580. [Google Scholar] [CrossRef]
- Matos, R.L.; Lu, T.; McConville, C.; Leeke, G.; Ingram, A. Analysis of curcumin precipitation and coating on lactose by the integrated supercritical antisolvent-fluidized bed process. J. Supercrit. Fluids 2018, 141, 143–156. [Google Scholar] [CrossRef]
- Chen, T.; Ma, Z.; Qiu, Z.; Zhong, Z.; Xing, L.; Guo, Q.; Luo, D.; Weng, Z.; Ge, F.; Huang, Y.; et al. Characterization of excipients to improve pharmaceutical properties of sirolimus in the supercritical anti-solvent fluidized process. Int. J. Pharm. 2022, 611, 121240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zhang, H.; Yang, S.; He, W.; Luan, Y. Redox-sensitive mPEG-SS-PTX/TPGS mixed micelles: An efficient drug delivery system for overcoming multidrug resistance. Int. J. Pharm. 2016, 515, 281–292. [Google Scholar] [CrossRef]
- Varma, M.V.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Deng, J.; Jia, X.; Zhou, J.; Lv, H. Solid dispersion of berberine-phospholipid complex/TPGS 1000/SiO2: Preparation, characterization and in vivo studies. Int. J. Pharm. 2014, 465, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, M.R.; Kumari, L.; Patel, K.K.; Vuddanda, P.R.; Vajanthri, K.Y.; Mahto, S.K.; Singh, S. Intravenous administration of trans–resveratrol–loaded TPGS–coated solid lipid nanoparticles for prolonged systemic circulation, passive brain targeting and improved in vitro cytotoxicity against C6 glioma cell lines. RSC Adv. 2016, 6, 50336–50348. [Google Scholar] [CrossRef]
- Dintaman, J.M.; Silverman, J.A. Inhibition of P-glycoprotein by D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm. Res. 1999, 16, 1550–1556. [Google Scholar] [CrossRef]
- Yan, H.; Du, X.; Wang, R.; Zhai, G. Progress in the study of D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) reversing multidrug resistance. Colloids Surf. B Biointerfaces 2021, 205, 111914. [Google Scholar] [CrossRef]
- Sun, J.; Bao, H.; Peng, Y.; Zhang, H.; Sun, Y.; Qi, J.; Zhang, H.; Gao, Y. Improvement of intestinal transport, absorption and anti-diabetic efficacy of berberine by using Gelucire44/14: In vitro, in situ and in vivo studies. Int. J. Pharm. 2018, 544, 46–54. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Duong, V.A.; Maeng, H.J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Vandana, V.; Teja, A.S. The solubility of paclitaxel in supercritical CO2 and N2O. Fluid Phase Equilibria 1997, 135, 83–87. [Google Scholar] [CrossRef]
- Liang, N.; Sun, S.; Li, X.; Piao, H.; Piao, H.; Cui, F.; Fang, L. α-Tocopherol succinate-modified chitosan as a micellar delivery system for paclitaxel: Preparation, characterization and in vitro/in vivo evaluations. Int. J. Pharm. 2012, 423, 480–488. [Google Scholar] [CrossRef]
- Park, J.H.; Yan, Y.D.; Chi, S.C.; Hwang, D.H.; Shanmugam, S.; Lyoo, W.S.; Woo, J.S.; Yong, C.S.; Choi, H.G. Preparation and evaluation of Cremophor-free paclitaxel solid dispersion by a supercritical antisolvent process. J. Pharm. Pharmacol. 2011, 63, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.A.; Huang, X.Y.; Jabeen, A.; Ahsan, A.; Seidu, T.A.; Kutoka, P.T.; Wang, B. Enhanced cellular uptake and cytotoxicity of vorinostat through encapsulation in TPGS-modified liposomes. Colloids Surf. B Biointerfaces 2021, 199, 111523. [Google Scholar] [CrossRef]
- Ebeid, K.; Meng, X.; Thiel, K.W.; Do, A.V.; Geary, S.M.; Morris, A.S.; Pham, E.L.; Wongrakpanich, A.; Chhonker, Y.S.; Murry, D.J.; et al. Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat. Nanotechnol. 2018, 13, 72–81. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, X.; Wang, H.; Fan, R.; Hu, Y.; Hu, X.; Li, J. Dasatinib self-assembled nanoparticles decorated with hyaluronic acid for targeted treatment of tumors to overcome multidrug resistance. Drug Deliv. 2021, 28, 670–679. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. No. | T °C | p Bar | Solvent | Cdrug mg/mL | Excipient/Drug (w/w) | Lactose g | Coating Efficiency % | SD % | Sample Name |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 35 | 80 | Acetone | 2 | - | 2 | 75.3 | 1.9 | - |
| 2 | 40 | 80 | Acetone | 2 | - | 2 | 79.1 | 1.0 | - |
| 3 | 45 | 80 | Acetone | 2 | - | 2 | 89.1 | 0.9 | SAS-FB (ACE) |
| 4 | 45 | 100 | Acetone | 2 | - | 2 | 79.0 | 6.2 | - |
| 5 | 45 | 120 | Acetone | 2 | - | 2 | 69.1 | 5.6 | - |
| 6 | 45 | 80 | Acetone | 2.5 | - | 2 | 85.6 | 3.0 | - |
| 7 | 45 | 80 | Acetone | 5 | - | 2 | 73.8 | 3.3 | - |
| 8 | 45 | 80 | MeOH | 2 | - | 2 | 47.2 | 4.8 | SAS-FB (MeOH) |
| 9 | 45 | 80 | DCM | 2 | - | 2 | 71.9 | 9.1 | SAS-FB (DCM) |
| 10 | 45 | 80 | EtOH | 2 | - | 2 | 82.3 | 7.0 | SAS-FB (EtOH) |
| 11 | 45 | 80 | ACE-DCM | 2 | - | 2 | 87.3 | 4.6 | SAS-FB (ACE-DCM) |
| 12 * | 45 | 80 | Acetone | 1 | - | 2 | 96.8 | 0.2 | SAS-FB |
| 13 | 45 | 80 | Acetone | 1 | TPGS (1:4) | 2 | 95.0 | 1.4 | SAS-FB (TPGS) |
| 14 | 45 | 80 | Acetone | 1 | Gelucire 44/14 (1:4) | 2 | 88.9 | 1.0 | SAS-FB (Gelucire 44/14) |
| 15 | 45 | 80 | Acetone | 1 | Pluronic F68 (1:4) | 2 | 88.1 | 1.4 | SAS-FB (Pluronic F68) |
| 16 | 45 | 80 | Acetone | 1 | Cou-6 (2:5) | 2 | 85.5 | 1.3 | SAS-FB (Cou-6) |
| 17 | 45 | 80 | Acetone | 1 | TPGS/ Cou-6 (1:4/2:5) | 2 | 83.6 | 1.7 | SAS-FB (Cou-6,TPGS) |
| 18 | 45 | 80 | Acetone | 1 | - | - | - | - | SAS |
| 19 | 45 | 80 | Acetone | 1 | TPGS (1:4) | - | - | - | SAS (TPGS) |
| 20 | 45 | 80 | Acetone | 1 | Cou-6 (2:5) | - | - | - | SAS (Cou-6) |
| 21 | 45 | 80 | Acetone | 0 | - | 2 | - | - | Lactose |
| Parameters | PTX | SAS-FB | SAS-FB (TPGS) |
|---|---|---|---|
| t1/2 (h) | 14.31 ± 2.21 | 17.02 ±5.00 | 14.26 ± 3.96 |
| Tmax (h) | 2.00 ± 2.23 | 2.00 ± 2.69 | 2.00 ± 2.34 |
| Cmax (ng/mL) | 21.01 ± 9.52 | 45.22 ± 25.98 | 99.08 ± 10.80 ** |
| AUC0→∞ (ng/mL × h) | 305.17 ± 60.16 | 456.67 ± 226.13 | 812.14 ± 336.85 ** |
| Relative bioavailability | 1 | 1.49 | 2.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, Z.; Lan, Y.; Chen, J.; Ping, L.; Li, X.; Wang, Q.; Zhuang, X.; Qiu, Z.; Yuan, T.; Guo, Q.; et al. Optimizing Paclitaxel Oral Absorption and Bioavailability: TPGS Co-Coating via Supercritical Anti-Solvent Fluidized Bed Technology. Pharmaceuticals 2024, 17, 412. https://doi.org/10.3390/ph17040412
Zhong Z, Lan Y, Chen J, Ping L, Li X, Wang Q, Zhuang X, Qiu Z, Yuan T, Guo Q, et al. Optimizing Paclitaxel Oral Absorption and Bioavailability: TPGS Co-Coating via Supercritical Anti-Solvent Fluidized Bed Technology. Pharmaceuticals. 2024; 17(4):412. https://doi.org/10.3390/ph17040412
Chicago/Turabian StyleZhong, Zicheng, Yanling Lan, Jinxing Chen, Lu Ping, Xuchun Li, Qing Wang, Xiaodong Zhuang, Zhenwen Qiu, Tianhui Yuan, Qiupin Guo, and et al. 2024. "Optimizing Paclitaxel Oral Absorption and Bioavailability: TPGS Co-Coating via Supercritical Anti-Solvent Fluidized Bed Technology" Pharmaceuticals 17, no. 4: 412. https://doi.org/10.3390/ph17040412
APA StyleZhong, Z., Lan, Y., Chen, J., Ping, L., Li, X., Wang, Q., Zhuang, X., Qiu, Z., Yuan, T., Guo, Q., Xi, L., Li, Q., & Luo, D. (2024). Optimizing Paclitaxel Oral Absorption and Bioavailability: TPGS Co-Coating via Supercritical Anti-Solvent Fluidized Bed Technology. Pharmaceuticals, 17(4), 412. https://doi.org/10.3390/ph17040412