
The Xanthine Derivative KMUP-1 Inhibits Hypoxia-Induced TRPC1 Expression and Store-Operated Ca2+ Entry in Pulmonary Arterial Smooth Muscle Cells
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
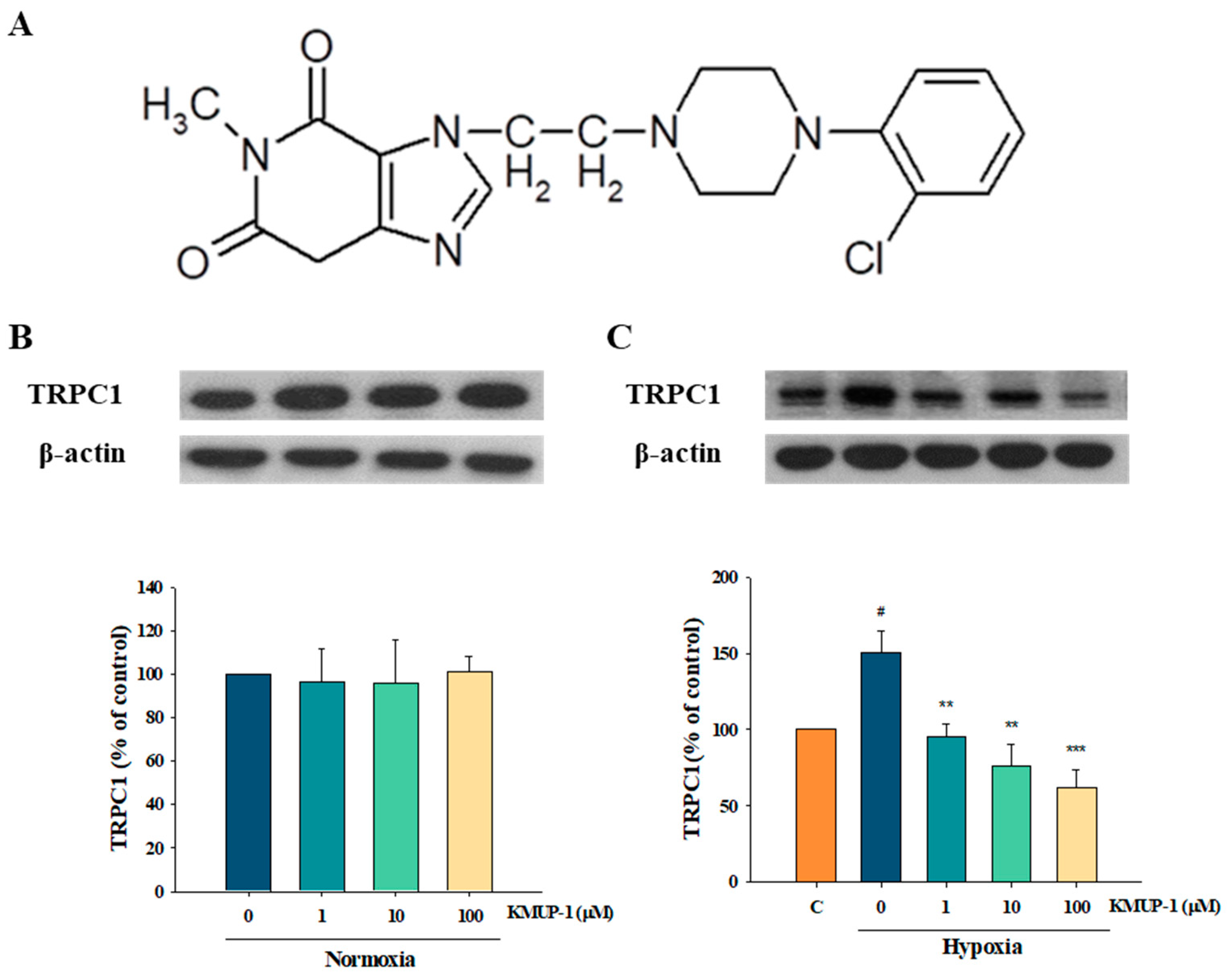
2.1. Effects of KMUP-1 in Normoxic and Hypoxic PASMCs
2.2. KMUP-1 Prevented Hypoxia-Enhanced TRPC1 Expression through cGMP/PKG Activation
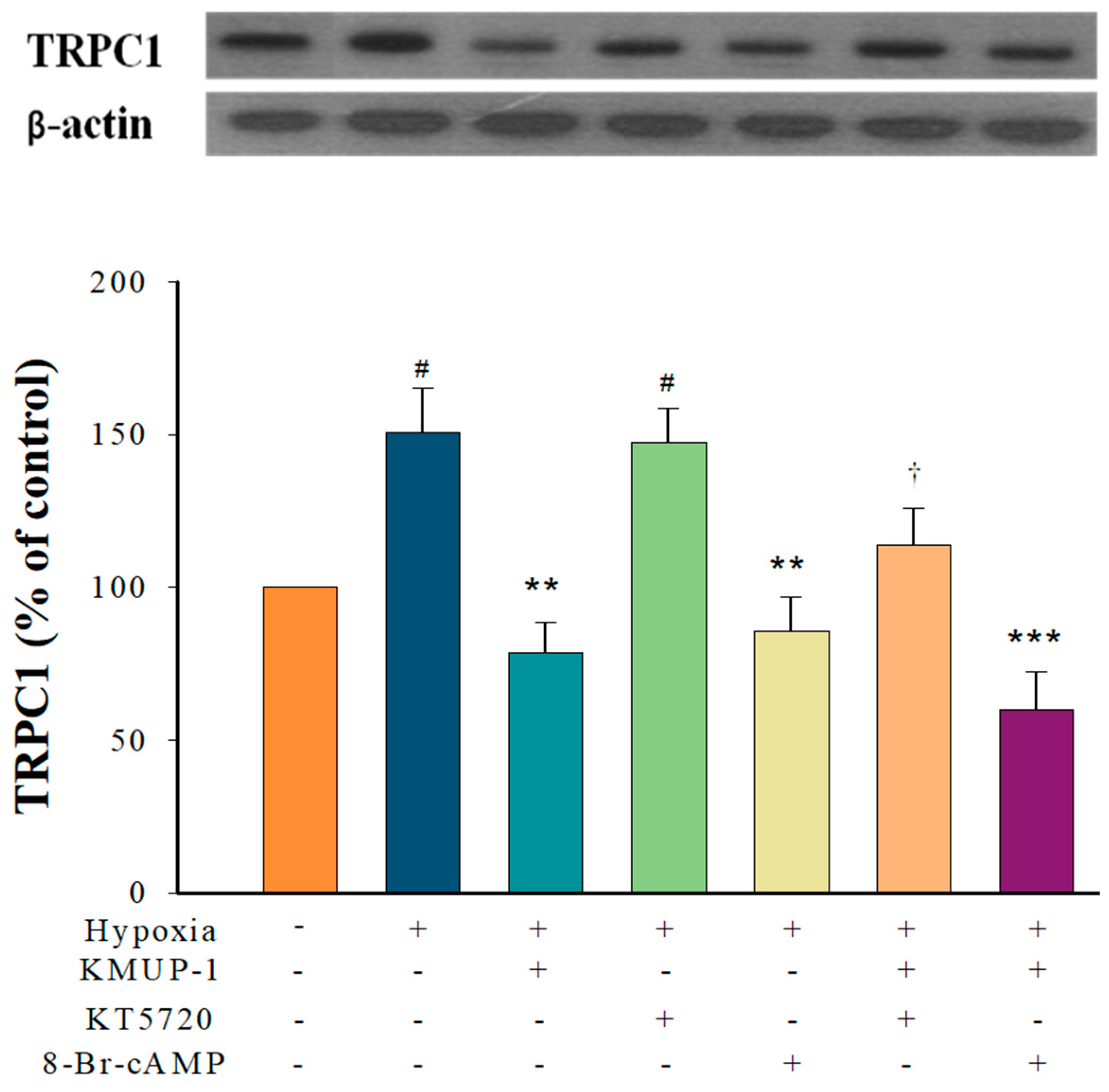
2.3. KMUP-1 Prevented Hypoxia-Enhanced TRPC1 Expression through cAMP/PKA Activation
2.4. KMUP-1 Prevented Hypoxia-Enhanced TRPC1 Expression through PKC Inhibition
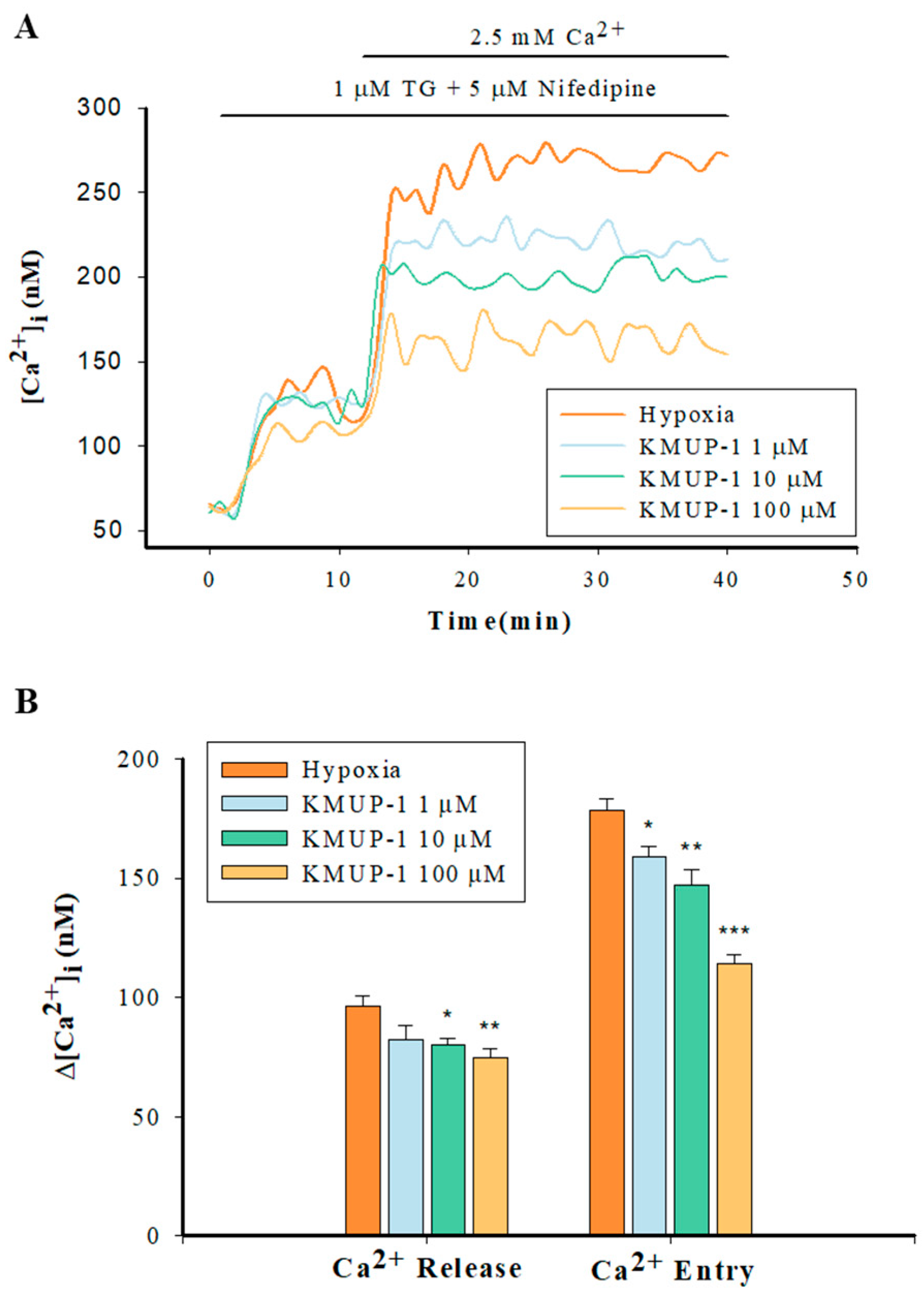
2.5. Enhanced Capacitative Ca2+ Entry (CCE) in Hypoxic PASMCs
2.6. KMUP-1 Attenuated SR Ca2+ Release and SOCs-Mediated CCE in Hypoxic PASMCs
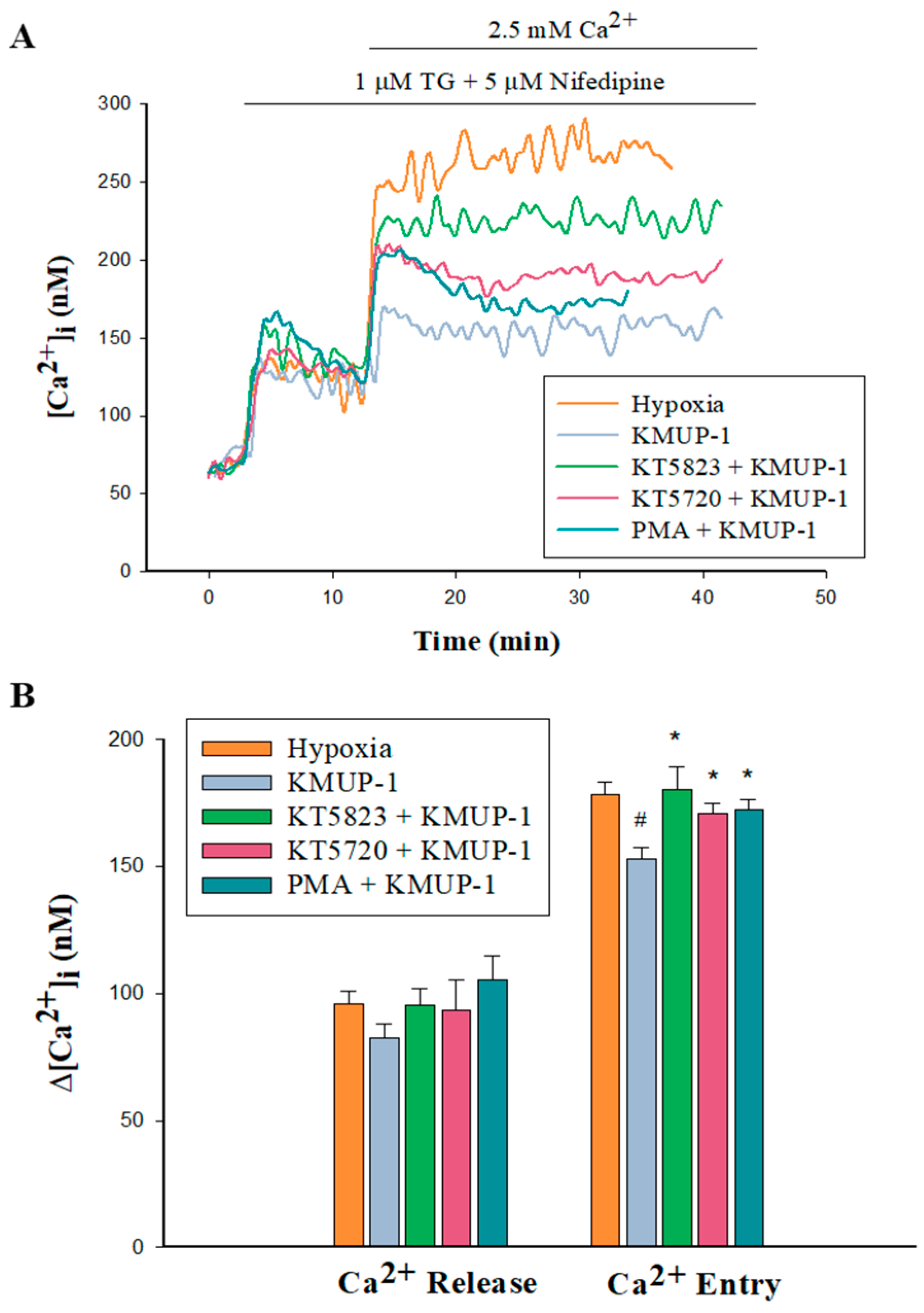
2.7. PKA/PKG/PKC Involvement in KMUP-1-Attenuated SOCs-Mediated CCE in Hypoxic PASMCs
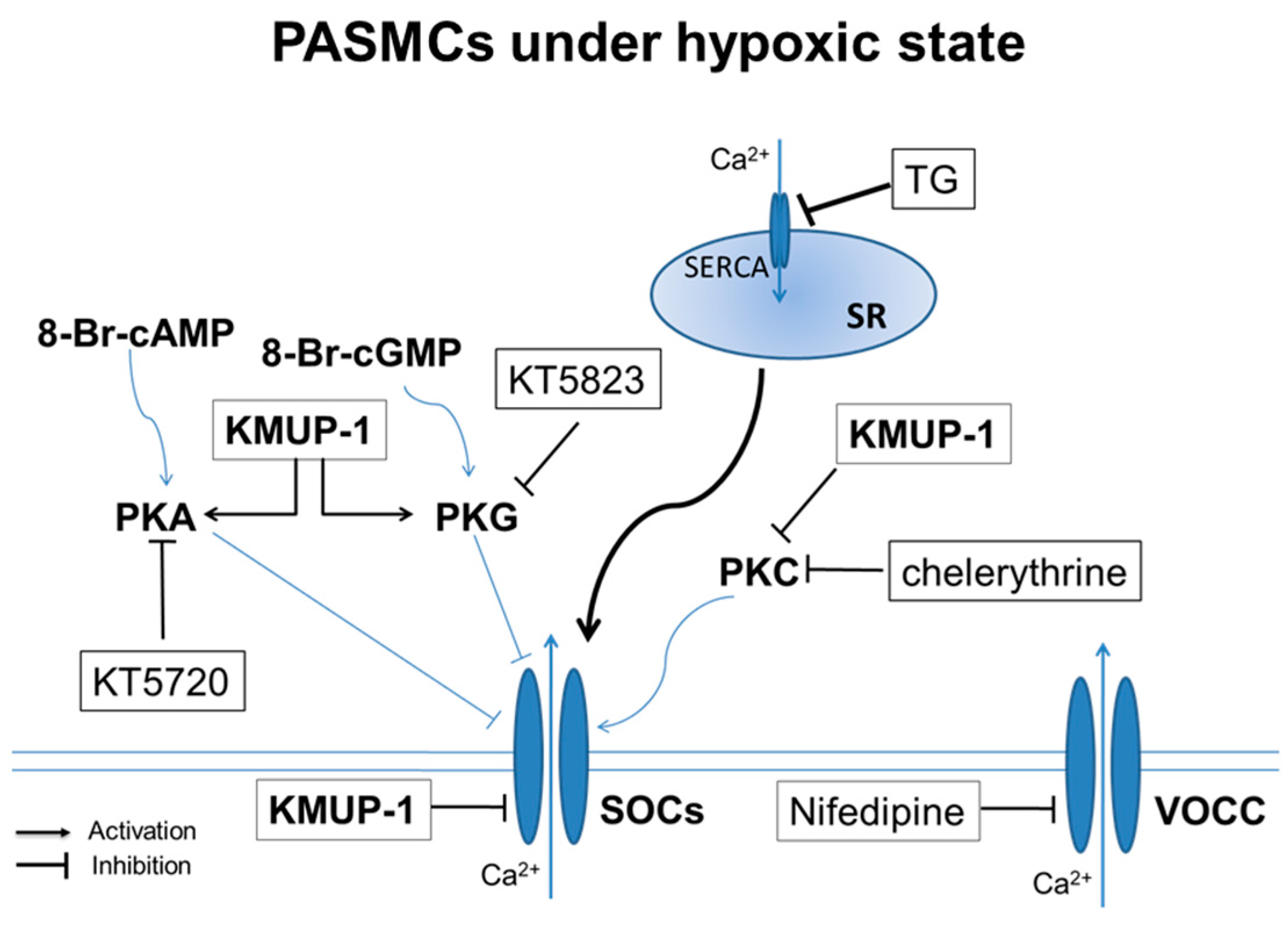
3. Discussion
4. Materials and Methods
4.1. Animal Procedures and Tissue Preparation
4.2. Primary Culture of PASMCs
4.3. Western Blot Analysis
4.4. Measurement of Capacitative Ca2+ Entry
4.5. Chemicals
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Otani, N.; Tomoe, T.; Kawabe, A.; Sugiyama, T.; Horie, Y.; Sugimura, H.; Yasu, T.; Nakamoto, T. Recent Advances in the Treatment of Pulmonary Arterial Hypertension. Pharmaceuticals 2022, 15, 1277. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Richter, M.J.; Rubin, L.J. The physiologic basis of pulmonary arterial hypertension. Eur. Respir. J. 2022, 59, 2102334. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Vonk Noordegraaf, A.; Chin, K.M.; Haddad, F.; Hassoun, P.M.; Hemnes, A.R.; Hopkins, S.R.; Kawut, S.M.; Langleben, D.; Lumens, J.; Naeije, R. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: An update. Eur. Respir. J. 2019, 53, 1801900. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Ruaro, B.; Salton, F.; Baratella, E.; Confalonieri, P.; Geri, P.; Pozzan, R.; Torregiani, C.; Bulla, R.; Confalonieri, M.; Matucci-Cerinic, M.; et al. An Overview of Different Techniques for Improving the Treatment of Pulmonary Hypertension Secondary in Systemic Sclerosis Patients. Diagnostics 2022, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Masson, B.; Montani, D.; Humbert, M.; Capuano, V.; Antigny, F. Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension. Biomolecules 2021, 11, 1781. [Google Scholar] [CrossRef]
- Smani, T.; Zakharov, S.I.; Csutora, P.; Leno, E.; Trepakova, E.S.; Bolotina, V.M. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 2004, 6, 113–120. [Google Scholar] [CrossRef]
- Landsberg, J.W.; Yuan, J.X. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol. Sci. 2004, 19, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Ran, P.; Zhang, D.; Peng, G.; Li, B.; Zhong, N.; Wang, J. Sildenafil inhibits chronically hypoxic upregulation of canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am. J. Physiol. Cell Physiol. 2010, 298, C114–C123. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.S.; Dai, Z.K.; Welsh, D.G.; Chen, I.J.; Wu, B.N. Protein kinases modulate store-operated channels in pulmonary artery smooth muscle cells. J. Biomed. Sci. 2011, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Kunichika, N.; Yu, Y.; Remillard, C.V.; Platoshyn, O.; Zhang, S.; Yuan, J.X. Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L962–L969. [Google Scholar] [CrossRef] [PubMed]
- Martín-Bórnez, M.; Galeano-Otero, I.; Del Toro, R.; Smani, T. TRPC and TRPV Channels’ Role in Vascular Remodeling and Disease. Int. J. Mol. Sci. 2020, 21, 6125. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, Y.; Su, Y.; Zhou, D.; Xu, Q. Role of store-operated Ca2+ entry in cardiovascular disease. Cell Commun. Signal. 2022, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.Y.; Wang, L.M.; Zhong, H.; Liu, Y.M.; Tang, N.; Zhu, L.P.; He, F.; Hu, Q.H. TRPC1 stimulates calcium-sensing receptor-induced store-operated Ca2+ entry and nitric oxide production in endothelial cells. Mol. Med. Rep. 2017, 16, 4613–4619. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Townsley, M.I.; Alexeyev, M.; Voelkel, N.F.; Stevens, T. Endothelial hyperpermeability in severe pulmonary arterial hypertension: Role of store-operated calcium entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L560–L569. [Google Scholar] [CrossRef]
- Miao, R.; Wan, J.; Liu, J.; Yuan, J.X.; Wang, J.; Xie, W.; Zhai, Z.; Wang, C. Bone Marrow-Derived Endothelial Progenitor Cells Contribute to Monocrotaline-Induced Pulmonary Arterial Hypertension in Rats via Inhibition of Store-Operated Ca2+ Channels. BioMed Res. Int. 2018, 2018, 4892349. [Google Scholar] [CrossRef]
- Chen, T.X.; Xu, X.Y.; Zhao, Z.; Zhao, F.Y.; Gao, Y.M.; Yan, X.H.; Wan, Y. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca2+ entry into rat pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L477–L487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zheng, Y.M. Role of ROS signaling in differential hypoxic Ca2+ and contractile responses in pulmonary and systemic vascular smooth muscle cells. Respir. Physiol. Neurobiol. 2010, 174, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Channick, R.N.; Frantz, R.P.; Grünig, E.; Jing, Z.C.; Moiseeva, O.; Preston, I.R.; Pulido, T.; Safdar, Z.; Tamura, Y.; et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801889. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.K.; Cheng, Y.J.; Chung, H.H.; Wu, J.R.; Chen, I.J.; Wu, B.N. KMUP-1 ameliorates monocrotaline-induced pulmonary arterial hypertension through the modulation of Ca2+ sensitization and K+-channel. Life Sci. 2010, 86, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Tseng, Y.T.; Liu, C.M.; Wu, B.N.; Wu, S.N. Actions of KMUP-1, a xanthine and piperazine derivative, on voltage-gated Na+ and Ca2+ -activated K+ currents in GH3 pituitary tumour cells. Br. J. Pharmacol. 2015, 172, 5110–5122. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Cheng, K.I.; Tsai, Y.L.; Hong, Y.R.; Howng, S.L.; Kwan, A.L.; Chen, I.J.; Wu, B.N. Potassium-channel openers KMUP-1 and pinacidil prevent subarachnoid hemorrhage-induced vasospasm by restoring the BKCa-channel activity. Shock 2012, 38, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, M.; Cendán, C.M.; Cobos, E.J.; Entrena, J.M.; Baeyens, J.M. Potassium channels and pain: Present realities and future opportunities. Eur. J. Pharmacol. 2004, 500, 203–219. [Google Scholar] [CrossRef]
- Cheng, K.I.; Yang, K.T.; Kung, C.L.; Cheng, Y.C.; Yeh, J.L.; Dai, Z.K.; Wu, B.N. BK(Ca) Channel Inhibition by Peripheral Nerve Injury Is Restored by the Xanthine Derivative KMUP-1 in Dorsal Root Ganglia. Cells 2021, 10, 949. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef]
- Albert, A.P.; Large, W.A. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium 2003, 33, 345–356. [Google Scholar] [CrossRef]
- Albert, A.P.; Saleh, S.N.; Peppiatt-Wildman, C.M.; Large, W.A. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J. Physiol. 2007, 583, 25–36. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Z.-K.; Chen, Y.-C.; Hsieh, S.-L.; Yeh, J.-L.; Hsu, J.-H.; Wu, B.-N. The Xanthine Derivative KMUP-1 Inhibits Hypoxia-Induced TRPC1 Expression and Store-Operated Ca2+ Entry in Pulmonary Arterial Smooth Muscle Cells. Pharmaceuticals 2024, 17, 440. https://doi.org/10.3390/ph17040440
Dai Z-K, Chen Y-C, Hsieh S-L, Yeh J-L, Hsu J-H, Wu B-N. The Xanthine Derivative KMUP-1 Inhibits Hypoxia-Induced TRPC1 Expression and Store-Operated Ca2+ Entry in Pulmonary Arterial Smooth Muscle Cells. Pharmaceuticals. 2024; 17(4):440. https://doi.org/10.3390/ph17040440
Chicago/Turabian StyleDai, Zen-Kong, Yi-Chen Chen, Su-Ling Hsieh, Jwu-Lai Yeh, Jong-Hau Hsu, and Bin-Nan Wu. 2024. "The Xanthine Derivative KMUP-1 Inhibits Hypoxia-Induced TRPC1 Expression and Store-Operated Ca2+ Entry in Pulmonary Arterial Smooth Muscle Cells" Pharmaceuticals 17, no. 4: 440. https://doi.org/10.3390/ph17040440