Abstract
The discovery and subsequent research on the MET oncogene’s role in cancer onset and progression have illuminated crucial insights into the molecular mechanisms driving malignancy. The identification of MET as the hepatocyte growth factor (HGF) receptor has paved the path for characterizing the MET tyrosine kinase activation mechanism and its downstream signaling cascade. Over the past thirty years, research has established the importance of HGF/MET signaling in normal cellular processes, such as cell dissociation, migration, proliferation, and cell survival. Notably, genetic alterations that lead to the continuous activation of MET, known as constitutive activation, have been identified as oncogenic drivers in various cancers. The genetic lesions affecting MET, such as exon skipping, gene amplification, and gene rearrangements, provide valuable targets for therapeutic intervention. Moreover, the implications of MET as a resistance mechanism to targeted therapies emphasize the need for combination treatments that include MET inhibitors. The intriguing “flare effect” phenomenon, wherein MET inhibition can lead to post-treatment increases in cancer cell proliferation, underscores the dynamic nature of cancer therapeutics. In human tumors, increased protein expression often occurs without gene amplification. Various mechanisms may cause an overexpression: transcriptional upregulation induced by other oncogenes; environmental factors (such as hypoxia or radiation); or substances produced by the reactive stroma, such as inflammatory cytokines, pro-angiogenic factors, and even HGF itself. In conclusion, the journey to understanding MET’s involvement in cancer onset and progression over the past three decades has not only deepened our knowledge, but has also paved the way for innovative therapeutic strategies. Selective pharmacological inactivation of MET stands as a promising avenue for achieving cancer remission, particularly in cases where MET alterations are the primary drivers of malignancy.
1. Introduction
Several laboratories have been engaged for about thirty years in the detailed study of an oncogene, identified with the acronym “MET”, that is capable of inducing and supporting the uncontrolled growth of cancer cells and—above all—the invasive and metastatic phenotype [1,2]. In 1984, Cooper and colleagues identified an oncogene in a human osteosarcoma cell line that had been induced chemically. They proposed the name “MET” for this oncogene, drawing inspiration from the mutagenic compound used in their study, N-methyl-N’-nitro-N-nitrosoguanidine [3]. The active oncoprotein was actually a fusion of two different loci from separate chromosomes [4]. This genetic alteration involved a segment from chromosome 1 at the 5′ end, known as TPR (translocated promoter region), and a part of the MET proto-oncogene from chromosome 7 at the 3′ end. This combination ensued in the expression of a chimeric mRNA, which resulted in the translation of a truncated cytoplasmic protein sharing similarities with tyrosine kinase families. The chimeric protein exhibited constitutive activation because of the spontaneous dimerization enabled by the leucine zipper domain of TPR. The MET-encoded protein was found to be a novel transmembrane tyrosine kinase featuring a dimeric structure of covalently linked alpha and beta chains [5]. Subsequently, it was proven that MET is the receptor tyrosine kinase (RTK) for HGF (hepatocyte growth factor), a cytokine associated with hepatocyte regeneration [6,7]. HGF was recognized to be identical to SF (scatter factor), a factor of cell motility [8,9]. The characterization of MET signaling commenced in 1994, revealing that MET undergoes dimerization and autophosphorylation at tyrosine residues Y1234 and Y1235 within its catalytic domain upon HGF stimulation [10]. Subsequently, the tyrosine residues 1349 and 1356 of the carboxy-terminal tail become phosphorylated, forming a tandem docking site that can attract a variety of SH2-containing signal transducers, such as Grb2 (growth factor receptor-bound protein 2), PI3K (phosphoinositide 3-kinase), PLCγ (phospholipase Cγ), and Src [10]. The GAB1 protein interacts with the activated MET receptor either directly [11] or indirectly via Grb2 [12], amplifying the MET signaling platform by providing additional docking sites for the attachment of downstream adaptor proteins. Using specific MET mutants that selectively activate either the Ras or PI3K pathways, research conducted by us and others has demonstrated that activating Ras is both essential and sufficient for cell proliferation, while targeting PI3K specifically enhances cell motility [13,14]. Further, using peptide inhibitors and dominant negative techniques, it has been found that activating STAT3 (signal transducer and activator of transcription 3) is necessary for cell polarization and the development of complex, branched tubular structures [15,16]. This indicates that it is possible to experimentally separate the complex processes involved in invasive growth and pinpoint the key players responsible for each process. The combined activation of the Ras and PI3K pathways, which stimulate cell growth and motility while inhibiting cell death, respectively, results in effective cell-cell dissociation, invasion into the extracellular matrix (ECM), and metastasis [17,18]. The oncogenic activity of MET results from the alteration of the proto-oncogene, which is present under normal conditions in all healthy organisms, whose functions have been usurped by malignant cells. The definitive link between abnormal MET activation and cancer was confirmed in 1997 through the discovery of MET mutations associated with inherited forms of renal carcinoma [19]. Pathologically, dysregulated MET activity is implicated in a wide range of cancers, such as renal [20], lung, liver, and gastric carcinomas, among others [1,2]. The risk of cancer associated with the MET gene emerges when there is aberrant activation of its signaling pathways. Mutations, rearrangements, or amplifications of the MET gene cause constitutive activation (without control) of its tyrosine–kinase activity and trigger malignant transformation. Genetic alterations of MET account for 3-5% of all cancers (a phenomenon called “addiction”) [1,21,22,23]. Yet, MET is over-expressed (excess production) in 90% of cancers, facilitating metastatic dissemination (a phenomenon known as “expedience”) [1,23,24]. Some tumors may produce HGF themselves or stimulate surrounding stromal cells to produce HGF, leading to autocrine or paracrine activation of MET. This creates a self-sustaining loop that encourages tumor growth and progression [25]. In particular, aberrant MET signaling can lead to increased cell motility, invasion, and disruption of normal tissue architecture, hallmarks of malignant progression. MET can also be activated through cross-talk with other receptors, even in the absence of its own ligand [26]. This can occur through heterodimerization with other receptor tyrosine kinases, which can amplify signaling pathways associated with tumorigenesis. The identification of MET as a cancer biomarker has played a significant role in the development of therapies in oncology such as MET tyrosine kinase inhibitors (TKIs), antibodies, and antibody–drug conjugates (ADCs) designed to target MET.
2. MET Structure
The receptor encoded by the MET gene is a dimeric protein proteolytically processed and glycosylated from a precursor of 170kDa (Figure 1A): The short α-chain (50kDa) is exposed to the surface of the cell and is covalently bound through disulfide bridges to the long transmembrane β-chain (145kDa) consisting of (i) an extracellular domain forming the functional domain called SEMA (Semaphorin), containing the binding site for the HGF factor (Figure 1B), with the α-chain [27,28,29,30]; (ii) a plexin–semaphorin–integrin (PSI) homology domain endowed with disulfide exchange isomerase activity [31]; and (iii) four IPT (immunoglobulin-like, plexins, transcription factors) domains, two of which (IPT3 and 4) contain a second high-affinity site for HGF binding [32].
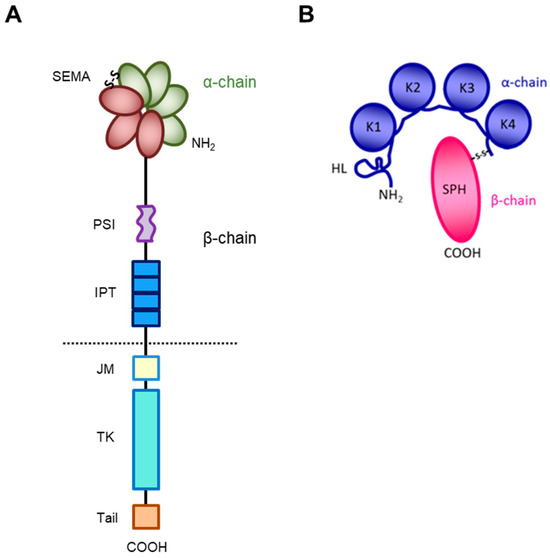
Figure 1.
Structure of HGF/MET couple. (A) MET tyrosine kinase receptor is formed by α and β chains, which together constitute the semaphorin (SEMA) domain involved in HGF ligand binding. Extracellularly, the MET β-chain is also composed of a plexin–semaphorin–integrin (PSI) domain and four immunoglobulin-like plexin transcription factors (IPT) domains. Intracellularly, it contains a regulatory juxtamembrane (JM) domain, a tyrosine kinase (TK) domain, and a C-terminal tail. (B) HGF is also formed by α and β chains. The α-chain of the HGF ligand is constituted by an N-terminal harpin loop (HL) and four kringle domains (K1-K4). Instead, the β-chain is composed of a serine protease homology domain (SPH) which lacks proteolytic activity.
A short transmembrane segment joins the extracellular to the intracellular portion that contains the functional domain endowed with tyrosine kinase activity [10]. MET/HGF interaction unleashes the kinase activity and the C-terminal tail of the receptor is phosphorylated, followed by signal transduction and MET degradation to terminate the signal (Figure 2A) [10,33]. Genetic alterations responsible for uncontrolled, constitutive activation of MET (Figure 2B–E) lead to abnormal cell growth (neoplastic transformation) and migration into tissues in an uncontrolled manner (invasion and metastasis) [34,35], featuring the invasive growth phenotype.
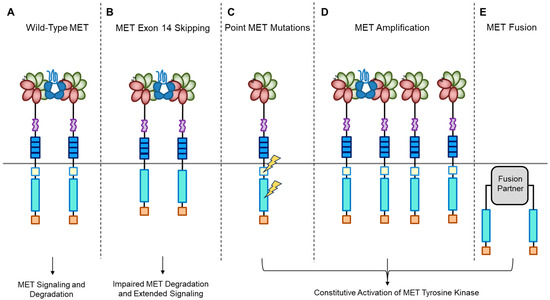
Figure 2.
MET oncogenic alterations leading to receptor and downstream signaling activation.
3. MET Cross-Talk with Other RTKs
The MET receptor can interact with other RTKs, among which are epidermal growth factor receptor (EGFR), human epidermal growth factors (HERs) 2 and 3 (also known as ERBB), and rearranged during transfection (RET) receptor [26]. The interaction between MET and EGFR is particularly important in non-small-cell lung carcinoma (NSCLC). EGFR signaling can lead to MET phosphorylation, which, especially when combined with the presence of ERBB3, can significantly increase the activity of these receptors [36]. Inhibition of EGFR or MAPK reduces MET activation and protein levels, highlighting the potential of combination therapy targeting EGFR and MET in NSCLC [36]. This relationship helps cancer cells survive and grow, and can also contribute to them being resistant to drugs. Some studies have shown that lung cancer cells resistant to gefitinib or erlotinib often have an increase in MET (MET amplification) [37,38,39]. MET amplification leads to ERBB3-dependent PI3K activation, traditionally associated with the EGFR/ERBB family. This indicates that MET’s role in resistance might extend beyond individual receptors, potentially affecting a range of ERBB-driven cancers, and this highlights the need for targeting this pathway in combinational treatment strategies [37]. A preference for HER3 among EGFR-family RTKs for MET-dependent tyrosine phosphorylation was observed in multiple MET-amplified cancer cell lines [40]. MET amplification is a consistent mechanism of acquired resistance in a number of other oncogene-driven molecular subsets of NSCLC post-tyrosine kinase inhibition [41]. A recent study conducted by Salokas et al. [42] revealed important interactions between MET and other receptors, such as the neurotrophic receptor tyrosine kinase 3 (NTRK3), platelet-derived growth factor receptor β (PDGFRβ), insulin receptor (INSR), and tyrosine protein kinase receptor (TYRO3). These unexpected MET interactions contribute to our understanding of the various cellular processes and signaling networks with key roles in enhanced cancer cell motility, invasion, and metastatic potential. Collective efforts to identify additional networks of MET interactions are crucial to enhancing our comprehension of oncogene signaling pathways and to develop new therapeutic strategies.
4. MET Exon 14 Skipping
Recently, we developed a bioinformatic tool to create an auto-updatable catalog (“MET observatory”) of the MET genetic alterations in cancer [43]. The catalog of genetic alterations results from the following data collection databases: The Cancer Genome Atlas (TCGA), Catalogue of Somatic Mutations in Cancer (COSMIC), and ClinVar datasets. The MET “observatory” revealed a peculiar mutational distribution. The most frequent lesions are not mutations affecting the tyrosine kinase domain (as in the case of similar oncogenes), but the sequences flanking exon 14 (Figure 2 and Figure 3) [44]. The point mutations occur in the splicing acceptor or donor sites, resulting in the “skipping” of the whole exon 14 (METΔ14). This exon encodes a protein tract immediately below the plasma membrane (JM, Figure 1A and Figure 2B). We deeply investigated this hotspot mutation and showed that the METΔ14 activation is ligand-dependent [45]. A lack of JM leads to receptor activation, exacerbating the invasive growth phenotype [46]. There are a number of considerations in the finding of MET exon 14 skipping mutation(s): (i) It represents a targetable alteration in cancer, particularly in NSCLC [44,47,48]; (ii) it occurs in 2-4% of NSCLC and, with less frequency, in gastrointestinal carcinomas, gliomas, sarcomas, and cancers of unknown primary origin (CUPs) [48,49,50]; (iii) drugs like crizotinib, capmatinib, ensartinib, and tepotinib have shown promising results in clinical trials for NSCLC patients with this mutation [51,52,53,54,55,56,57,58]; (iv) research on MET exon 14 skipping is ongoing, and new therapies are continually being developed and tested; and (v) while targeted therapies against MET, particularly METΔ14, have shown promise, resistance to these drugs can develop over time [59].
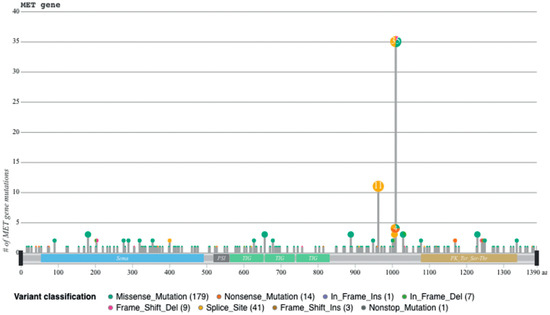
Figure 3.
Sequences flanking exon 14 are mutational hotspots, from [45].
5. MET Addiction
MET mutations affecting its catalytic or regulatory sites are sporadic, but they exist. The first activating mutations were identified in hereditary papillary renal carcinoma (HPRC), suggesting their causal role in this tumor [19]. Similar mutations were found in sporadic renal carcinoma, inducing constitutive kinase activation and oncogene “addiction” (Figure 2C), meaning that cancer cells rely heavily on a single hyperactive oncogene for their growth and survival. Transgenic mouse experiments confirmed the oncogenic potential of these mutations [60]. Activating mutations were later identified in hepatocellular carcinoma, head and neck cancers, oropharynx squamous cell cancer, gastric cancer, CUPs, and colorectal cancer [1,61]. Another commonly observed MET alteration occurs through gene amplification, with a prevalence rate of 3–5% across tumors [62,63,64,65,66,67,68,69]. Tumor cells become addicted to MET, justifying the use of targeted therapies (small molecules or antibodies [1]. MET targeting in cancer has proven its efficiency both in preclinical models and in patients [62,63,64,65,66,67,68,69,70]. MET gene amplification results in an increased number of MET receptors at the cell surface, which leads to constitutive kinase activation [22]. MET gene amplification can make cells independent of, or hypersensitive to, ligand stimulation (Figure 2D), enhancing MET signaling and driving cancer growth [63,71,72,73,74,75]. It is essential to consider MET amplification against the backdrop of widespread chromosomal aberrations seen in cancers, including the prevalent condition of cellular aneuploidy. Trisomy of chromosome 7 is a common occurrence in various cancers and can act as a pan-cancer genetic marker, potentially confounding the assessment of MET amplification. Unlike chromosome 7 trisomy, MET gene amplification is specifically selected during cancer development and functions as a cancer driver [76].
A recent study indicated that possessing at least five copies of the MET gene leads to a dependency on its signaling, thereby providing a rationale for targeted therapies [77]. While a specific threshold has not been universally agreed upon in clinical settings, accurate patient stratification for MET-directed treatments is critical. Techniques such as fluorescence in situ hybridization (FISH) can differentiate true MET gene amplification from chromosome 7 polysomy. In cases of polysomy, the ratio between the MET gene and the centromere of chromosome 7 (MET/CEN7) remains unchanged; however, an elevated ratio indicates true amplification of the MET gene. This distinction is essential for identifying patients who are most likely to respond to MET-targeted therapies [78]. Recent advancements have enabled next-generation sequencing (NGS) to establish criteria to differentiate between MET gene amplification and chromosome 7 polysomy in cancer, showing a high concordance with FISH analysis [79]. However, the performance of NGS in detecting polysomy in plasma samples is not as high as in tissue samples, indicating that there is room for improvement with non-invasive testing methods.
When MET amplification occurs in cancer cells already treated with targeted therapies, it can lead to treatment resistance [37,80]. This resistance driven by MET amplification highlights the adaptability of cancer cells. While targeted therapies designed to inhibit specific pathways can be highly effective initially, cancer cells often find ways to by-pass these interventions, making long-term treatment success challenging. One potential strategy to address resistance related to MET amplification is the development of combination therapies. Combining MET inhibitors with other targeted agents or immunotherapy may help overcome resistance by targeting multiple pathways simultaneously [81]. Biomarker testing for MET amplification is crucial for identifying patients who may benefit from MET-targeted therapies or combination treatments [81]. This underscores the importance of personalized medicine in cancer care.
6. The “Flare Effect”
When MET targeted therapies suffer from drug resistance, the line of treatment is discontinued. Previous studies have shown that withdrawal of MET tyrosine kinase receptor inhibition leads to a post-treatment increase in cancer cell proliferation due to a transient hyper-phosphorylation phase, which culminates in the “MET burst” [82], i.e., the “flare effect”. The molecular mechanisms behind this effect remain unclear, but are critically important for patients. Recently, our laboratory identified a positive feedback loop mediated by the AKT/mTOR pathway that leads to the “MET burst” after treatment withdrawal [83]. This feedback loop enhances MET translation through activation of p70S6K and 4EBP1 and causes MET hyper-phosphorylation via inactivation of the tyrosine–phosphatase PTP1B. These data suggest that the use of mTOR inhibitors during MET-targeted therapy may prevent the occurrence of the “flare effect”.
7. MET Fusion
Previously, TPR-MET was the only recognized MET gene rearrangement in human tumors predominantly found in gastric cancers. However, extensive analyses of the TCGA tumor database have revealed new hybrid proteins, where the intracellular domain of MET, or even the full-length MET, is fused with various partners (Figure 2E) [84]. These fusion events can result in novel hybrid proteins with altered functional properties and are a significant aspect of the genetic alterations observed in certain cancers. Some of these partners include proteins with a dimerization “coiled-coil” (CC) motif known for promoting dimerization, such as C8orf34, BAIAP2L1, TFG, or KIF5B. These fusions instigate ligand-independent dimerization of MET, causing continuous kinase activity that can lead to tumor formation. While these fusions are infrequent, they have been detected in lung adenocarcinomas, hepatocellular carcinomas, papillary renal carcinomas, and thyroid carcinomas [84].
Another notable gene rearrangement is between MET and PTPRZ1, a gene encoding a tyrosine phosphatase, prevalent in certain brain tumors like low-grade gliomas and pediatric glioblastomas [85]. The full-length MET coding sequence is present in the PTPRZ-MET fusion transcript, and the MET protein is overexpressed and endowed with enhanced kinase activity [86]. The exact mechanism of increased MET expression in tumors with PTPRZ-MET genes fusion is not fully understood. Yet, there is evidence suggesting that tumors harboring these gene fusions can be responsive to anti-MET monotherapy, as seen with PTPRZ1-MET in pediatric gliomas [87] and KIF5B-MET in lung cancers [88]. MET gene fusions also occur in melanomas, involving various N-terminal partners fused with the intracellular MET domain [89].
Different MET genetic alterations can induce either HGF-dependent or ligand-independent activation of the kinase, with a shared characteristic of driving invasive growth. Tumor cells become addicted to MET, making them susceptible to targeted therapies.
8. MET Oncogene Expedience
In multiple tumor types, the activation of MET is a subsequent event that intensifies the malignant characteristics of cells that have already undergone transformation. In such instances, the abnormal activation of MET may result from transcriptional upregulation induced by other oncogenes; environmental factors such as hypoxia or radiation [90,91]; or substances produced by the reactive stroma, such as inflammatory cytokines, pro-angiogenic factors [92,93], and even HGF itself [94,95]. Hypoxia is one condition that induces MET transcription [90]. This microenvironmental regulation of MET expression might explain why anti-angiogenic therapy leads to MET overexpression, constitutive kinase activation, MET-dependent invasive growth, and distant metastases [96,97,98]. Accordingly, concurrent MET and VEGF targeting mitigates tumor aggressiveness in pancreatic carcinoma [99]. When receptors are increased, they can cluster to form receptor oligomers. This interaction can trigger mutual activation, making cells sensitive to ligand concentrations that would normally be under the threshold to elicit a response. Unlike the concept of “oncogene addiction,” where the oncogene is a primary driver, the inappropriate activation of MET leading to “oncogene expedience” is a result, rather than the cause, of the transformed phenotype, and may facilitate the cancer’s progression to metastatic spreading. Therefore, alterations impacting the MET promoter merit attention. Re-analysis of the TCGA dataset may uncover a notable decrease in MET promoter methylation in cancer patients, indicating transcription activation and increased expression of MET [26]. This observation underscores the need to further understand central regulatory mechanisms. Gene expression is not solely governed at the transcription level; post-transcriptional control mechanisms affecting translation efficiency and messenger RNA stability may play crucial roles in observed cancer deregulations.
9. Clinical Impact of MET Oncogene
A clinical study of a large number of solid tumors tested at the same cancer center revealed MET amplification in 2.5% of 1115 patients with advanced cancers [100]. The prevalence was highest in renal cell carcinoma (RCC, 14%) followed by adrenocortical tumors (15%), gastroesophageal (6%), breast (5%), and ovarian cancers (4%) [100]. This is in agreement with previous works concentrated on a single tumor type [71,101,102,103,104,105,106]. MET-amplified tumors were associated with a higher histologic grade and development of more metastatic sites [100]. Approximately 2% of glioblastomas [107] and 12% of melanomas [75] exhibited MET amplification according to whole-genome analysis. Approximately 3% of advanced NSCLC cases harbor point mutations or deletions in MET exon 14 or its flanking introns [47], while around 1% of poor survival NSCLC patients are associated with de novo MET amplification [108]. Resistance to EGFR TKIs in lung cancer patients is commonly determined by MET amplification. This was reported in 5% of patients after first-generation EGFR TKIs and 10% of osimertinib (mutant T790M-selective EGFR TKI)-treated NSCLC patients [109,110]. Recently, MET amplification has also been detected in ALK-rearranged NSCLC patients treated with ALK TKI [111]. In the absence of MET amplification, MET has been found to be overexpressed in a variety of cancers, such as RCC [19], NSCLC [112], malignant pleural mesothelioma (MPM) [113], glioblastoma multiforme (GBM) [114], and gastric cancer (GC) [115]. High levels of MET expression are associated with poor survival outcomes in patients with gastrointestinal malignancies [116]. Numerous meta-analyses have demonstrated that abnormal activation of the MET pathway in cancerous tissue, characterized by overexpression of the MET gene, gene amplification, exon 14 skipping, and other activating mutations, consistently correlates with decreased survival and adverse outcomes (reviewed in [117]). These analyses primarily focus on NSCLC, as well as cancers of the breast, head and neck, colorectum, stomach, pancreas, and other parts of the gastrointestinal tract. The analyses of patient data in these studies have underscored a clear correlation between MET expression or mutation and survival rates. Patients exhibiting MET amplification or overexpression, indicative of heightened MET pathway activity, have consistently shown poorer prognoses across various cancer types. This trend is particularly pronounced in cancers with the highest prevalence of MET amplification, such as renal cell carcinoma and adrenocortical tumors. In NSCLC, MET exon 14 mutations or amplifications are significant predictors of reduced survival, reflecting the aggressive nature of MET-driven cancer phenotypes. Moreover, multiple studies have highlighted the usefulness of MET biomarkers for pinpointing patients who benefit most from targeted HGF/MET therapies, whether used alone or in combination [44,51,55,118,119]. The most significant predictive value for these biomarkers has been noted in responses to savolitinib in renal cancer [101] and tepotinib in NSCLC [51,55]. However, certain research, particularly that emphasizing MET expression, has not proven to be very useful in these categorizations, possibly due to a lack of standardized methods, especially in immunohistochemistry scoring systems, or because the cancer cells are not dependent on the MET pathway despite the presence of overexpression. Assessments based on amplification and mutation tend to be less affected by these issues.
In the last decade, several MET inhibitors, including monoclonal antibodies [120,121,122,123,124,125] and small molecules such as TKI [51,52,53,54,55,56,57,58,70,102,112,126,127,128,129,130], have been developed and tested in clinics. The lack of functional molecular stratification in patients with genetically susceptible tumors, such as MET∆14, has led to the failure of some trials. These studies have indicated the necessity of genetic assessment to identify oncogene-addicted tumors, thereby enriching the pool of patients who respond to treatment. In 2020, Japan and the USA approved two MET TKIs, tepotinib and capmatinib, respectively, for the treatment of advanced NSCLC with the MET exon 14 skipping mutation. As discussed above, MET inhibitors might be beneficial when used in combination with other targeted therapies to block HGF-dependent survival function [1,61,131,132,133]. Furthermore, other innovative therapeutic techniques, such as ADCs combining the specificity of antibodies with the potency of cytotoxic agents [134,135,136], have been tested. These therapeutic agents could be very functional in patients overexpressing the MET receptor.
10. Conclusions
In summary, since its discovery as an oncogene, MET has been found to undergo mutations, amplifications, or rearrangements in a wide range of cancers, spanning from the early stages of tumor initiation to instances of therapeutic resistance and recurrence. The analysis of genetic changes occurring within the MET receptor has revealed a distinct pattern of mutations, primarily affecting the sequences flanking exon 14. In clinical practice, the presence of MET genetic abnormalities is a critical factor in identifying tumors that rely on this oncogene (MET oncogene-addiction), making such patients potential candidates for targeted therapies. Where MET is genetically altered and is the first cause of transformation, pharmacological inactivation may result in complete cancer remission.
In physiological settings, fine-tuning the quantity of MET receptors is essential for wound healing and tissue regeneration. This “rescue” function is usurped by neoplastic cells to foster cell survival and facilitate invasion and the spread of metastases. Thus, wild-type MET can function as an “oncogene expedient”, enhancing the impact of other oncogenes and promoting malignant progression. Even if a tumor lacks MET mutations, it may still respond to treatments that target MET or its downstream pathways. Such targeted interventions may reduce the survival of the primary tumor, as well as its invasion and metastatic spread, thus impeding cancer progression.
Author Contributions
Conceptualization, T.C. and P.M.C.; writing—original draft preparation, T.C., S.G., and P.M.C.; writing—review and editing, T.C. and P.M.C.; visualization, S.G.; supervision, P.M.C.; project administration, P.M.C.; funding acquisition, T.C. and P.M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Association for Cancer Research (AIRC) AIRC-19-IG program, grant number 23820 to P.M.C.; Italian Ministry of Health “Ricerca corrente 2024”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
ADC, antibody–drug conjugate; CC, coiled-coil; CUP, cancer of unknown primary origin; COSMIC, Catalogue of Somatic Mutations in Cancer; ECM, extracellular matrix; FISH, fluorescence in situ hybridization; EGFR, epidermal growth factor receptor; GBM, glioblastoma multiforme; GC, gastric cancer; Grb2, growth factor receptor-bound protein 2; HGF, hepatocyte growth factor; HER, human epidermal growth factor; HL, harpin loop; HPRC, hereditary papillary renal carcinoma; INSR, insulin receptor; IPT, immunoglobulin-like, plexin transcription factors; JM, juxtamembrane domain; MPM, malignant pleural mesothelioma; NGS, next-generation sequencing; NTRK3, neurotrophic receptor tyrosine kinase 3; NSCLC, non-small-cell lung cancer; PDGFRβ, platelet-derived growth factor receptor β; PI3K, phosphoinositide 3-kinase; PLCγ, phospholipase Cγ; PSI, plexin–semaphorin–integrin; RET, rearranged during transfection; RCC, renal cell carcinoma; RTK, receptor tyrosine kinase; SEMA, semaphorin; SF, scatter factor; SPH, serine protease homology; STAT3, signal transducer and activator of transcription 3; TCGA, The Cancer Genome Atlas; TKI, tyrosine kinase inhibitor; TPR, translocated promoter region; TYRO3, tyrosine protein kinase receptor.
References
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Vande Woude, G.F. Mechanism of met oncogene activation. Cell 1986, 45, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; Di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991, 6, 501–504. Available online: https://pubmed.ncbi.nlm.nih.gov/1827664/ (accessed on 28 March 2024). [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Weidner, K.M.; Vigna, E.; Gaudino, G.; Bardelli, A.; Ponzetto, C.; Narsimhan, R.P.; Hartmann, G.; Zarnegar, R.; Michalopoulos, G.K. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991, 10, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.M.; Arakaki, N.; Hartmann, G.; Vandekerckhove, J.; Weingart, S.; Rieder, H.; Fonatsch, C.; Tsubouchi, H.; Hishida, T.; Daikuhara, Y. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc. Natl. Acad. Sci. USA 1991, 88, 7001–7005. [Google Scholar] [CrossRef]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; Dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Weidner, K.M.; Di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef]
- Nguyen, L.; Holgado-Madruga, M.; Maroun, C.; Fixman, E.D.; Kamikura, D.; Fournier, T.; Charest, A.; Tremblay, M.L.; Wong, A.J.; Park, M. Association of the multisubstrate docking protein Gab1 with the hepatocyte growth factor receptor requires a functional Grb2 binding site involving tyrosine. J. Biol. Chem. 1997, 272, 20811–20819. [Google Scholar] [CrossRef]
- Royal, I.; Park, M. Hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells requires phosphatidylinositol 3-kinase. J. Biol. Chem. 1995, 270, 27780–27787. [Google Scholar] [CrossRef]
- Bardelli, A.; Basile, M.L.; Audero, E.; Giordano, S.; Wennström, S.; Ménard, S.; Comoglio, P.M.; Ponzetto, C. Concomitant activation of pathways downstream of Grb2 and PI 3-kinase is required for MET-mediated metastasis. Oncogene 1999, 18, 1139–1146. [Google Scholar] [CrossRef]
- Boccaccio, C.; Andò, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; Vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef]
- Birchmeier, W.; Brinkmann, V.; Niemann, C.; Meiners, S.; DiCesare, S.; Naundorf, H.; Sachs, M. Role of HGF/SF and c-Met in morphogenesis and metastasis of epithelial cells. Ciba Found. Symp. 1997, 212, 230–240. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Kim, H.S.; Chon, H.J.; Kim, H.; Shin, S.J.; Wacheck, V.; Gruver, A.M.; Kim, J.S.; Rha, S.Y.; Chung, H.C. MET in gastric cancer with liver metastasis: The relationship between MET amplification and Met overexpression in primary stomach tumors and liver metastasis. J. Surg. Oncol. 2018, 117, 1679–1686. [Google Scholar] [CrossRef]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Zeng, Q.; Davis, L.J.; Hatch, H.; Hang, G.; Kohl, N.E.; Gibbs, J.B.; Pan, B.S. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007, 67, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Han, J.-Y.; Lee, G.K.; Shin, J.; Yun, S.A.; Oh, J.Y.; Lee, S.; Kim, H.T.; Lee, J.S. C-MET overexpression as a resistance biomarker to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer. J. Clin. Oncol. 2016, 34, e20660. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int. J. Cancer 2006, 119, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, J.; Okamoto, I.; Sakai, K.; Nakagawa, K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer 2011, 105, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Youles, M.E.; Miguel, R.N.; Blundell, T.L.; Iamele, L.; Gough, J.; Bandyopadhyay, A.; Hartmann, G.; Butler, P.J. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA 2003, 100, 12039–12044. [Google Scholar] [CrossRef]
- Gherardi, E.; Sandin, S.; Petoukhov, M.V.; Finch, J.; Youles, M.E.; Ofverstedt, L.G.; Miguel, R.N.; Blundell, T.L.; Vande Woude, G.F.; Skoglund, U.; et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc. Natl. Acad. Sci. USA 2006, 103, 4046–4051. [Google Scholar] [CrossRef]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF beta-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef]
- Uchikawa, E.; Chen, Z.; Xiao, G.Y.; Zhang, X.; Bai, X.C. Structural basis of the activation of c-MET receptor. Nat. Commun. 2021, 12, 4074. [Google Scholar] [CrossRef]
- Altintas, D.M.; Gallo, S.; Basilico, C.; Cerqua, M.; Bocedi, A.; Vitacolonna, A.; Botti, O.; Casanova, E.; Rancati, I.; Milanese, C.; et al. The PSI Domain of the MET oncogene encodes a functional disulfide isomerase essential for the maturation of the receptor precursor. Int. J. Mol. Sci. 2022, 23, 12427. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Arnesano, A.; Galluzzo, M.; Comoglio, P.M.; Michieli, P. A high affinity hepatocyte growth factor-binding site in the immunoglobulin-like region of Met. J. Biol. Chem. 2008, 283, 21267–21277. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Gilestro, G.F.; Lanzardo, S.; Comoglio, P.M.; Migone, N.; Giordano, S. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature 2002, 416, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Breindel, J.L.; Haskins, J.W.; Cowell, E.P.; Zhao, M.; Nguyen, D.X.; Stern, D.F. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013, 73, 5053–5065. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- van Veggel, B.; de Langen, A.J.; Hashemi, S.; Monkhorst, K.; Rosenberg, E.H.; Heideman, D.A.M.; Radonic, T.; Smit, E.F. Crizotinib treatment for patients with EGFR mutation positive NSCLC that acquire cMET amplification after EGFR TKI therapy results in short-lived and heterogeneous responses. Lung Cancer 2018, 124, 130–134. [Google Scholar] [CrossRef]
- Stern, Y.E.; Al-Ghabkari, A.; Monast, A.; Fiset, B.; Aboualizadeh, F.; Yao, Z.; Stagljar, I.; Walsh, L.A.; Duhamel, S.; Park, M. Met-HER3 crosstalk supports proliferation via MPZL3 in MET-amplified cancer cells. Cell. Mol. Life Sci. 2022, 79, 178. [Google Scholar] [CrossRef]
- Coleman, N.; Hong, L.; Zhang, J.; Heymach, J.; Hong, D.; Le, X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 2021, 6, 100319. [Google Scholar] [CrossRef] [PubMed]
- Salokas, K.; Liu, X.; Öhman, T.; Chowdhury, I.; Gawriyski, L.; Keskitalo, S.; Varjosalo, M. Physical and functional interactome atlas of human receptor tyrosine kinases. EMBO Rep. 2022, 23, e54041. [Google Scholar] [CrossRef] [PubMed]
- Altintas, D.M.; Comoglio, P.M. An Observatory for the MET Oncogene: A Guide for Targeted Therapies. Cancers 2023, 15, 4672. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, M.; Botti, O.; Arigoni, M.; Gioelli, N.; Serini, G.; Calogero, R.; Boccaccio, C.; Comoglio, P.M.; Altintas, D.M. MET∆14 promotes a ligand-dependent, AKT-driven invasive growth. Life Sci. Alliance 2022, 5, e202201409. [Google Scholar] [CrossRef] [PubMed]
- Vigna, E.; Gramaglia, D.; Longati, P.; Bardelli, A.; Comoglio, P.M. Loss of the exon encoding the juxtamembrane domain is essential for the oncogenic activation of TPR-MET. Oncogene 1999, 18, 4275–4281. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev. 2020, 87, 102022. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, Y.; Stoopler, M.B.; Shen, Y.; Cheng, H.; Chen, J.; Mansukhani, M.; Koul, S.; Halmos, B.; Borczuk, A.C. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J. Clin. Oncol. 2016, 34, 794–802. [Google Scholar] [CrossRef]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 patients with lung can- cer harboring MET exon 14 skipping alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Drilon, A.; Banna, G.L.; Peters, S.; Addeo, A. The METeoric rise of MET in lung cancer. Cancer 2020, 126, 4826–4837. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. GEOMETRY mono-1 Investigators. Capmatinib in MET Exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in non-small-cell lung cancer with MET Exon 14 skipping mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.G.Y.; Guo, R.; Drilon, A.; Tan, D.S.W. Refining patient selection of MET-activated non-small cell lung cancer through biomarker precision. Cancer Treat. Rev. 2022, 110, 102444. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jin, R.; Li, M.; Lan, F.; Zhu, H.; Yu, Y.; Miao, D.; Wang, Q.; Zhou, Y.; Selvaggi, G.; et al. Potent antitumor activity of ensartinib in MET exon 14 skipping-mutated non-small cell lung cancer. Cancer Lett. 2023, 561, 216140. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Paik, P.K.; Garassino, M.C.; Le, X.; Sakai, H.; Veillon, R.; Smit, E.F.; Cortot, A.B.; Raskin, J.; Viteri, S.; et al. Tepotinib treatment in patients with MET Exon 14-skipping non-small cell lung cancer: Long-term follow-up of the VISION Phase 2 nonrandomized clinical trial. JAMA Oncol. 2023, 9, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Offin, M.; Brannon, A.R.; Chang, J.; Chow, A.; Delasos, L.; Girshman, J.; Wilkins, O.; McCarthy, C.G.; Makhnin, A.; et al. MET Exon 14-altered lung cancers and MET inhibitor resistance. Clin. Cancer Res. 2021, 27, 799–806. [Google Scholar] [CrossRef]
- Graveel, C.; Su, Y.; Koeman, J.; Wang, L.M.; Tessarollo, L.; Fiscella, M.; Birchmeier, C.; Swiatek, P.; Bronson, R.; Vande Woude, G. Activating Met mutations produce unique tumor profiles in mice with selective duplication of the mutant allele. Proc. Natl. Acad. Sci. USA 2004, 101, 17198–17203. [Google Scholar] [CrossRef]
- Recondo, G.; Che, J.; Jänne, P.A.; Awad, M.M. Targeting MET dysregulation in cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Schildhaus, H.U.; Schultheis, A.M.; Rüschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.D.; Schlesinger, A.; Bos, M.; et al. MET amplification status in therapy-naïve adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res. 2015, 21, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.-W.; Seo, J.-W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Graziano, F.; Galluccio, N.; Lorenzini, P.; Ruzzo, A.; Canestrari, E.; D’Emidio, S.; Catalano, V.; Sisti, V.; Ligorio, C.; Andreoni, F.; et al. Genetic activation of the MET pathway and prognosis of patients with high-risk, radically resected gastric cancer. J. Clin. Oncol. 2011, 29, 4789–4795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, G.; Sun, X.; Ni, S.; Tan, C.; Xu, M.; Huang, D.; Ren, F.; Li, D.; Ping, W.; et al. MET amplification, expression, and exon 14 mutations in colorectal adenocarcinoma. Hum. Pathol. 2018, 77, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Ali, S.M.; Yakirevich, E.; Geynisman, D.M.; Karam, J.A.; Elvin, J.A.; Frampton, G.M.; Huang, X.; Lin, D.I.; Rosenzweig, M.; et al. Characterization of clinical cases of advanced papillary renal cell carcinoma via comprehensive genomic profiling. Eur. Urol. 2018, 73, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, K.Y.; Giubellino, A. The role of MET in melanoma and melanocytic lesions. Am. J. Pathol. 2019, 189, 2138–2148. [Google Scholar] [CrossRef]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006, 25, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET exon 14 mutations in non- small-cell lung cancer are associated with advanced age and stage- dependent MET genomic amplification and c- Met overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.; Chung, L.Y.; Chau, S.L.; Lung, R.W.; Tong, C.Y.; Chow, C.; Tin, E.K.; Yu, Y.H.; et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non- small cell lung carcinoma with poor prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole- genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef]
- Yin, W.; Cheng, J.; Tang, Z.; Toruner, G.; Hu, S.; Guo, M.; Robinson, M.; Medeiros, L.J.; Tang, G. MET Amplification (MET/CEP7 Ratio ≥ 1.8) is an independent poor prognostic marker in patients with treatment-naive non-small-cell lung cancer. Clin. Lung Cancer 2021, 22, e512–e518. [Google Scholar] [CrossRef]
- Sun, B.; Qiu, T.; Zeng, X.; Duan, J.; Bai, H.; Xu, J.; Li, J.; Li, J.; Hao, X.; Liu, Y.; et al. Detection of MET polysomy by next-generation sequencing and its clinical relevance for MET inhibitors. Cancer Res. Commun. 2023, 3, 532–539. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Kumaki, Y.; Oda, G.; Ikeda, S. Targeting MET amplification: Opportunities and obstacles in therapeutic approaches. Cancers 2023, 15, 4552. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Ducano, N.; Lupo, B.; Vigna, E.; Avanzato, D.; Perera, T.; Trusolino, L.; Lanzetti, L.; Comoglio, P.M. Rebound effects caused by withdrawal of MET kinase inhibitor are quenched by a MET therapeutic antibody. Cancer Res. 2016, 76, 5019–5029. [Google Scholar] [CrossRef] [PubMed]
- Altintas, D.M.; Cerqua, M.; De Laurentiis, A.; Trusolino, L.; Boccaccio, C.; Comoglio, P.M. An mTOR feedback loop mediates the ‘flare’ (‘rebound’) response to MET tyrosine kinase inhibition. Sci. Rep. 2023, 13, 1378. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Yu, K.; Tang, X.Y.; Bao, Z.S.; Jiang, T.; Fan, X.L.; Chen, X.W.; Su, X.D. Enhanced expression and phosphorylation of the MET oncoprotein by glioma-specific PTPRZ1-MET fusions. FEBS Lett. 2015, 589, 1437–1443. [Google Scholar] [CrossRef]
- International Cancer Genome Consortium PedBrain Tumor Project. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar] [CrossRef]
- Plenker, D.; Bertrand, M.; de Langen, A.J.; Riedel, R.; Lorenz, C.; Scheel, A.H.; Müller, J.; Brägelmann, J.; Daßler-Plenker, J.; Kobe, C.; et al. Structural alterations of MET trigger response to MET kinase inhibition in lung adenocarcinoma patients. Clin. Cancer Res. 2018, 24, 1337–1343. [Google Scholar] [CrossRef]
- Yeh, I.; Botton, T.; Talevich, E.; Shain, A.H.; Sparatta, A.J.; de la Fouchardiere, A.; Mully, T.W.; North, J.P.; Garrido, M.C.; Gagnon, A.; et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nature Commun. 2015, 6, 7174. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Kozuki, Y.; Yagasaki, K. Potentiation of invasive activity of hepatoma cells by reactive oxygen species is mediated by autocrine/paracrine loop of hepatocyte growth factor. Biochem. Biophys. Res. Commun. 2003, 305, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef]
- Sennino, B.; Ishiguro-Oonuma, T.; Schriver, B.J.; Christensen, J.G.; McDonald, D.M. Inhibition of cMet reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Res. 2013, 73, 3692–3703. [Google Scholar] [CrossRef]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Ebos, J.M.; Ebos, J.M.L.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after shortterm treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.-K.; Chapman, H.A.; Christensen, J.C.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef]
- Jardim, D.L.; Tang, C.; Gagliato, D.M.; Falchook, G.S.; Hess, K.; Janku, F.; Fu, S.; Wheler, J.J.; Zinner, R.G.; Naing, A.; et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin. Cancer Res. 2014, 20, 6336–6345. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; Rini, B.I.; Srinivas, S.; Stein, M.N.; Adams, L.M.; et al. Phase II and biomarker study of the dual MET/ VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Ooi, A.; Kobayashi, M.; Mai, M.; Yanagihara, K.; Nakanishi, I. Amplification of c-myc, K-sam, and c-met in gastric cancers: Detection by fluorescence in situ hybridization. Lab Investig. 1998, 78, 1143–1153. [Google Scholar] [PubMed]
- Houldsworth, J.; Cordon-Cardo, C.; Ladanyi, M.; Kelsen, D.P.; Chaganti, R.S. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res. 1990, 50, 6417–6422. [Google Scholar] [PubMed]
- Kuniyasu, H.; Yasui, W.; Kitadai, Y.; Yokozaki, H.; Ito, H.; Tahara, E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem. Biophys. Res. Commun. 1992, 189, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tsuda, H.; Miyai, K.; Takano, M.; Tamai, S.; Matsubara, O. Gene amplification and protein overexpression of MET are common events in ovarian clear-cell adenocarcinoma: Their roles in tumor progression and prognostication of the patient. Mod. Pathol. 2011, 24, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A., Jr.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J. Thorac. Oncol. 2019, 14, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib JAMA Oncol. 2018, 4, 1527–1534. 4. [CrossRef]
- Dagogo-Jack, I.; Yoda, S.; Lennerz, J.K.; Langenbucher, A.; Lin, J.J.; Rooney, M.M.; Prutisto-Chang, K.; Oh, A.; Adams, N.A.; Yeap, B.Y.; et al. MET Alterations are a Recurring and Actionable Resistance Mechanism in ALK-Positive Lung Cancer. Clin. Cancer Res. 2020, 26, 2535–2545. [Google Scholar] [CrossRef]
- Wu, Y.L.; Smit, E.F.; Bauer, T.M. Capmatinib for patients with non-small cell lung cancer with MET exon 14 skipping mutations: A review of preclinical and clinical studies. Cancer Treat. Rev. 2021, 95, 102173. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, R.; Ma, P.C.; Seiwert, T.Y.; Jagadeeswaran, S.; Zumba, O.; Nallasura, V.; Ahmed, S.; Filiberti, R.; Paganuzzi, M.; Puntoni, R.; et al. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 2006, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghabkari, A.; Huang, B.; Park, M. Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions. Cells 2024, 13, 218. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.T.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I.; et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1467–1482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miao, L.; Wang, S.; Zhao, Y.; Xie, Y.; Yun, H.; Ren, Z.; Wang, G.; Teng, M.; Li, Y. Study on the expression of c-Met in gastric cancer and its correlation with preoperative serum tumor markers and prognosis. World J. Surg. Oncol. 2022, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, F.; Giovannetti, E.; Saso, L.; Firuzi, O. HGF/MET pathway aberrations as diagnostic, prognostic, and predictive biomarkers in human cancers. Crit. Rev. Clin. Lab. Sci. 2019, 56, 533–566. [Google Scholar] [CrossRef] [PubMed]
- Bahcall, M.; Sim, T.; Paweletz, C.P.; Patel, J.D.; Alden, R.S.; Kuang, Y.; Sacher, A.G.; Kim, N.D.; Lydon, C.A.; Awad, M.M.; et al. Acquired MET D1228V mutation and resistance to MET inhibition in lung cancer. Cancer Discov. 2016, 6, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; Sa, J.K.; Cho, H.J.; Her, N.G.; Zhang, C.; Zhao, Z.; Zhang, Y.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV nonsmall-cell lung cancer: METLung. J. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef]
- Van der Horst, E.H.; Chinn, L.; Wang, M.; Velilla, T.; Tran, H.; Madrona, Y.; Lam, A.; Ji, M.; Hoey, T.C.; Sato, A.K. Discovery of fully human Anti-MET monoclonal antibodies with antitumor activity against colon cancer tumor models In Vivo. Neoplasia 2009, 11, 355–364. [Google Scholar] [CrossRef]
- Lee, J.M.; Kim, B.; Lee, S.B.; Jeong, Y.; Oh, Y.M.; Song, Y.J.; Jung, S.; Choi, J.; Lee, S.; Cheong, K.H.; et al. Cbl independent degradation of Met: Ways to avoid agonism of bivalent met-targeting antibody. Oncogene 2014, 33, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Kang, S.; Kim, K.-A.; Song, Y.-J.; Cheong, K.H.; Cha, H.-Y.; Kim, C.-H. Met degradation by SAIT301, a Met monoclonal antibody, reduces the invasion and migration of nasopharyngeal cancer cells via inhibition of EGR-1 expression. Cell Death Dis. 2014, 5, e1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manroet, J.R.; et al. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF independent MET activation and tumor growth. Clin. Cancer Res. 2014, 20, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, I.; Modica, C.; Chiriaco, C.; Basilico, C.; Hughes, J.M.; Corso, S.; Giordano, S.; Comoglio, P.M.; Vigna, E. hOA-DN30: A highly effective humanized single-arm MET antibody inducing remission of ‘MET-addicted’ cancers. J. Exp. Clin. Cancer Res. 2022, 41, 112. [Google Scholar] [CrossRef]
- Waqar, S.N.; Morgensztern, D.; Sehn, J. MET mutation associated with responsiveness to crizotinib. J. Thorac. Oncol. 2015, 10, e29–e31. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, M.A.; Goldman, J.W. MET-Mutated NSCLC with Major Response to Crizotinib. J. Thorac. Oncol. 2015, 10, e33–e34. [Google Scholar] [CrossRef] [PubMed]
- Mahjoubi, L.; Gazzah, A.; Besse, B.; Lacroix, L.; Soria, J.C. A never- smoker lung adenocarcinoma patient with a MET exon 14 mutation (D1028N) and a rapid partial response after crizotinib. Investig. New Drugs 2016, 34, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET- amplified recurrent glioblastoma with a mesenchymal- epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, B.-Y.; Cheng, A.-L.; Ren, Z.; Kim, T.-Y.; Pan, H.; Rau, K.-M.; Choi, H.J.; Park, J.-W.; Kim, J.H.; Yen, C.J.; et al. Randomised Phase 1b/2 trial of tepotinib vs sorafenib in Asian patients with advanced hepatocellular carcinoma with MET overexpression. Clinical Trial Br. J. Cancer 2021, 125, 200–208. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Harbinski, F.; Craig, V.J.; Sanghavi, S.; Jeffery, D.; Liu, L.; Sheppard, K.A.; Wagner, S.; Stamm, C.; Buness, A.; Chatenay-Rivauday, C.; et al. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012, 2, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Gymnopoulos, M.; Betancourt, O.; Blot, V.; Fujita, R.; Galvan, D.; Lieuw, V.; Nguyen, S.; Snedden, J.; Stewart, C.; Villicana, J.; et al. TR1801-ADC: A highly potent cMet antibody-drug conjugate with high activity in patient-derived xenograft models of solid tumors. Mol. Oncol. 2020, 14, 54–68. [Google Scholar] [CrossRef] [PubMed]
- DaSilva, J.O.; Yang, K.; Surriga, O.; Nittoli, T.; Kunz, A.; Franklin, M.C.; Delfino, F.J.; Mao, S.; Zhao, F.; Giurleo, J.T.; et al. Biparatopic antibody-drug conjugate to treat MET-expressing cancers, including those that are unresponsive to MET pathway blockade. Mol. Cancer. Ther. 2021, 20, 1966–1976. [Google Scholar] [CrossRef]
- Wang, J.; Anderson, M.G.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Tucker, L.; Zhang, Q.; Han, E.K.; Palma, J.P.; Naumovski, L.; et al. ABBV-399, a c-Met antibody-drug conjugate that targets both MET-amplified and c-Met-overexpressing tumors, irrespective of MET pathway dependence. Clin. Cancer Res. 2017, 23, 992–1000. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).