Abstract
Midazolam, a short-acting benzodiazepine, is widely used to alleviate patient anxiety, enhance compliance, and aid in anesthesia. While its side effects are typically dose-dependent and manageable with vigilant perioperative monitoring, serious cardiorespiratory complications, including fatalities and permanent neurological impairment, have been documented. Prolonged exposure to benzodiazepines, such as midazolam, has been associated with neurological changes in infants. Despite attempts to employ therapeutic drug monitoring for optimal sedation dosing, its efficacy has been limited. Consequently, efforts are underway to identify alternative predictive markers to guide individualized dosing and mitigate adverse effects. Understanding these factors is crucial for determining midazolam’s suitability for future administration, particularly after a severe adverse reaction. This article aims to elucidate the factors influencing midazolam’s pharmacokinetics and pharmacodynamics, potentially leading to adverse events. Finally, a case study is presented to exemplify the complex investigation into the causative factors of midazolam-related adverse events.
1. Introduction
Midazolam, a fast- and short-acting benzodiazepine with an imidazole structure, has anxiolytic, muscle relaxant, anticonvulsant, sedative, hypnotic, and amnesic properties. There are different formulations of midazolam for different medical needs. In its intravenous injection form, midazolam serves as a sedative, anxiolytic, and amnesic agent administered before or during diagnostic, therapeutic, or endoscopic procedures, as well as prior to surgery. It may also be used to induce general anesthesia, as part of a balanced anesthesia approach, or as a continuous intravenous infusion for sedation in intubated and mechanically ventilated patients in critical care settings or for palliative sedation therapy. Oral formulations are utilized for preoperative sedation, anxiolysis, and amnesia, or before diagnostic, therapeutic, or endoscopic procedures in monitored environments. In cases of prolonged, acute, convulsive seizures, intra-muscular, buccal, or nasal formulations of midazolam are available for treatment. Midazolam onset of action is rapid regardless of the route, within 2 min (intravenous/intramuscular) to 15 min (oral). Midazolam exists in a pH equilibrium between closed and open benzodiazepine ring structures. The benzodiazepine ring of midazolam opens at a lower pH. At physiologic pH, the ring closes and the molecule becomes lipid soluble, allowing rapid penetration across the blood–brain barrier.
Dosage should be individualized, and dose titration typically involves administering bolus doses ranging from 0.1 to 0.3 mg/kg, along with a maintenance dose of 0.03–0.2 mg/kg per hour, as seen in ventilated patients, for instance. Midazolam is commonly administered intravenously in single doses ranging from 0.5 mg to 2.5 mg to achieve conscious sedation of patients undergoing endoscopy or minor surgery. A single 10 mg dose can be administered via intramuscular injection for the treatment of status epilepticus in adults.
Midazolam has a wide therapeutic range; however, toxicities can occur, particularly with higher doses and when administered intravenously. Midazolam toxicity manifests as central nervous system depression, ranging from drowsiness to coma. In mild to moderate cases, symptoms may include drowsiness, confusion, difficulty speaking, lethargy, a hypnotic state, reduced reflexes, lack of coordination, and decreased muscle tone. Occasionally, paradoxical or disinhibitory reactions may arise. In severe cases of overdose, patients may experience respiratory depression and coma. Overdosing on benzodiazepines, especially when combined with other central nervous system depressants like opioids, can be fatal. Various case reports detail adverse events, such as hypoxemia, inadequate sedation, and delayed recovery, associated with midazolam use.
The attempt to use therapeutic drug monitoring (TDM) to determine the optimal individual dose for adequate sedation with midazolam was unfortunately not successful. Efforts have been made to identify other predictive markers that can be used to determine the optimal dose for individual patients to avoid major side effects and toxicities.
Interindividual differences in pharmacokinetics, such as the clearance and half-life of midazolam, can be substantial and differ by a factor of ≥10 [1,2]. The kinetics of midazolam are highly dependent on hepatic cytochrome P450 3A (CYP3A) enzyme activity, as evidenced, for example, by a ≥10-fold increase in the area under the plasma drug concentration–time curve (AUC) of midazolam when potent CYP3A inhibitors are administered concomitantly. The AUC for the CYP3A4 substrate midazolam varied up to 400-fold in healthy volunteers exposed sequentially to the CYP3A4 inhibitor itraconazole and its transcriptional inducer rifampicin [3]. Midazolam is therefore recommended by the Food and Drug Administration (FDA) as a “clinical index substrate” for drug–drug interaction studies.
Factors such as co-administered drugs, age, sex, or inflammation have been shown to affect CYP3A activity and midazolam kinetics. Despite the influence of these non-genetic factors, genetic makeup could also provide an important contribution to the interindividual variability in the pharmacokinetics and dynamics of midazolam.
The heritability of midazolam plasma clearance was estimated to be 96% using a repeated drug administration approach [4]. In a study on the heritability of non-induced CYP3A activity conducted on twins, genetic effects accounted for 15% and 73% of the variation in the midazolam AUC and the 1′-hydroxy-midazolam (1-OH-M) AUC, respectively [5]. Although studies differ greatly in their estimates of the genetic contribution to CYP3A activity, they do not exclude a significant influence of genetics. Analogous to the important pharmacogene CYP2D6, for which genotyping to determine metabolizer status has become part of routine care, pharmacogenetic markers have also been sought for CYP3A genes. However, to date, no consistent association between CYP3A4/5 genetic variation and pharmacokinetic/pharmacodynamic responses to midazolam that is useful for routine pharmacogenetic testing has been established [6,7,8]. Although many single nucleotide polymorphisms (SNPs) within the CYP3A locus have been identified, these variants appear to explain only a minority of the total heritability inferred indirectly from population data. Because of this “lack of heritability” problem, no clinical guideline has been established to guide midazolam prescription based on CYP3A genotype.
This review discusses the reasons why the TDM of midazolam is ineffective for dose individualization, for example, in long-term sedated patients, despite a clear medical need for a straightforward marker to adjust sedation depth and avoid toxicities, in light of potential drug–drug interactions and the effect of comorbidities of these often critically ill patients on pharmacokinetics. Furthermore, we describe several factors that contribute to the substantial interindividual variability in the pharmacokinetics and pharmacodynamics of midazolam. These factors include non-genetic variables such as sex, nutritional status, concomitant medication use, and the presence of comorbid conditions such as renal or hepatic dysfunction and inflammatory diseases. Genetic influences, including polymorphisms in genes encoding metabolizing enzymes or the GABAA receptor, as well as epigenetic factors, are also discussed. Finally, a case is presented that illustrates the problem of “lack of heritability”.
2. TDM of Midazolam
TDM assists clinicians in determining optimal drug dosages for individualized treatment, particularly in cases where medications with narrow therapeutic indexes are prescribed for long-term therapy. Monitoring of analgosedation in continuous infusion therapy of midazolam, such as for analgosedation in mechanically ventilated patients, is crucial to avoid inadequate sedation and its complications. Suboptimal sedation levels can lead to various adverse effects, including hypercatabolism, immunosuppression, and unintended extubation, while excessive sedation may prolong mechanical ventilation and increase susceptibility to pneumonia and neuro-psychological impairment [9]. TDM of midazolam (as either a replacement or complement to clinical scores or neurophysiological monitoring) has been proposed to facilitate personalized treatment, ensure an appropriate depth of sedation, minimize toxicity, and address potential drug–drug interactions and comorbidities that affect drug metabolism and elimination [9]. However, TDM for midazolam is complex due to its active metabolite, 1-OH-M, necessitating the measurement of both drug and metabolite concentrations for a comprehensive assessment of effect. Clinically useful TDM requires a strong correlation between plasma concentration and clinical effect. While population-level data show a general correlation between plasma concentrations of midazolam and its active metabolite and sedative effect [10], significant individual variability complicates the prediction of sedation levels at specific midazolam concentrations in individual patients [9,10,11]. Generally, achieving deep sedation, defined as patients being asleep and reacting to pain induction, requires midazolam levels spanning a wide range of 100–2400 ng/mL in individual patients [12]. Therefore, midazolam does not meet a prerequisite for effective TDM, namely low interindividual pharmacokinetic and pharmacodynamic variability, and thus a clear concentration–response relationship. As a result, midazolam is not a drug for which TDM has demonstrated clinical value and is frequently monitored.
The challenge in correlating targeted plasma levels of midazolam with specific degrees of sedation is attributed to inter-subject variability in midazolam pharmacokinetics and pharmacodynamics, influenced by factors such as polypharmacy with drug interactions and complex metabolism, particularly in critically ill patients [9,10,11]. The subsequent section of the review explores factors influencing the pharmacokinetics and pharmacodynamics of midazolam, collectively contributing to inter-subject variability.
3. Transport and Metabolism of Midazolam
The ATP-binding cassette transporter P-glycoprotein (P-gp) is expressed on the apical membrane of mucosal cells in the intestine. Functioning as an ATP-dependent efflux pump with broad substrate specificity, P-gp facilitates the normal excretion of xenobiotics back into the gut lumen, consequently reducing the oral bioavailability of certain pharmaceutical drugs. The substrate specificities of CYP3A and P-gp are thought to overlap. Therefore, experimental studies have been performed to determine whether midazolam is a P-gp substrate, with conflicting results [13,14]. Takano et al. observed no inhibitory effect of the P-gp inhibitor verapamil on the basal-to-apical transport of midazolam in Caco-2 cells and concluded that P-gp is not involved in the transport of midazolam in the intestinal epithelia [13]. A more detailed investigation using a P-gp ATPase assay, efflux inhibition studies, and transport studies of midazolam across MDR1-MDCK and 1-α,25-dihydroxy vitamin D3-induced Caco-2 monolayers with and without the P-gp inhibitor GF120918 revealed that midazolam has the characteristics of a highly permeable P-gp substrate [14]. Substrates with high permeability, such as midazolam, exhibit rapid transmembrane movement, leading to significantly faster permeation into intestinal epithelial cells than P-gp efflux [14]. Therefore, the transport of highly permeable drugs remains largely unpolarized, suggesting that P-gp does not significantly impact their oral bioavailability [15]. Another class of transport proteins, known as membrane solute carrier transporters, typically plays a critical role in the cellular uptake of drugs. However, experiments conducted in Xenopus laevis oocytes or HEK293 cells expressing solute carriers revealed that midazolam did not function as a substrate for any of the tested transporters, including the organic anion transporters OAT1, OAT2, and OAT3, the organic cation transporters OCT1 and OCT2, and the organic-anion-transporting polypeptides Oatp1b2, OATP1A2, OATP1B1, OATP2B1, and OATP1B3 [16,17]. No study to date has investigated whether transport proteins are involved in the distribution or excretion of midazolam metabolites, some of which are also pharmacologically active [18].
Despite its rapid absorption following oral administration, the bioavailability of midazolam is relatively low at 44% due to a significant first-pass effect. Midazolam exhibits 94% plasma protein binding, and only 0.5% is excreted unchanged [19]. Because of extensive hepatic metabolism, hepatic impairment reduces the clearance of midazolam, leading to a subsequent increase in its terminal half-life. In a study comparing patients with liver cirrhosis to healthy controls, oral bioavailability significantly increased (76% vs. 38%), while midazolam elimination was notably delayed, demonstrated by lower total clearance (3.34 vs. 5.63 mL/min/kg), a lower total elimination rate constant (0.40 vs. 0.72 h-1), and a longer elimination half-life (7.4 vs. 3.8 h) [20]. Consequently, drug labels include a warning that clinical effects in patients with hepatic impairment may be more pronounced and prolonged, recommending dose reductions and the careful monitoring of vital signs in such patients.
The first step in the metabolism of midazolam is hydroxylation. The 1-OH-M metabolite comprises 60% to 70% of the biotransformation products of midazolam, while 4-hydroxy-midazolam (4-OH-M) constitutes 5% or less. Small amounts of a dihydroxy derivative, 1,4-OH-M, have also been detected. Hydroxylation is catalyzed by the enzymes of the CYP3A subfamily, including CYP3A5, CYP3A4, and CYP3A7 (Figure 1). However, CYP3A7 is only expressed in fetal tissue and therefore only plays a minor role later in life. Studies on drug–drug interactions involving potent CYP3A inhibitor drugs such as clarithromycin or ketoconazole underscore the significance of CYP3A enzymes in midazolam metabolism. A single dose of midazolam co-administered with oral clarithromycin (500 mg twice daily for 4 days) increased the midazolam AUC0–∞ by 219% after intravenous midazolam administration and 550% after oral midazolam administration [21]. Co-administration of a single dose of midazolam with oral ketoconazole (400 mg daily for 5 days) resulted in a 1296% increase in midazolam AUC0–∞ after the oral administration of midazolam [22].
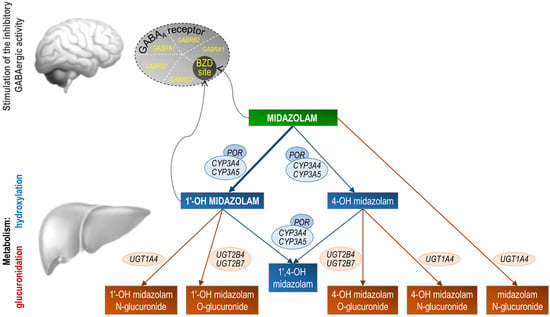
Figure 1.
Stylized representation of midazolam pharmacokinetics and pharmacodynamics. Midazolam is primarily metabolized in the liver and gut by human cytochrome P450 3A (CYP3A4) enzymes to its pharmacologic active metabolite, 1′-OH midazolam (which accounts for approximately 70% of the biotransformation products), followed by glucuronidation of the 1′-hydroxyl metabolite. Studies in humans suggest that 1′-OH midazolam is as potent as the parent compound at the GABAA receptor. Conversely, 4-OH-M’s low receptor affinity (7% compared to midazolam) and limited formation (less than 5% of midazolam’s biotransformation products) suggest it has negligible contribution to the overall activity of midazolam. Genes coding for the midazolam metabolic enzymes or the subunits that form the midazolam target receptor are shown. POR, cytochrome P450 oxidoreductase; UGT, UDP-glucuronosyltransferase; GABRA1, GABAA receptor α1 subunit; GABRB2, GABAA receptor β2 subunit; GABRG2, GABAA receptor γ2 subunit; BZD site, benzodiazepine binding site at the GABAA receptor.
The CYP3A4 and 3A5 enzymes, which share approximately 85% sequence homology, have overlapping substrate specificity. However, the affinity for CYP3A4 over CYP3A5 varies among CYP3A substrates. Using human liver microsomes, Tseng et al. showed in vitro that CYP3A5 contributes 55% and 44% to the 1′- and 4-hydroxylation of midazolam, respectively, suggesting that midazolam 1′-hydroxylation is more specific for CYP3A5 [23]. However, the in vitro data do not correspond to human in vivo data. In humans, there are individuals known as CYP3A5 expressors, who have functional CYP3A5 enzymes, and non-expressors, who lack functional CYP3A5 enzymes due to genetic variations. CYP3A5 non-expressors compared to expressors have been used to estimate the relative contribution of CYP3A4 and CYP3A5 for midazolam clearance. Among healthy volunteers, the pharmacokinetics of intravenously administered midazolam did not differ between CYP3A5 expressors and non-expressors, suggesting that hepatic CYP3A5 generally plays a minor role in midazolam metabolism [24]. However, the inhibition of CYP3A4 resulted in decreased midazolam clearance, specifically in CYP3A5 non-expressors compared to CYP3A4 expressors [24], indicating that CYP3A5 serves as an alternative metabolic pathway for midazolam, particularly when CYP3A4 is not functional.
1-OH-M is the major metabolite of midazolam and has been shown to account for 95% of the net intrinsic clearance of midazolam in human liver microsomes [25]. After hydroxylation, midazolam is further metabolized in the second step by the UDP-glucuronosyltransferases (UGTs) 1A4, 2B4, and 2B7, producing 1′-hydroxy-midazolam-glucuronide (1-OH-MG) as the main metabolite [26]. The amount of 1-OH-M excreted as a conjugate in the urine of healthy volunteers exceeded 75% of the initially administered dose compared to 4% for the minor primary metabolite 4-OH-M and 6% for the second minor secondary metabolite 1′,4-dihydroxy-midazolam. Approximately 1–2% is also excreted directly as glucuronylated midazolam generated by UGT1A4 [27,28].
4. Pharmacokinetics and Pharmacodynamics of Midazolam Metabolites
Several studies have explored the pharmacological activities of certain midazolam metabolites. The primary metabolite, 1-OH-M, has been shown to exhibit similar acute pharmacodynamic effects as midazolam in studies involving healthy volunteers [29,30], thus contributing to midazolam’s overall pharmacological activity. In a randomized, double-blind, crossover trial, Mandema et al. evaluated the pharmacodynamic response using saccadic eye movement and electroencephalographic (EEG) measurements in eight healthy volunteers following the intravenous administration of midazolam or 1-OH-M. Concentration–effect relationships of midazolam and its main metabolite, 1-OH-M, were found to be comparable [30]. Consistent with these findings, in vitro binding studies demonstrated that the affinity of 1-OH-M for the cerebral benzodiazepine receptor (with an affinity constant of 2.2 nmol/L) closely resembled that of midazolam (1.4 nmol/L) [31]. However, the elimination half-life of the metabolite 1-OH-M is shorter than that of midazolam, approximately 1 h versus 1.5 to 3.0 h [32,33]. This faster inactivation of 1-OH-M results in a quicker decrease in its pharmacodynamic effect compared to midazolam. This aligns with the findings of Johnson et al., who conducted a pharmacokinetic–pharmacodynamic analysis of single-dose oral midazolam and concluded that the metabolite 1-OH-M contributed approximately half to the sedative effect compared to the parent drug [34]. The analysis was based on study data on the pharmacokinetics of midazolam and its active 1-OH metabolite and their contribution to the sedative effect in 45 children undergoing day surgery [34].
The affinity of 4-OH-M for the benzodiazepine receptor was found to be only 7% compared to midazolam. Therefore, considering that 4-OH-M accounts for 5% or less of the biotransformation products of midazolam, this metabolite is unlikely to substantially contribute to the overall pharmacologic activity of midazolam. The binding affinity of 1-OH-MG to the cerebral benzodiazepine receptor was only about 10 times weaker than that of midazolam; therefore, this glucuronidated metabolite may also contribute to the overall pharmacological activity, especially if it accumulates [31].
Franken et al. demonstrated that the clearance of 1-OH-MG correlates with the estimated glomerular filtration rate, which serves as an indicator of renal function, suggesting that 1-OH-MG is mainly excreted via the kidneys [35]. Serum concentration monitoring in patients with severe renal failure showed high concentrations of 1-OH-MG, whereas the concentrations of the unconjugated metabolite and the parent drug were below the sedative range or even below the detection range. Moreover, the accumulation of 1-OH-MG has been associated with the prolonged sedative effects frequently seen in critically ill patients with renal impairment [31]. Accordingly, the drug labels include a warning that in patients with renal impairment, the excretion of midazolam and its metabolites may be slowed, which may result in prolonged sedation possibly including clinically relevant respiratory and cardiovascular depression, but they do not provide specific dose adjustment recommendations.
5. Effect of Sex and Nutritional Status on the Pharmacokinetics of Midazolam
Sex influences numerous pharmacokinetically important parameters, including the expression of drug-metabolizing enzymes and transporters [36,37]. Analyses of CYP3A4 in the human liver have shown that relative mRNA expression was significantly higher in females than in males, by approximately 26% [38], and that CYP3A4 protein levels are approximately 2-fold higher in female than in male liver tissue [38,39,40,41]. Men appear to have higher activity than women for some CYP isoenzymes such as CYP3A5, for the drug efflux transporter P-gp, and for some UGT isoforms such as UGT2B4 [42,43]. Many clinical studies have indicated that women metabolize CYP3A4 substrates such as midazolam more rapidly than men [44]. In a phase I clinical trial, Tsunoda et al. observed that female subjects exhibited a mean midazolam clearance 1.6-fold higher than male subjects when adjusted for weight [45]. Notably, this difference disappeared in the presence of the CYP3A inhibitor ketoconazole, confirming a contributory role of sex differences in CYP3A activity to the observed variations in midazolam kinetics [45]. One endogenous factor that regulates the sexually dimorphic expression of hepatic CYP3A4 is growth hormone (GH), which exhibits a sexually dimorphic pattern of secretion. The masculine GH profile is episodic, while the feminine profile is continuous. Masculine-like episodic GH profiles suppress CYP3A4 expression, whereas feminine-like continuous GH profiles induce it [46,47].
In a meta-analysis of ten studies involving 409 healthy volunteers, women showed a 16% higher weight-corrected midazolam oral clearance and 20% higher systemic clearance than men, although no significant difference in the AUC after the oral dosing of midazolam was noted between the sexes [48]. Population pharmacokinetic modeling, utilizing data from 13 studies including 138 healthy volunteers, indicated that women displayed an 11% higher mean weight-corrected total body midazolam clearance and a 28% higher oral clearance compared to men [49]. Despite the significantly greater midazolam clearance in women, the overall magnitude of observed disparity, however, may have negligible clinical implications.
Nutritional status is another factor that potentially influences CYP3A4 activity and thus midazolam metabolism [50]. In a randomized, controlled, cross-over study, 36 h fasting altered midazolam metabolism in different ways. Here, CYP3A4-mediated midazolam metabolism increased by 12%, while UGT-mediated metabolism decreased by 13% [51]. The findings indicate that short-term fasting diminishes the activity of UGT phase II drug-metabolizing enzymes, reflected by reduced UGT-mediated midazolam metabolism. Conversely, it augments the activity of the phase I drug metabolizing enzyme CYP3A4, as evidenced by the increased CYP3A4-mediated metabolism of midazolam. The human studies align with experimental findings showing that short-term fasting significantly increases mRNA expression of the orthologues of human CYP3A4 in both rats and mice [52,53]. A high-fat diet did not affect the systemic clearance of midazolam or its metabolites in a study with healthy volunteers [54]. In terminally ill patients receiving midazolam, hypalbuminemia, possibly due to a catabolic state, was significantly associated with decreased midazolam clearance [35].
Obesity is a factor that may influence the pharmacokinetics of midazolam, as demonstrated in a study comparing normal weight (n = 12) and obese patients (n = 20, body mass index 40–68 kg/m2) [55]. While the clearance of midazolam did not show a significant difference between the groups, the mean half-life was notably greater in the obese group (5.9 versus 2.3 h). This was attributed to an approximately 50% increase in the volume of distribution corrected for total body weight [55].
6. Impact of Inflammation on the Pharmacokinetics of Midazolam
During acute or chronic inflammation, released proinflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 act as signaling molecules to elicit marked changes in liver gene expression profiles and lead to the strong downregulation, and activity reduction, of many drug-metabolizing enzymes, including CYP3A4 [56,57,58,59]. A negative correlation between plasma TNF-α concentrations and midazolam clearance was found in patients with chronic inflammation [60], and midazolam clearance correlated with plasma levels of the inflammatory marker C-reactive protein in critically ill patients with acute inflammation [61]. In patients with severe inflammatory syndrome due to acute respiratory syndrome-coronavirus 2, C-reactive protein elevation was significantly associated with a decrease in the 1-OH-M/midazolam plasma ratio, suggesting reduced metabolism of midazolam by CYP3A [62].
The mechanism behind these effects is at least partly attributed to the transcriptional repression of CYP3A4 [57,58,63]. For instance, the treatment of human hepatocytes with IL-6 in vitro resulted in a decreased expression of CYP3A4 at both mRNA and protein levels, along with reduced enzymatic activity [64]. Experimental studies suggest that IL-6 down-regulates CYP3A4 through translational induction of the CCAAT/enhancer binding protein (C/EBP) β-LIP isoform, which competes with and antagonizes constitutive C/EBP transactivators [63]. Protein expression of the transcription factor C/EBPβ occurs by alternative in-frame translation initiation at consecutive start sites from a single transcript to generate three isoforms, two long and one truncated liver inhibitory protein (LIP). The long C/EBPβ isoforms are considered gene activators, whereas the LIP isoform reportedly acts as a dominant-negative repressor [63].
The inflammatory cytokines IL-1β and TNF-α or bacterial lipopolysaccharides activate nuclear factor kappa B (NF-κB), which can strongly inhibit human CYP3A4 expression [65]. Gene silencing experiments in primary human hepatocytes indicate that retinoid X receptor α (RXRα) plays a central role in mediating transcriptional downregulation [56,66]. During inflammation, endotoxin prompts the rapid loss of nuclear-localized RXRα [66]. RXRα serves as a necessary heterodimerization partner for several key ligand-activated DNA-binding transcription factors, including pregnane X receptor (PXR), which regulates the expression of its target gene, CYP3A4, by binding to the gene’s promoter [56]. Experimental studies showed that the p65 subunit of NF-κB directly interacts with the DNA-binding domain of RXRα and prevents its binding to the consensus DNA sequences, thus inhibiting transactivation by the PXR/RXRα complex [65].
7. Genetic Factors—CYP3A Polymorphisms
The substrate specificities of CYP3A4 and CYP3A5 generally overlap, but the relative contribution of the metabolic capability of CYP3A5 compared with that of CYP3A4 is either reduced or, at best, equal depending on the substrate. CYP3A4 is the major contributor to midazolam metabolism, and CYP3A5 becomes relevant only when CYP3A4 activity is impaired [24]. Studies in human liver microsomes have shown that both CYP3A4 and CYP3A5 contribute to the metabolism of midazolam, but biotransformation with CYP3A5 is metabolically less active than that with CYP3A4 [67].
CYP3A5 expression is polymorphic, with 25 allelic variants of CYP3A5 listed on the Pharmacogene Variation Consortium website [68,69]. The most common nonfunctional variant, CYP3A5*3 (rs776746), results from a change from A to G at position 6986 of the gene, creating a cryptic splice site in intron 3. This alteration leads to abnormal mRNA splicing, generating an alternatively spliced isoform with an insertion from intron 3. Consequently, a premature termination codon occurs, rendering the protein nonfunctional [69]. Individuals with the CYP3A5*3/*3 genotype are considered non-expressors. The frequency of this genotype varies significantly among populations. In White Europeans, the allele frequency of CYP3A5*3 is approximately 0.94, with around 89% of the population carrying two 3 alleles and thus being non-expressors. In contrast, only 42% of Asians and 29% of Africans are non-expressors, i.e., carry the CYP3A5*3/*3 genotype. There is no difference in 1-OH-M formation between CYP3A5 expressers and non-expressers, suggesting that the 1′-OH hydroxylation of midazolam is primarily catalyzed by CYP3A4 [70,71,72]. Moreover, the pharmacodynamics of midazolam are not affected by the CYP3A5*3 variant. This was demonstrated in a prospective study involving 100 Korean patients undergoing upper gastrointestinal endoscopy under sedation with midazolam, where the sedation grade showed no association with the CYP3A5*3 genotype [71]. Similarly, in a cohort of 71 Caucasian patients receiving prolonged sedation during intensive care treatment, no link between the CYP3A5*3 genotype and sedation depth, as assessed by the Ramsay score, was identified [72].
Two other commonly studied CYP3A5 loss-of-function alleles, *6 (rs10264272) and *7 (rs41303343), result in nonfunctional proteins due to aberrant splicing, with the absence of exon 7 and a frameshift leading to premature termination, respectively. These alleles are relatively common in African populations (0.12–0.15) but are rare or absent in White and Asian populations (0–0.003) [73].
CYP3A4 activity can vary significantly among individuals, by up to a factor of 100 [74]. However, unlike the distribution of CYP2D6 activity in the population, which exhibits multiple peaks allowing for clear differentiation between extensive and poor or rapid metabolizers, the distribution of CYP3A4 activity in the population is continuous and unimodal [75,76]. This finding suggests that the regulation of CYP3A4 activity involves multiple genes, with individual genetic factors playing a minor role [75]. As a result, current FDA drug labeling does not provide information on genomic biomarkers of CYP3A4 [77].
To date, approximately 9800 SNPs have been identified in the CYP3A4 gene (dbSNP database), with 35 core alleles currently listed by PharmVar [78]. The functional implications of some of these core alleles are not yet clear. Using the allele definitions issued by PharmVar, the Royal Dutch Association for the Advancement of Pharmacy—Pharmacogenetics Working Group listed the following allele function assignments and phenotype mapping results: normal function of *1 suballeles including *1A, *1B, and *1G; decreased function of alleles *8, *11, *12, *13, *16, *17, *18, and *22; complete loss of function of alleles *6, *20, and *26 [2,76,79].
The extensively studied subvariant CYP3A4*1B appears to have no or very limited functional impact on CYP3A4 [80], contrary to some initial reports influenced by its linkage disequilibrium with CYP3A5*1. These early reports suggest that the CYP3A4*1B allele (defined by the promoter SNP rs2740574, −392A>G) exhibits greater enzymatic activity than the wild-type allele, CYP3A4*1A, resulting in an increased systemic clearance of midazolam [81]. However, in approximately 80% of Caucasians carrying CYP3A4*1B, this allele is in linkage disequilibrium with the fully active *1 allele of CYP3A5. Therefore, the clinical phenotype initially associated with CYP3A4*1B is likely attributable to CYP3A5 activity [82].
The CYP3A4*2 allele, characterized by the c.664T>C SNP, results in a serine-to-proline amino acid residue exchange at codon 222, potentially altering the three-dimensional structure of CYP3A4 because the proline residue is a known helix breaker [83]. Two in vitro studies have linked this allele with a marked functional defect in CYP3A4 activity [84,85], demonstrating reduced activity toward midazolam in vitro, although its clinical relevance remains undemonstrated.
For CYP3A4*3, an allelic frequency of approximately 1% has been reported, but unlike CYP3A4*2, no correlation with enzyme activity has been identified [83,86,87]. There have been no clinically significant associations reported for CYP3A4*3, raising doubts about its functional role, particularly when compared to CYP3A4*2.
The CYP3A4*4, CYP3A4*5, and CYP3A4*6 alleles were initially discovered in 102 Chinese subjects, with allelic frequencies of 1.5%, 0.98%, and 0.5%, respectively, all linked to reduced CYP3A4 activity as indicated by the urinary 6β-hydroxycortisol-to-free cortisol ratio [88]. Like Hsieh et al.’s study [88], Wang et al. found that the CYP3A4*4 allele was associated with decreased CYP3A4 activity, resulting in an enhanced lipid-lowering effect of the CYP3A4 substrate simvastatin [89]. To date, the CYP3A4*4 allele has only been identified in the Chinese population [2,90,91]. The CYP3A4*6 allele (rs4646438, insertion c.830dupT) causes a frameshift and loss of enzymatic activity and is found in all populations, albeit with a very low allele frequency.
CYP3A4*7 (p.Gly56Asp), CYP3A4*8 (p.Arg130Gln), CYP3A4*9 (p.Val170Ile), CYP3A4*10 (p.Asp174Asn), CYP3A4*11 (p.Thr363Lys), CYP3A4*12 (p.Leu373Phe), CYP3A4*13 (p.Pro416Arg), and CYP3A4*20 (a frameshift variant with a premature stop codon) have been identified in Caucasian DNA samples. However, while some of these alleles have been associated with altered CYP3A4 activity in vitro, their reported allelic frequencies are very low [2,86,87,92]. Other coding SNPs such as CYP3A4*14 (p.Leu15Pro), CYP3A4*15 (p.Arg162Gln), CYP3A4*16 (p.Thr185Ser), CYP3A4*19 (p.Pro467Ser), and CYP3A4*21 (p.Tyr319Cys) are rare, particularly in Caucasians [2,86,93].
The CYP3A4*17 (p.Phe189Ser) allele is linked to reduced clearance of certain substrates, including midazolam, testosterone, and nifedipine, compared to the CYP3A4 wild-type allele [83,94]. The two total loss-of-function variants CYP3A4*20 (frameshift variant, c.1461dup) and CYP3A4*26 (premature stop codon, p.Arg268Ter) were initially identified due to anomalous pharmacokinetics in certain individuals [92,95]. These genetic variants have a very low frequency in the global population (less than 0.01%), but their prevalence varies across different subpopulations. For instance, CYP3A4*20 is present in the Spanish population with an allele frequency as high as 1.2% [96].
The decreased-function CYP3A4*22 allele was first reported in 2011 [97] and is characterized by a C>T substitution at position 15,389 (rs35599367) in intron 6 of CYP3A4, resulting in alternative RNA splicing [97]. The effect of the CYP3A4*22 allele on hepatic expression was confirmed by measuring total CYP3A4 mRNA levels in liver samples. Livers with the wild-type genotype, CYP3A4*1/*1 (CC), exhibited 1.7-fold higher mRNA levels than those from carriers of the CYP3A4*22 (CT or TT) variant allele. The CYP3A4*22 allele accounted for 7% of the variability in CYP3A4 mRNA expression. In additional microsomal liver samples, CYP3A4 activity, as assessed by the testosterone 6β-hydroxylation rate, was 2.4-fold higher in individuals with CYP3A4*1/*1 than in heterozygous CYP3A4*22 samples. The authors concluded that the presence of the CYP3A4*22 allele reduces both CYP3A4 liver expression levels and enzyme activity [97]. The SNP explained up to 12% of the variability observed in CYP3A4 activity. The association of CYP3A4*22 with reduced metabolic activity has been demonstrated for several CYP3A4 substrates, including midazolam [70,76]. Midazolam levels increased by 32% in individuals carrying the CYP3A4*22 variant compared to those in patients with the homozygous wild-type genotype [70]. This genotype-specific difference in CYP3A4 activity also affects metabolite formation. Plasma concentrations of the metabolite 1-OH-M and the corresponding metabolic ratio (the ratio of the metabolite to unchanged midazolam) were 19.8% and 21.8% lower, respectively, in carriers of the CYP3A4*22 variant compared to wild-type patients [70]. Overall, the intronic CYP3A4*22 variant appears to be the most relevant single CYP3A4 variant influencing CYP3A4 activity, with a minor allele frequency of 5% in Europeans and approximately 3% in admixed Americans, but it is rare in many other populations [98]. Accordingly, the Association for Molecular Pathology Clinical Practice Committee’s Pharmacogenomics Working Group currently recommends the inclusion of CYP3A4*22 together with CYP3A4*20, CYP3A5*3, *6, and *7 alleles as a panel of variant alleles in clinical pharmacogenetic genotyping assays for CYP3A substrate drugs [99]. While the impact of certain CYP3A4 genetic polymorphisms on midazolam pharmacokinetics, notably the effect of CYP3A4*22 on the 1-OH-M/midazolam metabolic ratio, has been demonstrated, their effect on pharmacodynamics, such as the depth of sedation, has not yet been investigated and requires further establishment.
Although many SNPs within the CYP3A locus have been identified, these variants appear to explain only a minority of the phenotypic variability. This was demonstrated in a study involving 21 healthy subjects genotyped for a broad panel of CYP3A4 and CYP3A5 alleles, where CYP3A activity was measured using midazolam as a probe substrate [100]. Importantly, the CYP3A genotypes did not adequately account for the interindividual variability in the activity of these two isozymes, highlighting the issue of “missing heritability” of CYP3A [100].
8. Transcriptional Regulation of CYP3A Genes
Significant (~50-fold) interindividual differences in CYP3A4 expression levels are observed in the human liver. There is growing consensus that this variability cannot be solely attributed to genetic polymorphisms in CYP3A4 but is instead influenced by polymorphisms/variability in CYP3A4 regulators [8,101,102]. The expression of CYP3A enzymes is known to be controlled by numerous signaling pathways and transcription factors. Constitutive transcriptional regulation involves transcription factors such as CCAAT/enhancer-binding protein α and β (C/EBPα and C/EBPβ) [63,103,104], hepatocyte nuclear factors (HNFs), particularly HNF1α, HNF3γ [103], and HNF4α [105,106,107], upstream stimulatory factors (USFs) [108], and estrogen receptor alpha (ESR1) [109]. ESR1 has been shown to account for 63% and 42% of the variability in the constitutive expression of CYP3A4 and CYP3A5 in human liver samples, respectively, indicating its significant role as a regulator of CYP3A expression [110].
CYP3A4 induction by endo- and xenobiotics primarily relies on the pregnane X receptor (PXR, alternate names: SXR, PAR, and NR1I2), and, to a lesser extent, the constitutive androstane receptor (CAR, NR1I3). Upon binding to its ligand (which can be a drug or xenobiotic), PXR or CAR undergoes a conformational change enabling it to form a heterodimer with the RXR. This dimeric complex, upon translocation from the cytoplasm into the nucleus, binds to responsive elements in the CYP3A4 promoter region, thereby regulating its transcription. These responsive elements, including the proximal PXR responsive element, the distal xenobiotic-responsive enhancer module (XREM), and the far distal enhancer module (FDEM), consist of various repeats of the consensus motif, AG(G/T)TCA [107,111,112,113]. The repeats can be arranged in different configurations such as direct repeats (DR) separated by 3–5 nucleotides (DR3, DR4, or DR5) or everted repeats (ER) separated by 6 or 8 nucleotides (ER6 or ER8). The proximal PXR responsive element contains an ER6 [114], while the distal XREM contains a DR3 [115] and the FDEM contains an ER6 motif [116].
PXR, predominantly expressed in the liver and small intestine, serves as a crucial xenosensor, playing a significant role in the pharmacokinetics of CYP3A4 substrate drugs such as midazolam. For instance, in healthy volunteers exposed to the known PXR agonist and CYP3A4 transcriptional inducer rifampicin, the AUC for the CYP3A4 substrate midazolam decreased to 2.3% of the AUC of unexposed individuals [3].
Genetic variants within transcriptional regulator genes also play a role in modulating CYP3A expression levels. Lamba et al. screened potential regulatory regions in the PXR gene NR1I2 for sequence variations that could explain the observed variability in the hepatic expression of PXR and its target CYP3A4 [117]. NR1I2 SNPs were genotyped in human liver donors characterized for CYP3A4 mRNA expression and in primary human hepatocytes assessed for both basal and rifampin-inducible CYP3A4 activity. Among the identified SNPs, those consistently associated with phenotypic measures of CYP3A4 included a promoter SNP 44477T>C (−1359), SNP 63396C>T within intron 1, and SNPs 56348C>A, 69789A>G, and 66034T>C [117]. Liver donors harboring variant NR1I2 alleles exhibited altered hepatic expression of the PXR-regulated gene, CYP3A4, in comparison to those with wild-type NR1I2 alleles. However, the impact of these polymorphisms on interindividual pharmacokinetic variations of midazolam remains unexplored.
The influence of the ligand-activated transcription factors PXR and CAR extends beyond CYP3A4, as they serve as transcriptional activators for at least 40 genes involved in drug transport and phase I/II metabolism [94]. These factors have been implicated in the transcriptional activation of genes crucial to the pharmacokinetics of midazolam, including cytochrome P450 oxidoreductase (POR) [118], CYP3A4 [119], CYP3A5 [120], UGT1A4 [121], and UGT2B7 [122]. PXR is activated by a wide range of xenobiotics, including antibiotics, antimycotics, herbal components [123], and benzodiazepines such as medazepam and midazolam [124]. PXR activation has been shown to induce not only the midazolam-metabolizing phase I enzyme CYP3A4 but also the phase II enzymes UGT1A4 and UGT2B7. While most benzodiazepines do not affect PXRs, midazolam exhibits slight PXR activation and weak induction of CYP3A4 mRNA in human hepatocytes [124].
Other ligand-dependent transcriptional regulators of CYP3A4 include the bile acid receptor FXR [125], the glucocorticoid receptor [126], the liver X receptors (LXRs) [127], and the vitamin D receptor (VDR) [128]. Several studies have suggested that peroxisome proliferator-activated receptor alpha (PPARα) also contributes to the constitutive and inducible regulation of CYP3A4. PPARα SNPs have been shown to influence hepatic CYP3A4 phenotypes [129,130]. Additional experiments demonstrated approximately a 3-fold induction of CYP3A4 in human hepatocytes by a potent PPARα agonist, while repression occurred with a PPARα antagonist and short hairpin RNA (shRNA)-mediated PPARα knockdown. A transcriptional profiling study [131] also underscored the impact of the lipid homeostasis regulator PPARα on CYP3A4.
In addition, NF-κB activation by inflammatory cytokines can potently suppress human CYP3A4 expression [65], and, conversely, PXR activation suppresses inflammation through interaction with the NF-κB pathway [132]. The cytokine-mediated downregulation of CYP3A4 during inflammation via the JAK/STAT pathway is clinically significant [63]. This process is particularly relevant in cancer patients because tumors can serve as sources of cytokines, leading to the substantial downregulation of CYP3A4 and other drug-metabolizing enzymes and transporters [58,133].
9. Regulation of CYP3A4 by Short Non-Coding Regulatory MicroRNAs
MicroRNAs (miRNAs) are approximately 22-nucleotide-long RNA molecules encoded by the genome, playing pivotal roles as gene expression regulators in eukaryotes. They exert post-transcriptional control by binding to target mRNAs, thereby modulating their translation or stability. Through targeting multiple mRNAs, individual miRNAs possess the ability to finely adjust or regulate gene expression within specific pathways. This regulatory versatility of miRNAs is particularly significant in xenobiotic metabolism, where the dynamic adaptation to environmental cues is crucial for metabolizing and eliminating substances. Indeed, studies have demonstrated the involvement of miRNAs in modulating the expression of genes involved in drug metabolism [119,120]. For example, CYP3A4 was shown to be directly regulated by the miRNA miR-27b [134] and lower miR-27b levels were associated with higher CYP3A activity [135]. Nuclear receptors also serve as targets for miRNAs, as evidenced by the control of PXR by miR-148a, which influences CYP3A4 and CYP2B6 expression levels and the metabolism of xenobiotic drug substrates [136]. Additionally, HNF4α is regulated by miR-24 and miR-34a, with the overexpression of these miRNAs leading to reduced HNF4α levels and the downregulation of its target genes [137]. The vitamin D receptor (VDR), another transcriptional regulator of CYP3A4, is subject to regulation by the CYP3A4-targeting miR-27b, thereby establishing both indirect and direct mechanisms for the miRNA regulation of CYP3A4 [134]. The same miRNA, miR-27b, also modulates PPARγ [138,139], while LXR has been shown to be regulated by miR-613 [140], further supporting the important role of miRNAs in hepatic gene regulation. Of particular interest for pharmacogenetic considerations are SNPs in miRNAs and miRNA binding sites, as well as miRNA copy number variations, which could impact their expression and function, thereby influencing target gene expression [141,142].
10. Genetic Factors—UGT1A Polymorphisms
The variability in midazolam clearance among subjects may not solely stem from variations in CYP3A4 but also from differences in N-glucuronidation capacity. UGT1A4 is primarily expressed in the liver and participates in the phase II metabolism of midazolam [27,28]. Pharmacokinetic modeling has indicated that UGT1A4 plays a role in metabolism, especially in scenarios where CYP3A is inhibited, like CYP3A5 deficiency or the inhibition of CYP3A4 by interacting drugs [27]. Consequently, direct N-glucuronidation of midazolam might partly counterbalance the decrease in metabolic clearance caused by impaired CYP3A4 and/or CYP3A5 function [27,28].
Variability in the expression or activity of UGT1A4 could result in differences in drug response and toxicity among individuals [143]. Aueviriyavit et al. observed 2.5-fold interindividual differences in the expression levels of UGT1A4 mRNA in human livers, attributed in part to genetic polymorphisms [144]. Currently, the Pharmacogenomics Laboratory (Université Laval) website lists eight core alleles, *1 to *8, for UGT1A4 [145]. Among the extensively studied alleles are UGT1A4*2, characterized by a proline-to-threonine amino acid change at codon 24 due to a single C>A substitution, and UGT1A4*3, which leads to a leucine-to-valine amino acid change at codon 48 caused by a single T>G substitution [146]. These two allele-defining variants are located in the first exon of the UGT1A4 gene and could alter enzyme activity, with UGT1A4*3 potentially increasing and UGT1A4*2 decreasing glucuronidation.
In vitro studies examining the effect of UGT1A4*2 on the metabolism of UGT1A4 substrates, such as olanzapine, clozapine, and lamotrigine, suggested that UGT1A4*2 might reduce enzyme activity [147,148,149]. Clinical investigations into the effects of UGT1A4*2 on the metabolism of UGT1A4 substrates have yielded less definitive results, showing, at most, a trend toward higher serum concentrations of the substrate lamotrigine [150]. Conversely, clinical studies have found that the UGT1A4*3 variant is linked to increased enzyme activity, resulting in decreased serum concentrations or an elevation in the glucuronidation rate of substrate drugs [150,151,152,153]. To date, no clinical study has explored the impact of UGT1A4*2 or UGT1A4*3 polymorphisms on midazolam or 1-OH-M metabolism.
11. Genetic Factors—Polymorphisms in GABAA Receptor Genes
Benzodiazepines primarily exert their pharmacodynamic effects by augmenting the activity of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) in the central nervous system. This enhancement results in increased GABAergic neurotransmission, leading to sedative, anxiolytic, muscle relaxant, and anticonvulsant effects. Benzodiazepines bind to specific sites on GABAA receptors, which are ligand-gated ion channels, thereby increasing the frequency of chloride channel opening. This hyperpolarizes neurons and suppresses neuronal excitability. GABAA receptors in the brain generally consist of two α, two β, and one γ subunit. While the GABA binding site is located at the α-subunits, precisely at the contact site with the β-regions, benzodiazepines bind at the contact site of the α-subunit with the γ-subunit. However, not every α-subunit can form a binding site for benzodiazepines with the γ-subunit; only α1, α2, α3, and α5 can do so. Approximately 60% of GABAA receptors have the α1β2γ2 configuration, while the combination α2β3γ2 accounts for approximately 15–20%, and the α3βnγ2 complex occurs in approximately 10–15%. Together, these three combinations make up 90% of all GABAA receptors in the CNS. Limited knowledge exists regarding the potential influence of polymorphisms in the GABAA receptor-encoding genes on the pharmacodynamics of midazolam. A study in an Asian population found that the intronic polymorphism rs4263535 in the GABAA receptor subunit α1 gene (GABRA1) was significantly associated with deeper sedation induced by intravenous midazolam [154]. However, this observation has not yet been validated in an independent cohort, and the underlying biological mechanism responsible for the genotype–phenotype association of this noncoding intronic variant has not been investigated. Consequently, the effect of the rs4263535 polymorphism or other polymorphisms in linkage disequilibrium on the expression or function of the GABAA receptor α1 subunit remains uncertain.
12. Epigenetics
Of particular interest is the epigenetic regulation of the important transcription factor and xenosensor PXR [136], whose functions are regulated at three epigenetic levels: by noncoding miRNAs, as previously described, through chromatin modifications, and via DNA methylation. Chromatin modifications are carried out, in part, through interaction with coregulator complexes, which include steroid receptor coactivators, corepressors (NcoR/SMRT), HNF4α, PPARγ coactivator 1α, and protein arginine methyltransferase 1. PXR can undergo post-translational modifications such as acetylation, phosphorylation, and sumoylation, while its promoter region can be subject to methylation at the CpG island. These factors collectively determine PXR activity, thereby affecting the magnitude and duration of PXR-regulated drug metabolic responses [155].
13. Case Example
The following case report of prolonged midazolam-associated sedation is intended to illustrate the clinical and pharmacogenetic factors that should be considered in the work-up. A Caucasian normal weight male child aged 4 years and 7 months underwent elective adenotonsillectomy and was sedated preanesthetically with midazolam followed by a standard anesthesia protocol. Midazolam syrup (0.65 mg/kg) was administered orally 40 min prior to surgery. Anesthesia was induced with 3.3 mg/kg of propofol before fentanyl administration (3.3 μg/kg). Muscle relaxation was achieved with mivacurium (0.2 mg/kg). General anesthesia was accomplished with desflurane supplemented with remifentanil (0.1 μg/kg/min). Additionally, 0.1 mg/kg ondansetron/0.15 mg/kg dexamethasone was used for the prophylaxis of postoperative nausea and vomiting, and 14 mg/kg dipyrone was used for postoperative pain management. The patient showed delayed recovery and prolonged respiratory depression for more than 5 h. Respiratory depression and sedation were reversed with 0.01 mg/kg flumazenil, suggesting that the delayed postanesthetic recovery was due to the effects of midazolam.
Delayed recovery from sedation is occasionally observed in critically ill patients due to liver failure and a decrease in the hepatic clearance of midazolam. Furthermore, it is well documented that the clearance of midazolam can be reduced by interaction with CYP3A-inhibiting drugs or food components (e.g., macrolide antibiotics, azole antimycotics, or grapefruit), resulting in prolonged sedation [156,157]. Our patient did not fall into either of the two categories described above, which poses challenges for differential diagnosis in such a rare case.
Despite the influence of environmental or clinical factors, genetics may influence the pharmacokinetics and pharmacodynamics of midazolam, as discussed above. In fact, a high degree of heritability was observed for hepatic CYP3A4 activity, as measured by midazolam plasma clearance [4]. We examined the patient’s germline DNA using whole-exome and targeted Sanger sequencing for the presence of functional SNPs or Indel variants that could explain the altered midazolam pharmacokinetics or pharmacodynamics. Candidate genes that may affect the pharmacokinetics or pharmacodynamics of midazolam include CYP3A, POR (the gene product, cytochrome P450 oxidoreductase, is a flavoprotein that donates electrons to microsomal P450 enzymes), UGTs, and genes encoding the five subunits of the GABAA receptor, the molecular target of midazolam [158] (Figure 1).
The patient was identified as a homozygous carrier of the CYP3A5*3 allele, i.e., as a CYP3A5 non-expressor. Therefore, the phase I metabolism of midazolam in our patient, as in 90–95% of all Europeans, depends on CYP3A4 activity [159]. Our patient, however, did not carry any of the previously described CYP3A4 decrease or loss-of-function alleles (e.g., *8, *11, *12, *13, *16, *17, *20, *22, *26). Moreover, no functional variants in POR or GABAA receptor-encoding genes were identified. However, we detected the missense variant rs139927449 in UGT1A4 (c.53T>A), which causes a Leu-to-His substitution at position 18 of the UGT1A4 precursor protein. This SNP is rare, with a minor allele frequency of only 0.00018 in the European population. The p.Leu18His polymorphic variant of UGT1A4 occurs within the signal peptide, 11 amino acids upstream of the cleavage site and the start of the mature protein. A bioinformatic analysis using SignalP 4.1 software indicated that the UGT1A4 p.Leu18His variant is associated with a marked decrease in the probability of signal peptide cleavage (41% vs. 83% of the wild-type UGT1A4 precursor protein). As shown for the more common p.Pro24Thr (UGT1A4 c.70C>T) variant, altered signal peptide cleavage may decrease the glucuronidation activity of the mutant UGT1A4 [160].
In conclusion, this case of prolonged midazolam-associated sedation highlights the factors crucial to consider during evaluation. Patient-specific variables, such as obesity, renal dysfunction, or hepatic dysfunction, can contribute to an extended elimination half-life of midazolam. Additionally, significant causes of prolonged midazolam-associated sedation include drug–drug interactions resulting from medications co-administered with midazolam, which are known to inhibit the CYP3A4 enzyme system. These medications belong to classes such as azole antimycotics, protease inhibitors, calcium channel antagonists, and macrolide antibiotics. For example, drugs like erythromycin or ketoconazole have been shown to markedly increase the AUC of orally administered midazolam by 3-fold or 15-fold, respectively. In the absence of the aforementioned factors, pharmacogenetic testing may be warranted. This case example is the first description of the application of whole-exome sequencing to identify genetic susceptibility factors associated with an adverse reaction to midazolam. Our results suggest that a rare mutation in UGT1A4 may be responsible for the decreased clearance of midazolam and its active metabolite 1-OH-M and thus for prolonged midazolam-associated sedation and respiratory depression. Because the metabolic pathways of benzodiazepines differ, it is possible that the patient could tolerate another benzodiazepine well. The functional relevance of the identified mutation, however, remains to be confirmed by experimental studies.
14. Conclusions
Although there is a correlation between the plasma concentrations of midazolam and its active metabolite 1-OH-M and the degree of sedation at the population level, predicting the extent of sedation in an individual based solely on these concentrations is impossible [161]. This unpredictability for individual patients stems from the high interpatient variability in midazolam pharmacokinetics. This review emphasizes the numerous factors influencing midazolam pharmacokinetics. The case example illustrates the complexity of identifying the cause of unexpected side effects of midazolam, often without reaching a definitive conclusion. Furthermore, the case highlights the issue of “missing heritability”, which is characteristic of pharmacogenetics involving CYP3A4.
Author Contributions
Conceptualization, O.Z. and J.-U.P.; case report, P.D.;. case genotyping, O.Z.; writing—original draft preparation, J.-U.P., P.D. and O.Z.; writing—review and editing J.-U.P. and O.Z. All authors have read and agreed to the published version of the manuscript.
Funding
Funded by the Brandenburg Medical School publication fund supported by the Ministry of Science, Research and Cultural Affairs of the State of Brandenburg.
Institutional Review Board Statement
Ethical review and approval were waived for this study because patient data were used for the case study after de-identification and anonymization.
Informed Consent Statement
Patient consent was waived because the participating patient cannot be identified.
Data Availability Statement
The data used to support the findings of the case report are available on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Altamimi, M.I.; Sammons, H.; Choonara, I. Inter-individual variation in midazolam clearance in children. Arch. Dis. Child. 2015, 100, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef]
- Backman, J.T.; Kivisto, K.T.; Olkkola, K.T.; Neuvonen, P.J. The area under the plasma concentration-time curve for oral midazolam is 400-fold larger during treatment with itraconazole than with rifampicin. Eur. J. Clin. Pharmacol. 1998, 54, 53–58. [Google Scholar] [CrossRef]
- Ozdemir, V.; Kalow, W.; Tang, B.K.; Paterson, A.D.; Walker, S.E.; Endrenyi, L.; Kashuba, A.D. Evaluation of the genetic component of variability in CYP3A4 activity: A repeated drug administration method. Pharmacogenetics 2000, 10, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, J.; Bonat, W.H.; Kerb, R.; Tzvetkov, M.V.; Strube, J.; Brunke, S.; Sachse-Seeboth, C.; Sehrt, D.; Hofmann, U.; von Bornemann Hjelmborg, J.; et al. Inherited and Acquired Determinants of Hepatic CYP3A Activity in Humans. Front. Genet. 2020, 11, 944. [Google Scholar] [CrossRef]
- Miao, J.; Jin, Y.; Marunde, R.L.; Gorski, C.J.; Kim, S.; Quinney, S.; Radovich, M.; Li, L.; Hall, S.D. Association of genotypes of the CYP3A cluster with midazolam disposition in vivo. Pharmacogenom. J. 2009, 9, 319–326. [Google Scholar] [CrossRef]
- He, P.; Court, M.H.; Greenblatt, D.J.; Von Moltke, L.L. Genotype-phenotype associations of cytochrome P450 3A4 and 3A5 polymorphism with midazolam clearance in vivo. Clin. Pharmacol. Ther. 2005, 77, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Floyd, M.D.; Gervasini, G.; Masica, A.L.; Mayo, G.; George, A.L., Jr.; Bhat, K.; Kim, R.B.; Wilkinson, G.R. Genotype-phenotype associations for common CYP3A4 and CYP3A5 variants in the basal and induced metabolism of midazolam in European- and African-American men and women. Pharmacogenetics 2003, 13, 595–606. [Google Scholar] [CrossRef]
- Nies, R.J.; Muller, C.; Pfister, R.; Binder, P.S.; Nosseir, N.; Nettersheim, F.S.; Kuhr, K.; Wiesen, M.H.J.; Kochanek, M.; Michels, G. Monitoring of sedation depth in intensive care unit by therapeutic drug monitoring? A prospective observation study of medical intensive care patients. J. Intensive Care 2018, 6, 62. [Google Scholar] [CrossRef]
- Bremer, F.; Reulbach, U.; Schwilden, H.; Schuttler, J. Midazolam therapeutic drug monitoring in intensive care sedation: A 5-year survey. Ther. Drug Monit. 2004, 26, 643–649. [Google Scholar] [CrossRef]
- de Wildt, S.N.; de Hoog, M.; Vinks, A.A.; Joosten, K.F.; van Dijk, M.; van den Anker, J.N. Pharmacodynamics of midazolam in pediatric intensive care patients. Ther. Drug Monit. 2005, 27, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Oldenhof, H.; de Jong, M.; Steenhoek, A.; Janknegt, R. Clinical pharmacokinetics of midazolam in intensive care patients, a wide interpatient variability? Clin. Pharmacol. Ther. 1988, 43, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Hasegawa, R.; Fukuda, T.; Yumoto, R.; Nagai, J.; Murakami, T. Interaction with P-glycoprotein and transport of erythromycin, midazolam and ketoconazole in Caco-2 cells. Eur. J. Pharmacol. 1998, 358, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Tolle-Sander, S.; Rautio, J.; Wring, S.; Polli, J.W.; Polli, J.E. Midazolam exhibits characteristics of a highly permeable P-glycoprotein substrate. Pharm. Res. 2003, 20, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Available online: https://www.fda.gov/media/134582/download (accessed on 17 March 2024).
- Franke, R.M.; Baker, S.D.; Mathijssen, R.H.; Schuetz, E.G.; Sparreboom, A. Influence of solute carriers on the pharmacokinetics of CYP3A4 probes. Clin. Pharmacol. Ther. 2008, 84, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Imanaga, J.; Kotegawa, T.; Imai, H.; Tsutsumi, K.; Yoshizato, T.; Ohyama, T.; Shirasaka, Y.; Tamai, I.; Tateishi, T.; Ohashi, K. The effects of the SLCO2B1 c.1457C > T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet. Genom. 2011, 21, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, E.; Deng, F.; Kiander, W.; Sinokki, A.; Kidron, H.; Sjostedt, N. The Role of Uptake and Efflux Transporters in the Disposition of Glucuronide and Sulfate Conjugates. Front. Pharmacol. 2021, 12, 802539. [Google Scholar] [CrossRef] [PubMed]
- Allonen, H.; Ziegler, G.; Klotz, U. Midazolam kinetics. Clin. Pharmacol. Ther. 1981, 30, 653–661. [Google Scholar] [CrossRef]
- Pentikainen, P.J.; Valisalmi, L.; Himberg, J.J.; Crevoisier, C. Pharmacokinetics of midazolam following intravenous and oral administration in patients with chronic liver disease and in healthy subjects. J. Clin. Pharmacol. 1989, 29, 272–277. [Google Scholar] [CrossRef]
- Lee, S.; Lee, Y.; Kim, A.H.; Yoon, S.; Lee, J.; Ji, S.C.; Yoon, S.H.; Lee, S.; Yu, K.S.; Jang, I.J.; et al. Urinary metabolic markers reflect on hepatic, not intestinal, CYP3A activity in healthy subjects. Drug Metab. Pharmacokinet. 2021, 36, 100374. [Google Scholar] [CrossRef]
- Stoch, S.A.; Friedman, E.; Maes, A.; Yee, K.; Xu, Y.; Larson, P.; Fitzgerald, M.; Chodakewitz, J.; Wagner, J.A. Effect of different durations of ketoconazole dosing on the single-dose pharmacokinetics of midazolam: Shortening the paradigm. J. Clin. Pharmacol. 2009, 49, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Tseng, E.; Walsky, R.L.; Luzietti, R.A., Jr.; Harris, J.J.; Kosa, R.E.; Goosen, T.C.; Zientek, M.A.; Obach, R.S. Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab. Dispos. 2014, 42, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.S.; Cho, J.Y.; Jang, I.J.; Hong, K.S.; Chung, J.Y.; Kim, J.R.; Lim, H.S.; Oh, D.S.; Yi, S.Y.; Liu, K.H.; et al. Effect of the CYP3A5 genotype on the pharmacokinetics of intravenous midazolam during inhibited and induced metabolic states. Clin. Pharmacol. Ther. 2004, 76, 104–112. [Google Scholar] [CrossRef] [PubMed]
- von Moltke, L.L.; Greenblatt, D.J.; Schmider, J.; Duan, S.X.; Wright, C.E.; Harmatz, J.S.; Shader, R.I. Midazolam hydroxylation by human liver microsomes in vitro: Inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J. Clin. Pharmacol. 1996, 36, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.A.; Bae, S.K.; Choi, Y.K.; Choi, C.S.; Liu, K.H.; Shin, J.G. Metabolism of 1′- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab. Dispos. 2010, 38, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Klieber, S.; Hugla, S.; Ngo, R.; Arabeyre-Fabre, C.; Meunier, V.; Sadoun, F.; Fedeli, O.; Rival, M.; Bourrie, M.; Guillou, F.; et al. Contribution of the N-glucuronidation pathway to the overall in vitro metabolic clearance of midazolam in humans. Drug Metab. Dispos. 2008, 36, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Hyland, R.; Osborne, T.; Payne, A.; Kempshall, S.; Logan, Y.R.; Ezzeddine, K.; Jones, B. In vitro and in vivo glucuronidation of midazolam in humans. Br. J. Clin. Pharmacol. 2009, 67, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, W.H.; Schalch, E.; Leishman, B.; Eckert, M. Comparison of the effects of intravenously administered midazolam, triazolam and their hydroxy metabolites. Br. J. Clin. Pharmacol. 1983, 16 (Suppl. S1), 63S–69S. [Google Scholar] [CrossRef] [PubMed]
- Mandema, J.W.; Tuk, B.; van Steveninck, A.L.; Breimer, D.D.; Cohen, A.F.; Danhof, M. Pharmacokinetic-pharmacodynamic modeling of the central nervous system effects of midazolam and its main metabolite alpha-hydroxymidazolam in healthy volunteers. Clin. Pharmacol. Ther. 1992, 51, 715–728. [Google Scholar] [CrossRef]
- Bauer, T.M.; Ritz, R.; Haberthur, C.; Ha, H.R.; Hunkeler, W.; Sleight, A.J.; Scollo-Lavizzari, G.; Haefeli, W.E. Prolonged sedation due to accumulation of conjugated metabolites of midazolam. Lancet 1995, 346, 145–147. [Google Scholar] [CrossRef]
- Stanski, D.; Hudson, R. Midazolam pharmacology and pharmacokinetics. Anesth. Rev. 1985, 12, 21–23. [Google Scholar]
- Kanto, J.H. Midazolam: The first water-soluble benzodiazepine. Pharmacology, pharmacokinetics and efficacy in insomnia and anesthesia. Pharmacotherapy 1985, 5, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Rostami-Hodjegan, A.; Goddard, J.M.; Tanner, M.S.; Tucker, G.T. Contribution of midazolam and its 1-hydroxy metabolite to preoperative sedation in children: A pharmacokinetic-pharmacodynamic analysis. Br. J. Anaesth. 2002, 89, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.G.; Masman, A.D.; de Winter, B.C.M.; Baar, F.P.M.; Tibboel, D.; van Gelder, T.; Koch, B.C.P.; Mathot, R.A.A. Hypoalbuminaemia and decreased midazolam clearance in terminally ill adult patients, an inflammatory effect? Br. J. Clin. Pharmacol. 2017, 83, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Beierle, I.; Meibohm, B.; Derendorf, H. Gender differences in pharmacokinetics and pharmacodynamics. Int. J. Clin. Pharmacol. Ther. 1999, 37, 529–547. [Google Scholar] [PubMed]
- Gandhi, M.; Aweeka, F.; Greenblatt, R.M.; Blaschke, T.F. Sex differences in pharmacokinetics and pharmacodynamics. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 499–523. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Panetta, J.C.; Strom, S.; Schuetz, E.G. Genetic predictors of interindividual variability in hepatic CYP3A4 expression. J. Pharmacol. Exp. Ther. 2010, 332, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Baumann, F.; Hanschmann, H.; Geissler, F.; Preiss, R. Gender difference in ifosfamide metabolism by human liver microsomes. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 193–200. [Google Scholar] [CrossRef]
- Wolbold, R.; Klein, K.; Burk, O.; Nussler, A.K.; Neuhaus, P.; Eichelbaum, M.; Schwab, M.; Zanger, U.M. Sex is a major determinant of CYP3A4 expression in human liver. Hepatology 2003, 38, 978–988. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef]
- Gallagher, C.J.; Balliet, R.M.; Sun, D.; Chen, G.; Lazarus, P. Sex differences in UDP-glucuronosyltransferase 2B17 expression and activity. Drug Metab. Dispos. 2010, 38, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Meibohm, B.; Beierle, I.; Derendorf, H. How important are gender differences in pharmacokinetics? Clin. Pharmacokinet. 2002, 41, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Cotreau, M.M.; von Moltke, L.L.; Greenblatt, D.J. The influence of age and sex on the clearance of cytochrome P450 3A substrates. Clin. Pharmacokinet. 2005, 44, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.M.; Velez, R.L.; von Moltke, L.L.; Greenblatt, D.J. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: Effect of ketoconazole. Clin. Pharmacol. Ther. 1999, 66, 461–471. [Google Scholar] [CrossRef]
- Thangavel, C.; Boopathi, E.; Shapiro, B.H. Intrinsic sexually dimorphic expression of the principal human CYP3A4 correlated with suboptimal activation of GH/glucocorticoid-dependent transcriptional pathways in men. Endocrinology 2011, 152, 4813–4824. [Google Scholar] [CrossRef] [PubMed]
- Dhir, R.N.; Dworakowski, W.; Thangavel, C.; Shapiro, B.H. Sexually dimorphic regulation of hepatic isoforms of human cytochrome p450 by growth hormone. J. Pharmacol. Exp. Ther. 2006, 316, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.Y.; Zhao, Y.S. Sex-dependent differences in cytochrome P450 3A activity as assessed by midazolam disposition in humans: A meta-analysis. Drug Metab. Dispos. 2010, 38, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ma, L.; Drusano, G.L.; Bertino, J.S., Jr.; Nafziger, A.N. Sex differences in CYP3A activity using intravenous and oral midazolam. Clin. Pharmacol. Ther. 2006, 80, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Zarezadeh, M.; Saedisomeolia, A.; Shekarabi, M.; Khorshidi, M.; Emami, M.R.; Muller, D.J. The effect of obesity, macronutrients, fasting and nutritional status on drug-metabolizing cytochrome P450s: A systematic review of current evidence on human studies. Eur. J. Nutr. 2021, 60, 2905–2921. [Google Scholar] [CrossRef]
- Lammers, L.A.; Achterbergh, R.; Romijn, J.A.; Mathot, R.A.A. Nutritional Status Differentially Alters Cytochrome P450 3A4 (CYP3A4) and Uridine 5′-Diphospho-Glucuronosyltransferase (UGT) Mediated Drug Metabolism: Effect of Short-Term Fasting and High Fat Diet on Midazolam Metabolism. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 751–767. [Google Scholar] [CrossRef]
- de Vries, E.M.; Lammers, L.A.; Achterbergh, R.; Klumpen, H.J.; Mathot, R.A.; Boelen, A.; Romijn, J.A. Fasting-Induced Changes in Hepatic P450 Mediated Drug Metabolism Are Largely Independent of the Constitutive Androstane Receptor CAR. PLoS ONE 2016, 11, e0159552. [Google Scholar] [CrossRef]
- Lammers, L.A.; Achterbergh, R.; de Vries, E.M.; van Nierop, F.S.; Klumpen, H.J.; Soeters, M.R.; Boelen, A.; Romijn, J.A.; Mathot, R.A. Short-term fasting alters cytochrome P450-mediated drug metabolism in humans. Drug Metab. Dispos. 2015, 43, 819–828. [Google Scholar] [CrossRef]
- Lammers, L.A.; Achterbergh, R.; van Schaik, R.H.N.; Romijn, J.A.; Mathot, R.A.A. Effect of Short-Term Fasting on Systemic Cytochrome P450-Mediated Drug Metabolism in Healthy Subjects: A Randomized, Controlled, Crossover Study Using a Cocktail Approach. Clin. Pharmacokinet. 2017, 56, 1231–1244. [Google Scholar] [CrossRef]
- Brill, M.J.; van Rongen, A.; Houwink, A.P.; Burggraaf, J.; van Ramshorst, B.; Wiezer, R.J.; van Dongen, E.P.; Knibbe, C.A. Midazolam pharmacokinetics in morbidly obese patients following semi-simultaneous oral and intravenous administration: A comparison with healthy volunteers. Clin. Pharmacokinet. 2014, 53, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, C.; Rodieux, F.; Desmeules, J.A.; Rollason, V.; Samer, C.F. Impact of Inflammation on Cytochromes P450 Activity in Pediatrics: A Systematic Review. Clin. Pharmacokinet. 2021, 60, 1537–1555. [Google Scholar] [CrossRef]
- Aitken, A.E.; Morgan, E.T. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 2007, 35, 1687–1693. [Google Scholar] [CrossRef]
- Aitken, A.E.; Richardson, T.A.; Morgan, E.T. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 123–149. [Google Scholar] [CrossRef]
- Stanke-Labesque, F.; Gautier-Veyret, E.; Chhun, S.; Guilhaumou, R.; French Society of Pharmacology and Therapeutics. Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol. Ther. 2020, 215, 107627. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.E.; Brown, K.C.; Werner, R.E.; Gotzkowsky, K.; Gaedigk, A.; Blake, M.; Hein, D.W.; van der Horst, C.; Kashuba, A.D. Variability in drug metabolizing enzyme activity in HIV-infected patients. Eur. J. Clin. Pharmacol. 2010, 66, 475–485. [Google Scholar] [CrossRef]
- Brussee, J.M.; Vet, N.J.; Krekels, E.H.J.; Valkenburg, A.J.; Jacqz-Aigrain, E.; van Gerven, J.M.A.; Swart, E.L.; van den Anker, J.N.; Tibboel, D.; de Hoog, M.; et al. Predicting CYP3A-mediated midazolam metabolism in critically ill neonates, infants, children and adults with inflammation and organ failure. Br. J. Clin. Pharmacol. 2018, 84, 358–368. [Google Scholar] [CrossRef]
- Le Carpentier, E.C.; Canet, E.; Masson, D.; Martin, M.; Deslandes, G.; Gaultier, A.; Dailly, E.; Bellouard, R.; Gregoire, M. Impact of Inflammation on Midazolam Metabolism in Severe COVID-19 Patients. Clin. Pharmacol. Ther. 2022, 112, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Bort, R.; Gomez-Lechon, M.J.; Castell, J.V. Down-regulation of human CYP3A4 by the inflammatory signal interleukin-6: Molecular mechanism and transcription factors involved. FASEB J. 2002, 16, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hao, C.; Yang, D.; Shi, D.; Song, X.; Luan, X.; Hu, G.; Yan, B. Pregnane X receptor is required for interleukin-6-mediated down-regulation of cytochrome P450 3A4 in human hepatocytes. Toxicol. Lett. 2010, 197, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ke, S.; Liu, D.; Sheng, T.; Thomas, P.E.; Rabson, A.B.; Gallo, M.A.; Xie, W.; Tian, Y. Role of NF-kappaB in regulation of PXR-mediated gene expression: A mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 2006, 281, 17882–17889. [Google Scholar] [CrossRef]
- Keller, R.; Klein, M.; Thomas, M.; Drager, A.; Metzger, U.; Templin, M.F.; Joos, T.O.; Thasler, W.E.; Zell, A.; Zanger, U.M. Coordinating Role of RXRalpha in Downregulating Hepatic Detoxification during Inflammation Revealed by Fuzzy-Logic Modeling. PLoS Comput. Biol. 2016, 12, e1004431. [Google Scholar] [CrossRef] [PubMed]
- Patki, K.C.; Von Moltke, L.L.; Greenblatt, D.J. In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: Role of cyp3a4 and cyp3a5. Drug Metab. Dispos. 2003, 31, 938–944. [Google Scholar] [CrossRef]
- Pharmacogene Variation Consortium. CYP3A5. Available online: https://www.pharmvar.org/gene/CYP3A5 (accessed on 15 February 2024).
- Rodriguez-Antona, C.; Savieo, J.L.; Lauschke, V.M.; Sangkuhl, K.; Drogemoller, B.I.; Wang, D.; van Schaik, R.H.N.; Gilep, A.A.; Peter, A.P.; Boone, E.C.; et al. PharmVar GeneFocus: CYP3A5. Clin. Pharmacol. Ther. 2022, 112, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Elens, L.; van Gelder, T.; Hesselink, D.A.; Haufroid, V.; van Schaik, R.H. CYP3A4*22: Promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013, 14, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, B.J.; Lee, S.W.; Kang, H.; Kim, J.W.; Jang, I.J.; Kim, J.G. Influence of midazolam-related genetic polymorphism on conscious sedation during upper gastrointestinal endoscopy in a Korean population. Sci. Rep. 2019, 9, 16001. [Google Scholar] [CrossRef]
- Fromm, M.F.; Schwilden, H.; Bachmakov, I.; Konig, J.; Bremer, F.; Schuttler, J. Impact of the CYP3A5 genotype on midazolam pharmacokinetics and pharmacodynamics during intensive care sedation. Eur. J. Clin. Pharmacol. 2007, 63, 1129–1133. [Google Scholar] [CrossRef]
- MacPhee, I.A. Pharmacogenetic biomarkers: Cytochrome P450 3A5. Clin. Chim. Acta 2012, 413, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Westlind-Johnsson, A.; Malmebo, S.; Johansson, A.; Otter, C.; Andersson, T.B.; Johansson, I.; Edwards, R.J.; Boobis, A.R.; Ingelman-Sundberg, M. Comparative analysis of CYP3A expression in human liver suggests only a minor role for CYP3A5 in drug metabolism. Drug Metab. Dispos. 2003, 31, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 15 February 2024).
- Pharmacogene Variation Consortium. CYP3A4. Available online: https://www.pharmvar.org/gene/CYP3A4 (accessed on 15 February 2024).
- PHARMGKB. Curation of DPWG Content into PharmGKB. Available online: https://www.pharmgkb.org/page/dpwgMapping#cyp3a4 (accessed on 15 February 2024).
- Garcia-Martin, E.; Martinez, C.; Pizarro, R.M.; Garcia-Gamito, F.J.; Gullsten, H.; Raunio, H.; Agundez, J.A. CYP3A4 variant alleles in white individuals with low CYP3A4 enzyme activity. Clin. Pharmacol. Ther. 2002, 71, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Alkattan, A.; Alsalameen, E. Polymorphisms of genes related to phase-I metabolic enzymes affecting the clinical efficacy and safety of clopidogrel treatment. Expert Opin. Drug Metab. Toxicol. 2021, 17, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Wojnowski, L.; Hustert, E.; Klein, K.; Goldammer, M.; Haberl, M.; Kirchheiner, J.; Koch, I.; Klattig, J.; Zanger, U.; Brockmoller, J. Re: Modification of clinical presentation of prostate tumors by a novel genetic variant in CYP3A4. J. Natl. Cancer Inst. 2002, 94, 630–631; author reply 631–632. [Google Scholar] [CrossRef][Green Version]
- Lee, S.J.; Goldstein, J.A. Functionally defective or altered CYP3A4 and CYP3A5 single nucleotide polymorphisms and their detection with genotyping tests. Pharmacogenomics 2005, 6, 357–371. [Google Scholar] [CrossRef]
- Miyazaki, M.; Nakamura, K.; Fujita, Y.; Guengerich, F.P.; Horiuchi, R.; Yamamoto, K. Defective activity of recombinant cytochromes P450 3A4.2 and 3A4.16 in oxidation of midazolam, nifedipine, and testosterone. Drug Metab. Dispos. 2008, 36, 2287–2291. [Google Scholar] [CrossRef]
- Sata, F.; Sapone, A.; Elizondo, G.; Stocker, P.; Miller, V.P.; Zheng, W.; Raunio, H.; Crespi, C.L.; Gonzalez, F.J. CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: Evidence for an allelic variant with altered catalytic activity. Clin. Pharmacol. Ther. 2000, 67, 48–56. [Google Scholar] [CrossRef]
- Dai, D.; Tang, J.; Rose, R.; Hodgson, E.; Bienstock, R.J.; Mohrenweiser, H.W.; Goldstein, J.A. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J. Pharmacol. Exp. Ther. 2001, 299, 825–831. [Google Scholar]
- Eiselt, R.; Domanski, T.L.; Zibat, A.; Mueller, R.; Presecan-Siedel, E.; Hustert, E.; Zanger, U.M.; Brockmoller, J.; Klenk, H.P.; Meyer, U.A.; et al. Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics 2001, 11, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, K.P.; Lin, Y.Y.; Cheng, C.L.; Lai, M.L.; Lin, M.S.; Siest, J.P.; Huang, J.D. Novel mutations of CYP3A4 in Chinese. Drug Metab. Dispos. 2001, 29, 268–273. [Google Scholar] [PubMed]
- Wang, A.; Yu, B.N.; Luo, C.H.; Tan, Z.R.; Zhou, G.; Wang, L.S.; Zhang, W.; Li, Z.; Liu, J.; Zhou, H.H. Ile118Val genetic polymorphism of CYP3A4 and its effects on lipid-lowering efficacy of simvastatin in Chinese hyperlipidemic patients. Eur. J. Clin. Pharmacol. 2005, 60, 843–848. [Google Scholar] [CrossRef]
- Al Maruf, A.; Ahmed, M.U.; Yasmin, H.; Ullah, M.A.; Azad, M.A.; Daly, A.K.; Hasnat, A. Genotypes and phenotypes of CYP3A in Bangladeshi population. Clin. Chim. Acta 2011, 412, 531–536. [Google Scholar] [CrossRef]
- Rais, N.; Chawla, Y.K.; Kohli, K.K. CYP3A phenotypes and genotypes in North Indians. Eur. J. Clin. Pharmacol. 2006, 62, 417–422. [Google Scholar] [CrossRef]
- Westlind-Johnsson, A.; Hermann, R.; Huennemeyer, A.; Hauns, B.; Lahu, G.; Nassr, N.; Zech, K.; Ingelman-Sundberg, M.; von Richter, O. Identification and characterization of CYP3A4*20, a novel rare CYP3A4 allele without functional activity. Clin. Pharmacol. Ther. 2006, 79, 339–349. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, X.; Shu, C.; Cai, Y.; Gong, W.; Wang, X.; Wang, D.M.; Hu, S. Analysis of CYP3A4 genetic polymorphisms in Han Chinese. J. Hum. Genet. 2011, 56, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kumondai, M.; Gutierrez Rico, E.M.; Hishinuma, E.; Ueda, A.; Saito, S.; Saigusa, D.; Tadaka, S.; Kinoshita, K.; Nakayoshi, T.; Oda, A.; et al. Functional Characterization of 40 CYP3A4 Variants by Assessing Midazolam 1′-Hydroxylation and Testosterone 6beta-Hydroxylation. Drug Metab. Dispos. 2021, 49, 212–220. [Google Scholar] [CrossRef]
- Werk, A.N.; Lefeldt, S.; Bruckmueller, H.; Hemmrich-Stanisak, G.; Franke, A.; Roos, M.; Kuchle, C.; Steubl, D.; Schmaderer, C.; Brasen, J.H.; et al. Identification and characterization of a defective CYP3A4 genotype in a kidney transplant patient with severely diminished tacrolimus clearance. Clin. Pharmacol. Ther. 2014, 95, 416–422. [Google Scholar] [CrossRef]
- Apellaniz-Ruiz, M.; Inglada-Perez, L.; Naranjo, M.E.; Sanchez, L.; Mancikova, V.; Curras-Freixes, M.; de Cubas, A.A.; Comino-Mendez, I.; Triki, S.; Rebai, A.; et al. High frequency and founder effect of the CYP3A4*20 loss-of-function allele in the Spanish population classifies CYP3A4 as a polymorphic enzyme. Pharmacogenom. J. 2015, 15, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenom. J. 2011, 11, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Pratt, V.M.; Cavallari, L.H.; Fulmer, M.L.; Gaedigk, A.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; et al. CYP3A4 and CYP3A5 Genotyping Recommendations: A Joint Consensus Recommendation of the Association for Molecular Pathology, Clinical Pharmacogenetics Implementation Consortium, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, European Society for Pharmacogenomics and Personalized Therapy, and Pharmacogenomics Knowledgebase. J. Mol. Diagn. 2023, 25, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Eap, C.B.; Buclin, T.; Hustert, E.; Bleiber, G.; Golay, K.P.; Aubert, A.C.; Baumann, P.; Telenti, A.; Kerb, R. Pharmacokinetics of midazolam in CYP3A4- and CYP3A5-genotyped subjects. Eur. J. Clin. Pharmacol. 2004, 60, 231–236. [Google Scholar] [CrossRef]
- Schuetz, E.G. Lessons from the CYP3A4 promoter. Mol. Pharmacol. 2004, 65, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, C.P.; Jover, R.; Donato, M.T.; Castell, J.V.; Gomez-Lechon, M.J. Transcriptional regulation and expression of CYP3A4 in hepatocytes. Curr. Drug Metab. 2007, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Bort, R.; Jover, R.; Tindberg, N.; Ingelman-Sundberg, M.; Gomez-Lechon, M.J.; Castell, J.V. Transcriptional regulation of human CYP3A4 basal expression by CCAAT enhancer-binding protein alpha and hepatocyte nuclear factor-3 gamma. Mol. Pharmacol. 2003, 63, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, C.P.; Gomez-Lechon, M.J.; Castell, J.V.; Jover, R. Transcriptional regulation of the human hepatic CYP3A4: Identification of a new distal enhancer region responsive to CCAAT/enhancer-binding protein beta isoforms (liver activating protein and liver inhibitory protein). Mol. Pharmacol. 2005, 67, 2088–2101. [Google Scholar] [CrossRef]
- Tirona, R.G.; Lee, W.; Leake, B.F.; Lan, L.B.; Cline, C.B.; Lamba, V.; Parviz, F.; Duncan, S.A.; Inoue, Y.; Gonzalez, F.J.; et al. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 2003, 9, 220–224. [Google Scholar] [CrossRef]
- Tegude, H.; Schnabel, A.; Zanger, U.M.; Klein, K.; Eichelbaum, M.; Burk, O. Molecular mechanism of basal CYP3A4 regulation by hepatocyte nuclear factor 4alpha: Evidence for direct regulation in the intestine. Drug Metab. Dispos. 2007, 35, 946–954. [Google Scholar] [CrossRef]
- Jover, R.; Moya, M.; Gomez-Lechon, M.J. Transcriptional regulation of cytochrome p450 genes by the nuclear receptor hepatocyte nuclear factor 4-alpha. Curr. Drug Metab. 2009, 10, 508–519. [Google Scholar] [CrossRef]
- Biggs, J.S.; Wan, J.; Cutler, N.S.; Hakkola, J.; Uusimaki, P.; Raunio, H.; Yost, G.S. Transcription factor binding to a putative double E-box motif represses CYP3A4 expression in human lung cells. Mol. Pharmacol. 2007, 72, 514–525. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Collins, J.M.; Wang, D. Co-expression of drug metabolizing cytochrome P450 enzymes and estrogen receptor alpha (ESR1) in human liver: Racial differences and the regulatory role of ESR1. Drug Metab. Pers. Ther. 2021, 36, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; D’Costa, D.J.; Robertson, G.R.; Liddle, C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol. 2002, 62, 359–365. [Google Scholar] [CrossRef]
- Matsumura, K.; Saito, T.; Takahashi, Y.; Ozeki, T.; Kiyotani, K.; Fujieda, M.; Yamazaki, H.; Kunitoh, H.; Kamataki, T. Identification of a novel polymorphic enhancer of the human CYP3A4 gene. Mol. Pharmacol. 2004, 65, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Mathas, M.; Nestler, S.; Bengel, C.; Nem, D.; Godtel-Armbrust, U.; Lang, T.; Taudien, S.; Burk, O.; Wojnowski, L. The unique complexity of the CYP3A4 upstream region suggests a nongenetic explanation of its expression variability. Pharmacogenet. Genom. 2010, 20, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol. Pharmacol. 1999, 56, 1329–1339. [Google Scholar] [CrossRef]
- Liu, F.J.; Song, X.; Yang, D.; Deng, R.; Yan, B. The far and distal enhancers in the CYP3A4 gene co-ordinate the proximal promoter in responding similarly to the pregnane X receptor but differentially to hepatocyte nuclear factor-4alpha. Biochem. J. 2008, 409, 243–250. [Google Scholar] [CrossRef]
- Lamba, J.; Lamba, V.; Strom, S.; Venkataramanan, R.; Schuetz, E. Novel single nucleotide polymorphisms in the promoter and intron 1 of human pregnane X receptor/NR1I2 and their association with CYP3A4 expression. Drug Metab. Dispos. 2008, 36, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Li, Z.H.; Liu, J.Y.; Liu, T.T.; Wang, P.; Fang, Y.; Zhou, J.; Cui, M.Z.; Gao, N.; Tian, X.; et al. Correlation of Cytochrome P450 Oxidoreductase Expression with the Expression of 10 Isoforms of Cytochrome P450 in Human Liver. Drug Metab. Dispos. 2016, 44, 1193–1200. [Google Scholar] [CrossRef]
- El-Sankary, W.; Plant, N.J.; Gibson, G.G.; Moore, D.J. Regulation of the CYP3A4 gene by hydrocortisone and xenobiotics: Role of the glucocorticoid and pregnane X receptors. Drug Metab. Dispos. 2000, 28, 493–496. [Google Scholar]
- Burk, O.; Koch, I.; Raucy, J.; Hustert, E.; Eichelbaum, M.; Brockmoller, J.; Zanger, U.M.; Wojnowski, L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J. Biol. Chem. 2004, 279, 38379–38385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wong, T.; Hashizume, T.; Dickmann, L.Z.; Scian, M.; Koszewski, N.J.; Goff, J.P.; Horst, R.L.; Chaudhry, A.S.; Schuetz, E.G.; et al. Human UGT1A4 and UGT1A3 conjugate 25-hydroxyvitamin D3: Metabolite structure, kinetics, inducibility, and interindividual variability. Endocrinology 2014, 155, 2052–2063. [Google Scholar] [CrossRef]
- Xu, C.; Gao, J.; Zhang, H.F.; Gao, N.; Guo, Y.Y.; Fang, Y.; Wen, Q.; Qiao, H.L. Content and Activities of UGT2B7 in Human Liver In Vitro and Predicted In Vivo: A Bottom-Up Approach. Drug Metab. Dispos. 2018, 46, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef]
- Vrzal, R.; Kubesova, K.; Pavek, P.; Dvorak, Z. Benzodiazepines medazepam and midazolam are activators of pregnane X receptor and weak inducers of CYP3A4: Investigation in primary cultures of human hepatocytes and hepatocarcinoma cell lines. Toxicol. Lett. 2010, 193, 183–188. [Google Scholar] [CrossRef]
- Gnerre, C.; Blattler, S.; Kaufmann, M.R.; Looser, R.; Meyer, U.A. Regulation of CYP3A4 by the bile acid receptor FXR: Evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 2004, 14, 635–645. [Google Scholar] [CrossRef]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Duret, C.; Daujat-Chavanieu, M.; Vilarem, M.J.; Maurel, P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: Crosstalk and consequences. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Duniec-Dmuchowski, Z.; Ellis, E.; Strom, S.C.; Kocarek, T.A. Regulation of CYP3A4 and CYP2B6 expression by liver X receptor agonists. Biochem. Pharmacol. 2007, 74, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Yoshinari, K.; Aoyama, K.; Sugawara, M.; Sekiya, Y.; Nagata, K.; Yamazoe, Y. Role of vitamin D receptor in the lithocholic acid-mediated CYP3A induction in vitro and in vivo. Drug Metab. Dispos. 2008, 36, 2058–2063. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Thomas, M.; Winter, S.; Nussler, A.K.; Niemi, M.; Schwab, M.; Zanger, U.M. PPARA: A novel genetic determinant of CYP3A4 in vitro and in vivo. Clin. Pharmacol. Ther. 2012, 91, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.; Wollnik, J.; Wrzodek, C.; Drager, A.; Bonin, M.; Burk, O.; Thomas, M.; Thasler, W.E.; Zanger, U.M.; Zell, A. Inferring statin-induced gene regulatory relationships in primary human hepatocytes. Bioinformatics 2011, 27, 2473–2477. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grun, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Slaviero, K.A.; Clarke, S.J.; Rivory, L.P. Inflammatory response: An unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol. 2003, 4, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.Z.; Gao, W.; Yu, A.M. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab. Dispos. 2009, 37, 2112–2117. [Google Scholar] [CrossRef]
- Ekstrom, L.; Skilving, I.; Ovesjo, M.L.; Aklillu, E.; Nylen, H.; Rane, A.; Diczfalusy, U.; Bjorkhem-Bergman, L. miRNA-27b levels are associated with CYP3A activity in vitro and in vivo. Pharmacol. Res. Perspect. 2015, 3, e00192. [Google Scholar] [CrossRef]
- Takagi, S.; Nakajima, M.; Mohri, T.; Yokoi, T. Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J. Biol. Chem. 2008, 283, 9674–9680. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Nakajima, M.; Kida, K.; Yamaura, Y.; Fukami, T.; Yokoi, T. MicroRNAs regulate human hepatocyte nuclear factor 4alpha, modulating the expression of metabolic enzymes and cell cycle. J. Biol. Chem. 2010, 285, 4415–4422. [Google Scholar] [CrossRef] [PubMed]
- Karbiener, M.; Fischer, C.; Nowitsch, S.; Opriessnig, P.; Papak, C.; Ailhaud, G.; Dani, C.; Amri, E.Z.; Scheideler, M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARgamma. Biochem. Biophys. Res. Commun. 2009, 390, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, C.; von Knethen, A.; Schmid, T.; Brune, B. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA destabilization. J. Biol. Chem. 2010, 285, 11846–11853. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Wada, T.; Gramignoli, R.; Li, S.; Strom, S.C.; Huang, M.; Xie, W. MicroRNA hsa-miR-613 targets the human LXRalpha gene and mediates a feedback loop of LXRalpha autoregulation. Mol. Endocrinol. 2011, 25, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Schmeier, S.; Schaefer, U.; MacPherson, C.R.; Bajic, V.B. dPORE-miRNA: Polymorphic regulation of microRNA genes. PLoS ONE 2011, 6, e16657. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Yang, F.; Urban, T.J.; Li, L.; Chalasani, N.; Flockhart, D.A.; Liu, W. Impact of the Interaction between 3′-UTR SNPs and microRNA on the Expression of Human Xenobiotic Metabolism Enzyme and Transporter Genes. Front. Genet. 2012, 3, 248. [Google Scholar] [CrossRef]
- Kasteel, E.E.J.; Darney, K.; Kramer, N.I.; Dorne, J.; Lautz, L.S. Human variability in isoform-specific UDP-glucuronosyltransferases: Markers of acute and chronic exposure, polymorphisms and uncertainty factors. Arch. Toxicol. 2020, 94, 2637–2661. [Google Scholar] [CrossRef]
- Aueviriyavit, S.; Furihata, T.; Morimoto, K.; Kobayashi, K.; Chiba, K. Hepatocyte nuclear factor 1 alpha and 4 alpha are factors involved in interindividual variability in the expression of UGT1A6 and UGT1A9 but not UGT1A1, UGT1A3 and UGT1A4 mRNA in human livers. Drug Metab. Pharmacokinet. 2007, 22, 391–398. [Google Scholar] [CrossRef]
- Pharmacogenomics Laboratory at Université Laval. Available online: https://www.pharmacogenomics.pha.ulaval.ca/wp-content/uploads/2015/04/HAP-UGT1A4.htm (accessed on 15 February 2024).
- Ehmer, U.; Vogel, A.; Schutte, J.K.; Krone, B.; Manns, M.P.; Strassburg, C.P. Variation of hepatic glucuronidation: Novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology 2004, 39, 970–977. [Google Scholar] [CrossRef]
- Erickson-Ridout, K.K.; Sun, D.; Lazarus, P. Glucuronidation of the second-generation antipsychotic clozapine and its active metabolite N-desmethylclozapine. Potential importance of the UGT1A1 A(TA)(7)TAA and UGT1A4 L48V polymorphisms. Pharmacogenetics Genom. 2012, 22, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Ridout, K.K.; Zhu, J.; Lazarus, P. Olanzapine metabolism and the significance of UGT1A448V and UGT2B1067Y variants. Pharmacogenetics Genom. 2011, 21, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Argikar, U.A.; Remmel, R.P. Functional analysis of UGT1A4(P24T) and UGT1A4(L48V) variant enzymes. Pharmacogenomics 2011, 12, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Reimers, A.; Sjursen, W.; Helde, G.; Brodtkorb, E. Frequencies of UGT1A4*2 (P24T) and *3 (L48V) and their effects on serum concentrations of lamotrigine. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Gulcebi, M.I.; Ozkaynakci, A.; Goren, M.Z.; Aker, R.G.; Ozkara, C.; Onat, F.Y. The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy Res. 2011, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Yang, L.Y.; Zhang, M.C.; Liu, S.Y. Correlation of the UGT1A4 gene polymorphism with serum concentration and therapeutic efficacy of lamotrigine in Han Chinese of Northern China. Eur. J. Clin. Pharmacol. 2014, 70, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Haslemo, T.; Loryan, I.; Ueda, N.; Mannheimer, B.; Bertilsson, L.; Ingelman-Sundberg, M.; Molden, E.; Eliasson, E. UGT1A4*3 encodes significantly increased glucuronidation of olanzapine in patients on maintenance treatment and in recombinant systems. Clin. Pharmacol. Ther. 2012, 92, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lee, S.Y.; Yang, K.S.; Park, J.Y.; Yoon, S.Z.; Yoon, S.M. Polymorphism rs4263535 in GABRA1 intron 4 was related to deeper sedation by intravenous midazolam. J. Int. Med. Res. 2015, 43, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y. Epigenetic regulation of pregnane X receptor activity. Drug Metab. Rev. 2013, 45, 166–172. [Google Scholar] [CrossRef]
- Meierhans, R.; Stover, J.F.; Bechir, M.; Keel, M.; Stocker, R. Reduced midazolam clearance must be considered in prolonged coma. Anaesth. Intensive Care 2008, 36, 915–916. [Google Scholar]
- Babu, T.A. Prolonged sedation following administration of oral midazolam. Indian Pediatr. 2013, 50, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Ma, W.; Guo, Q.; Liu, J.; Li, W.; McLeod, H.L.; He, Y. The pharmacogenetics of medications used in general anesthesia. Pharmacogenomics 2018, 19, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Lockwood, G.F.; Graham, M.A.; Brian, W.R.; Loi, C.M.; Dobrinska, M.R.; Shen, D.D.; Watkins, P.B.; Wilkinson, G.R.; Kharasch, E.D.; et al. In-vivo phenotyping for CYP3A by a single-point determination of midazolam plasma concentration. Pharmacogenetics 2001, 11, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Troberg, J.; Finel, M. The Polymorphic Variant P24T of UDP-Glucuronosyltransferase 1A4 and Its Unusual Consequences. Drug Metab. Dispos. 2015, 43, 1769–1772. [Google Scholar] [CrossRef]
- Spina, S.P.; Ensom, M.H. Clinical pharmacokinetic monitoring of midazolam in critically ill patients. Pharmacotherapy 2007, 27, 389–398. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).