
Solid Self-Nanoemulsifying Drug Delivery Systems of Furosemide: In Vivo Proof of Concept for Enhanced Predictable Therapeutic Response

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
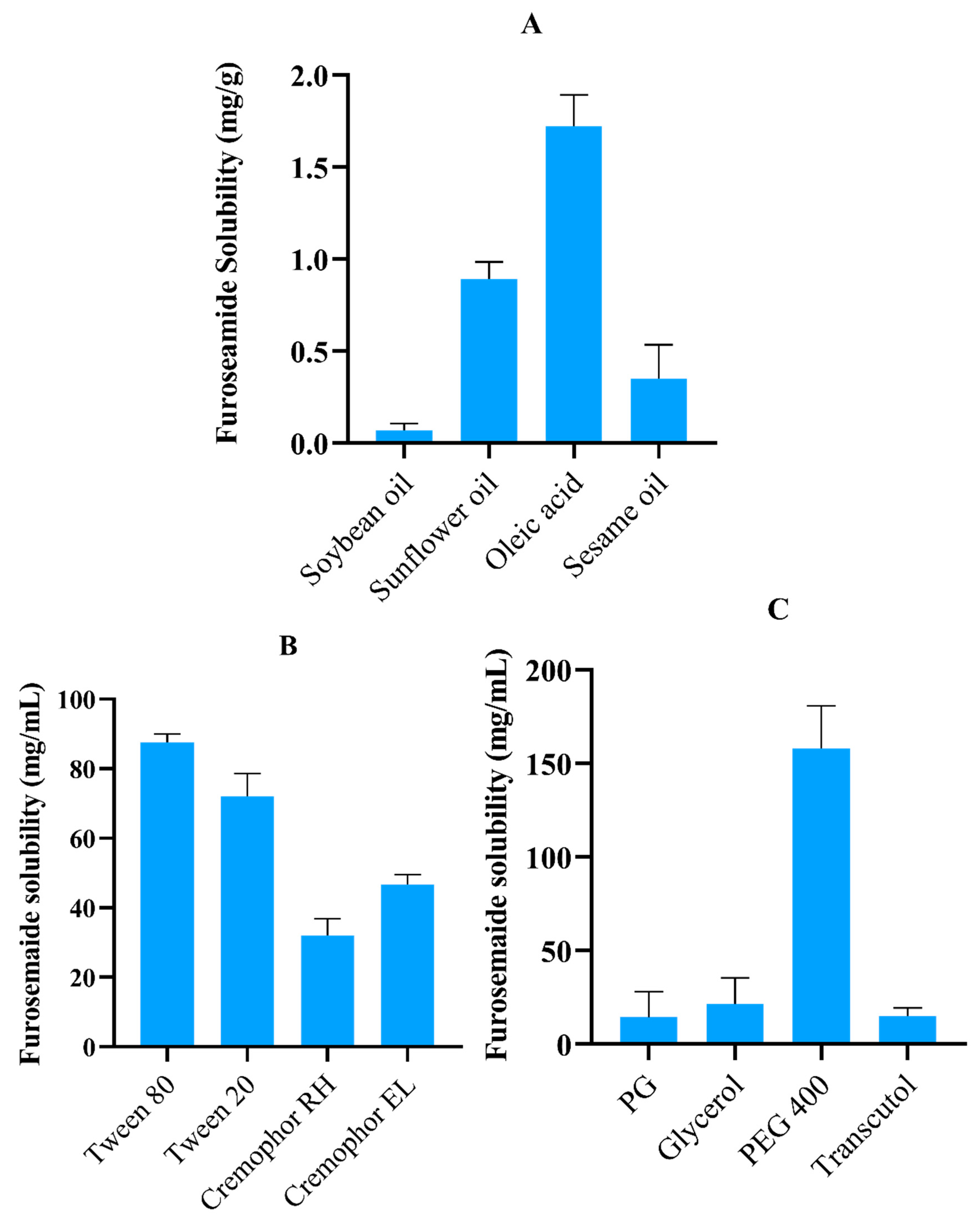
2.1. FSM Solubility Studies
2.2. Surfactant Emulsification Study
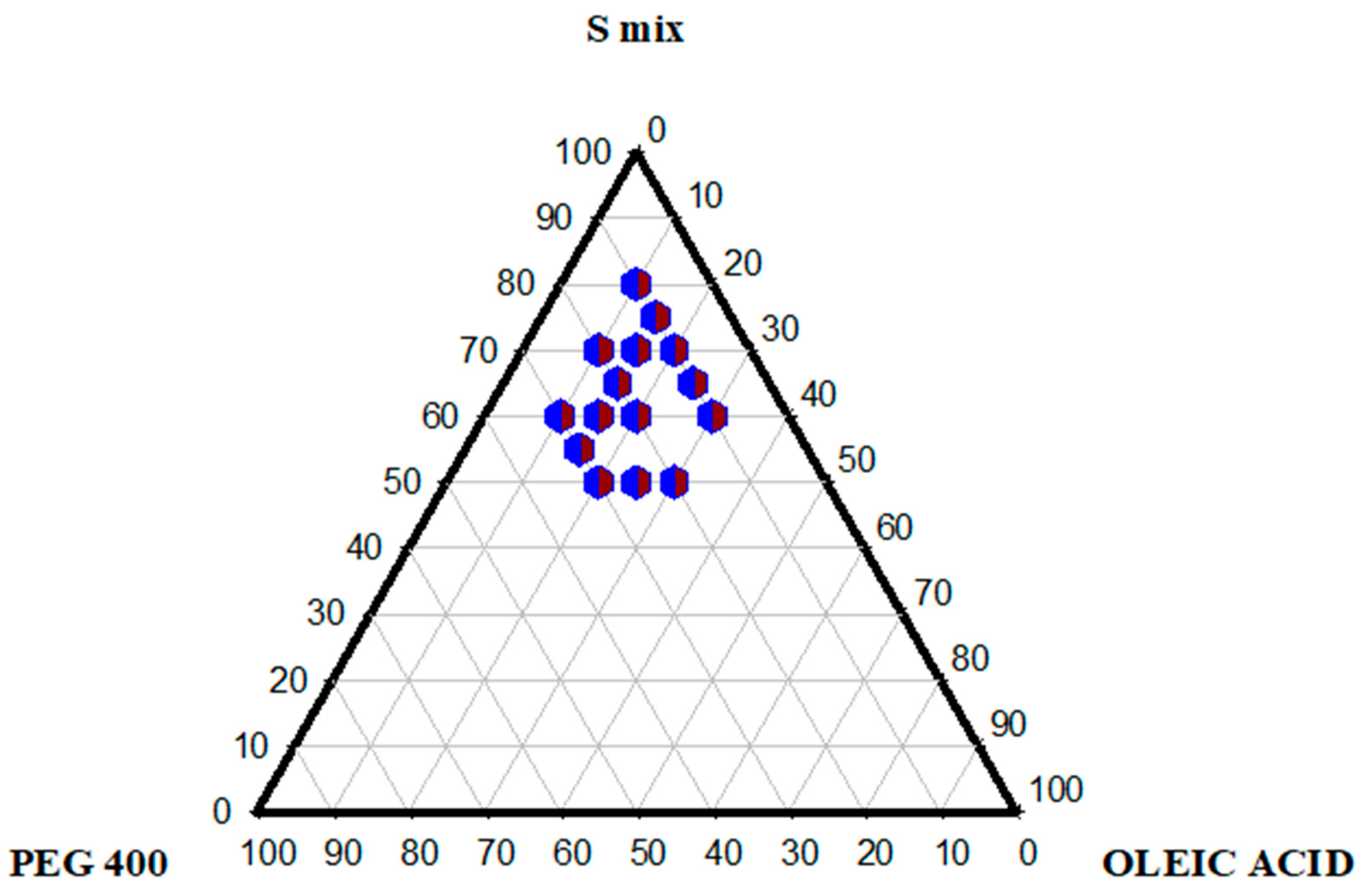
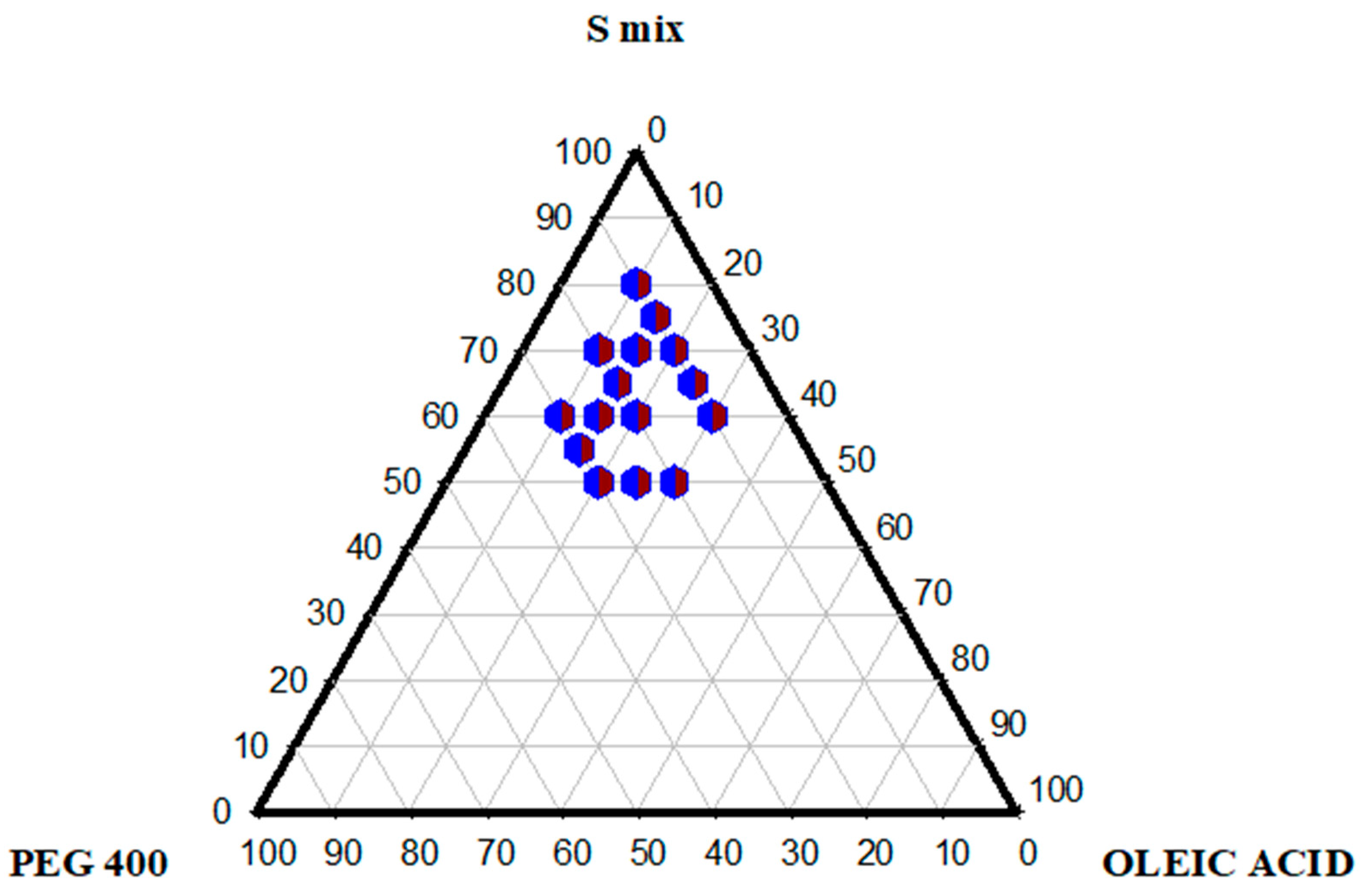
2.3. Pseudo-Ternary Phase Diagram
2.4. Preparation of SNEDDS
2.5. SNEDDS Characterization
2.5.1. Droplet Size, PDI, and Zeta Potential
2.5.2. Thermodynamic Stability
2.5.3. Percentage Transmittance
2.5.4. pH and Dilution Stability
2.5.5. ATR-FTIR
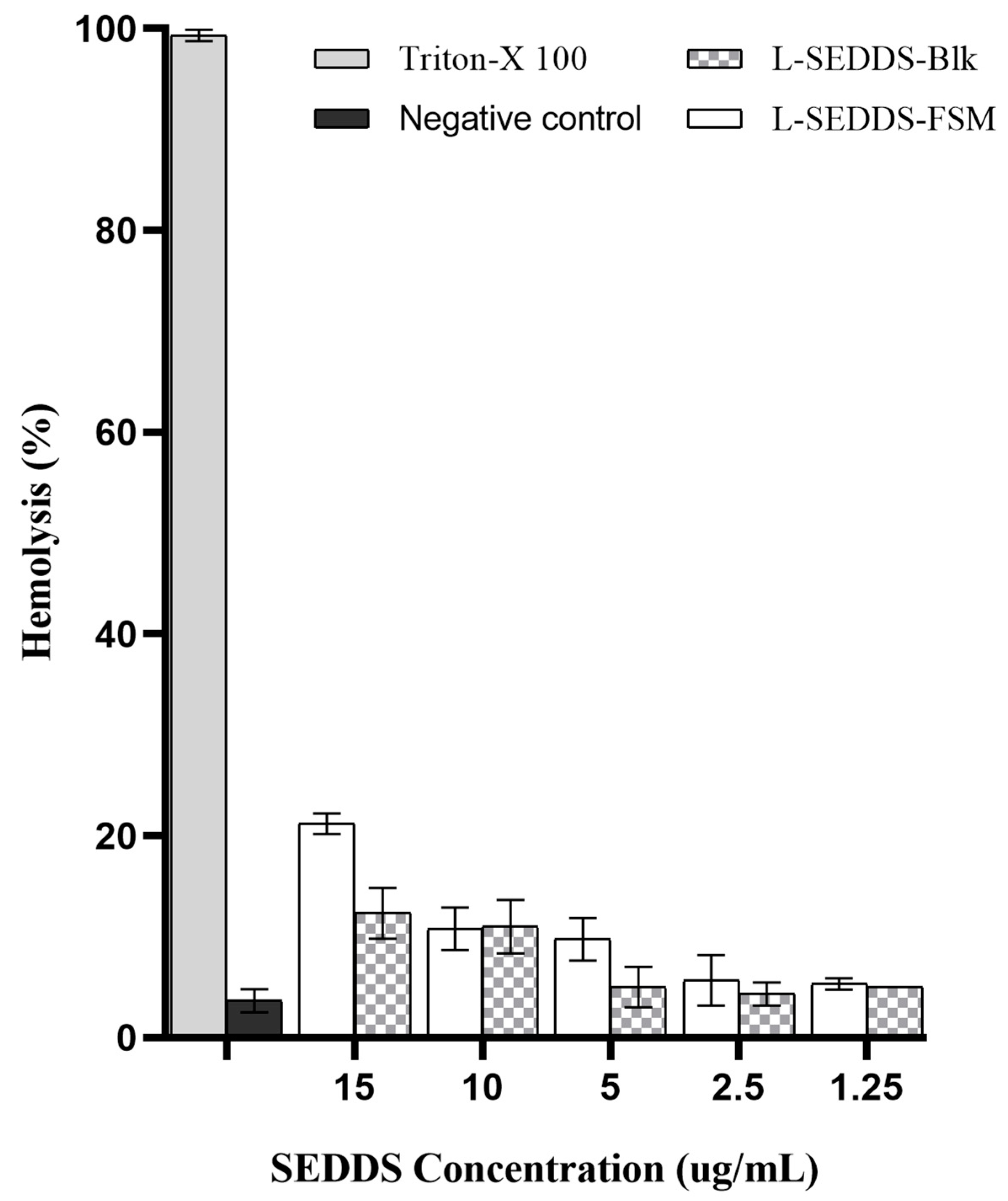
2.6. Hemolysis Studies
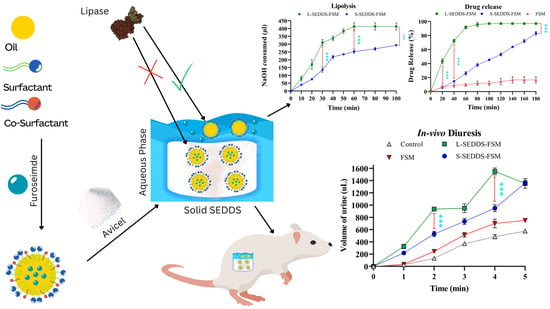
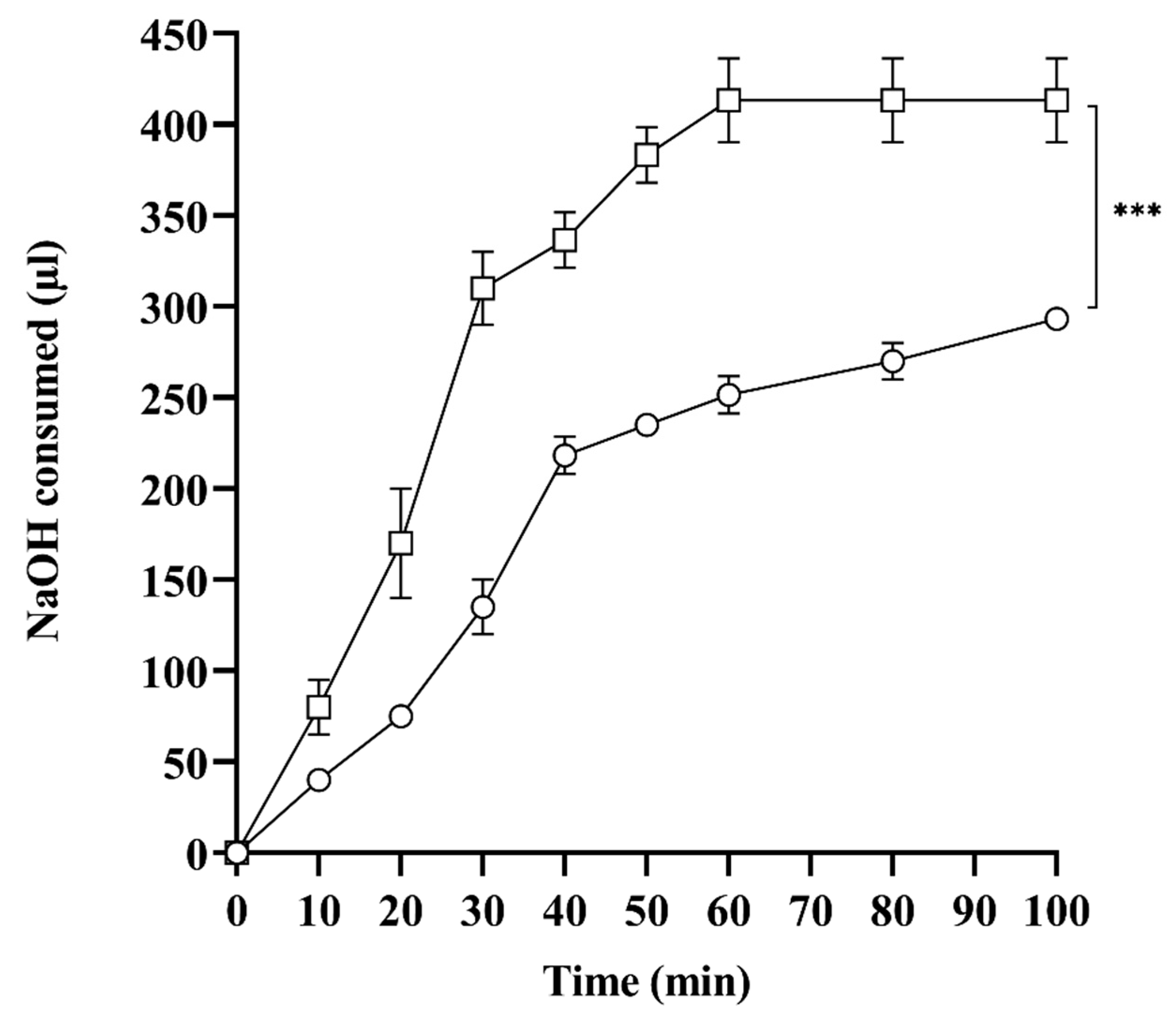
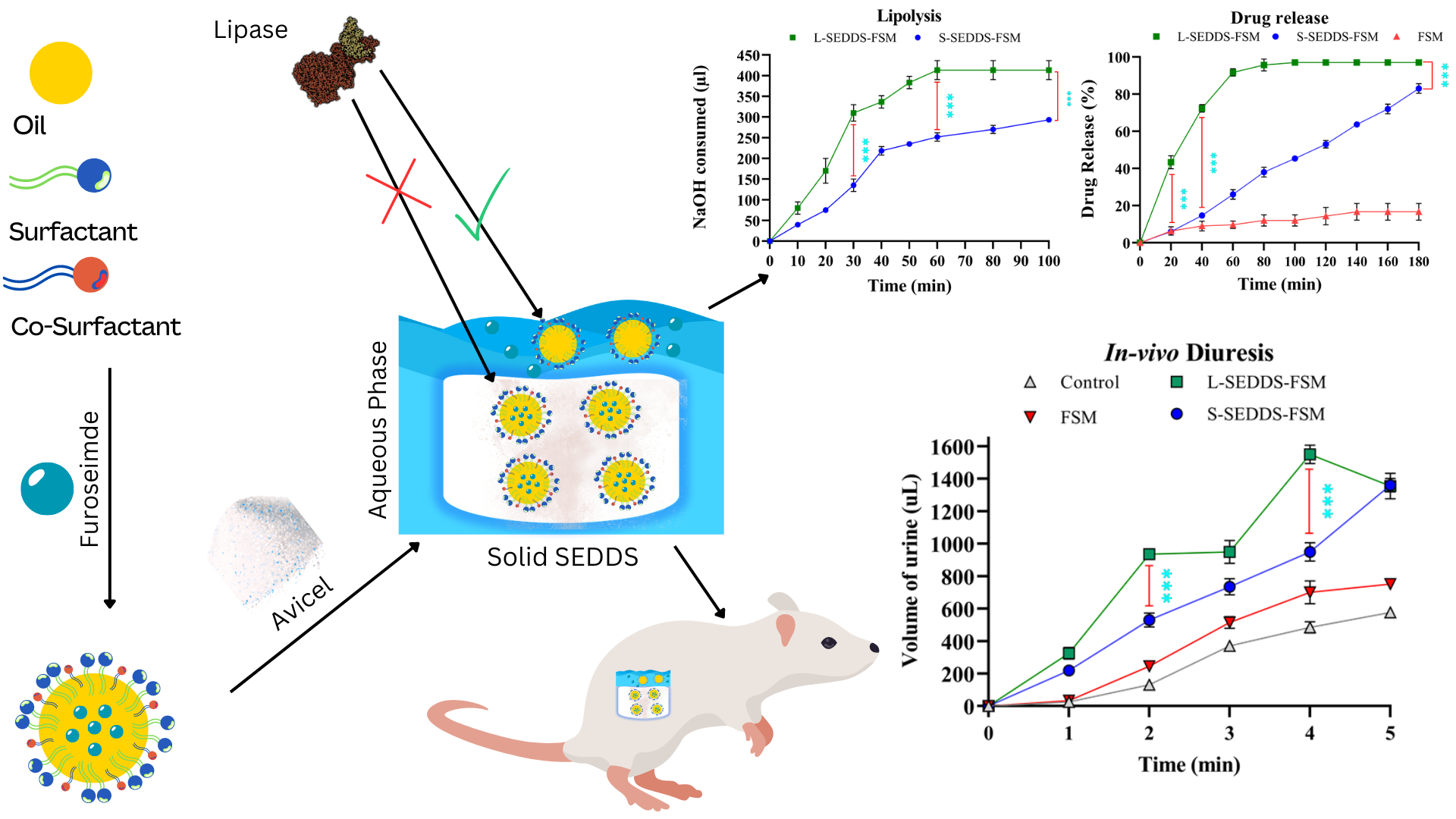
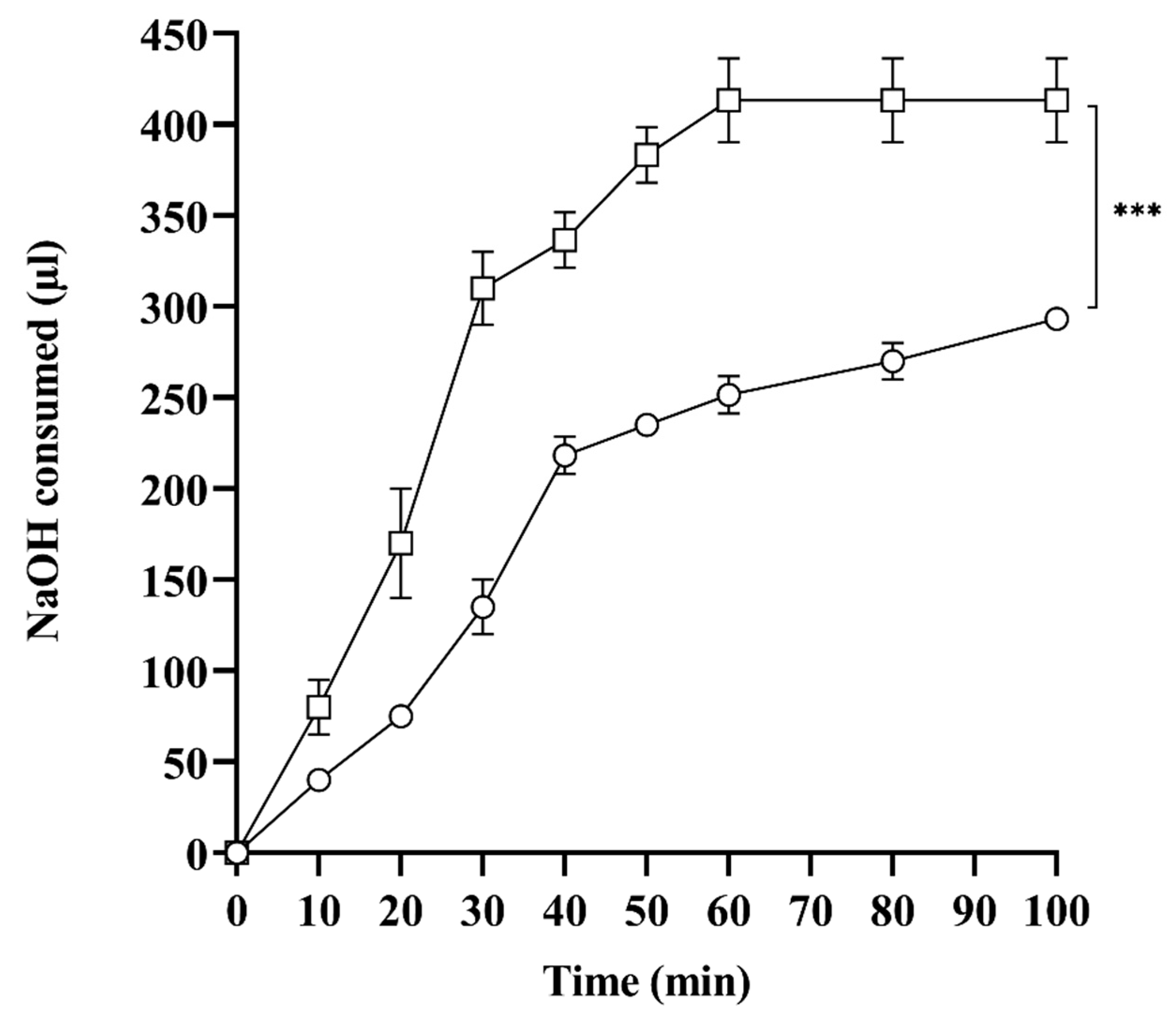
2.7. In Vitro Lipase Assay
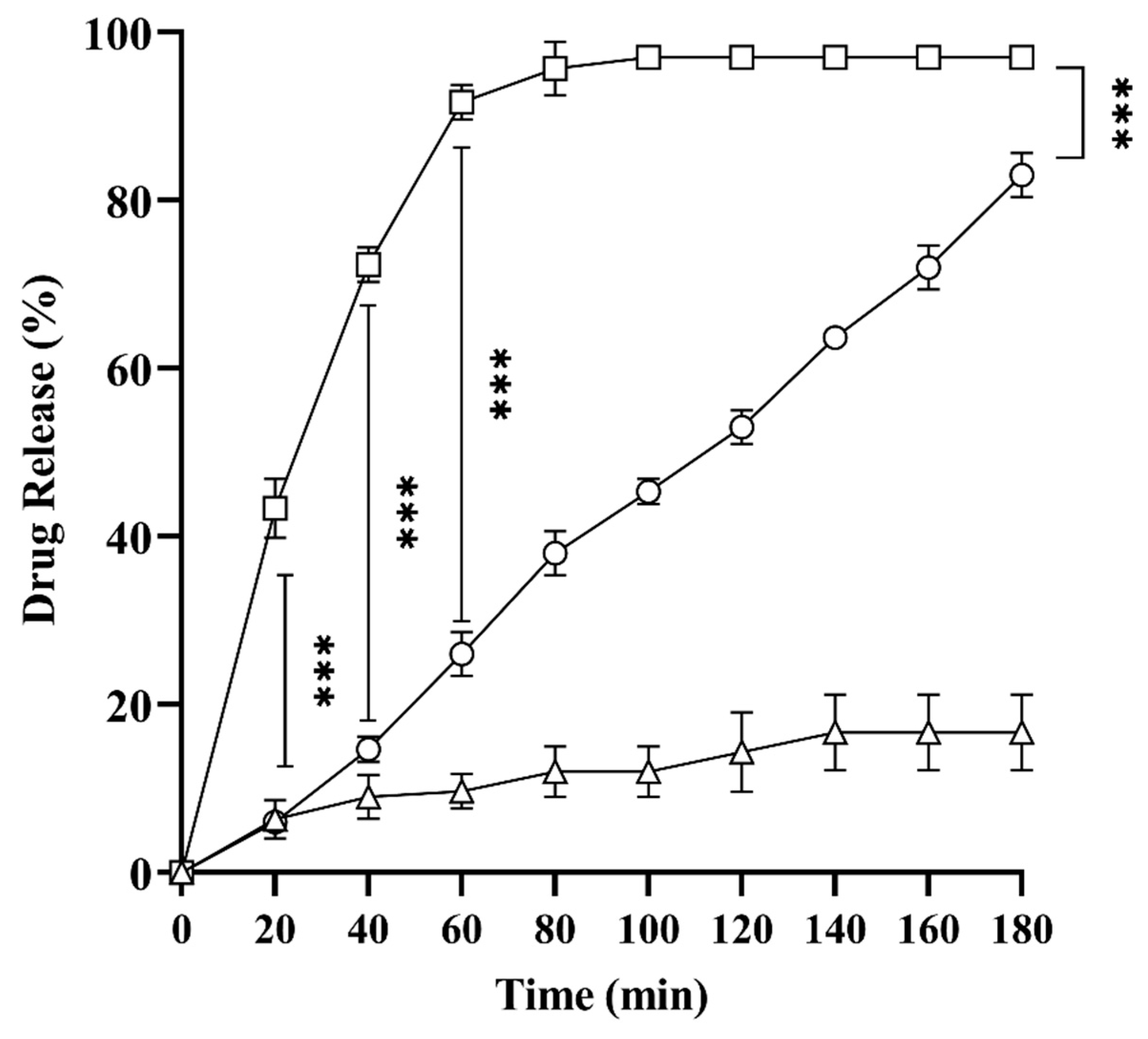
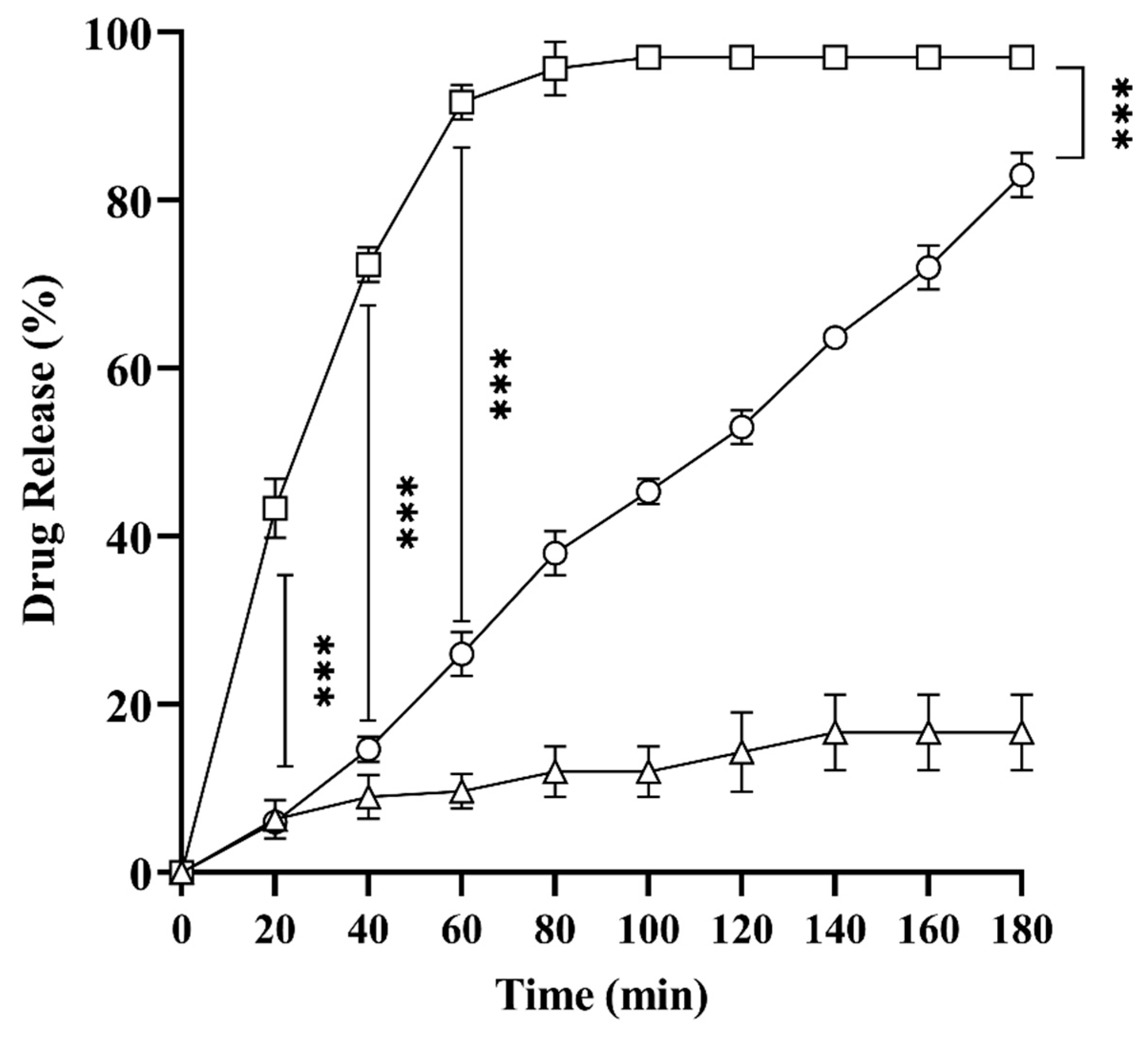
2.8. In Vitro Drug Release Studies
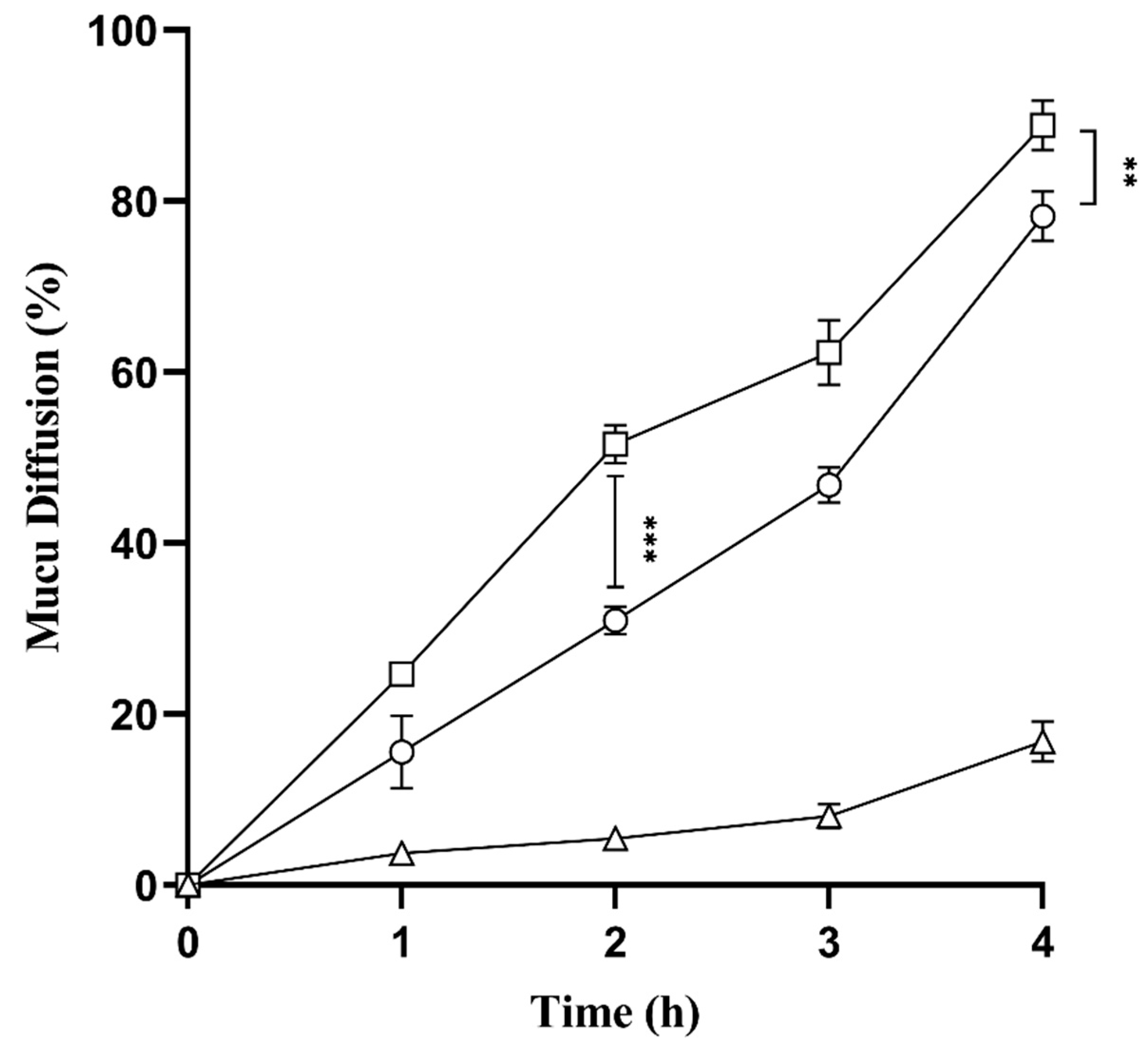
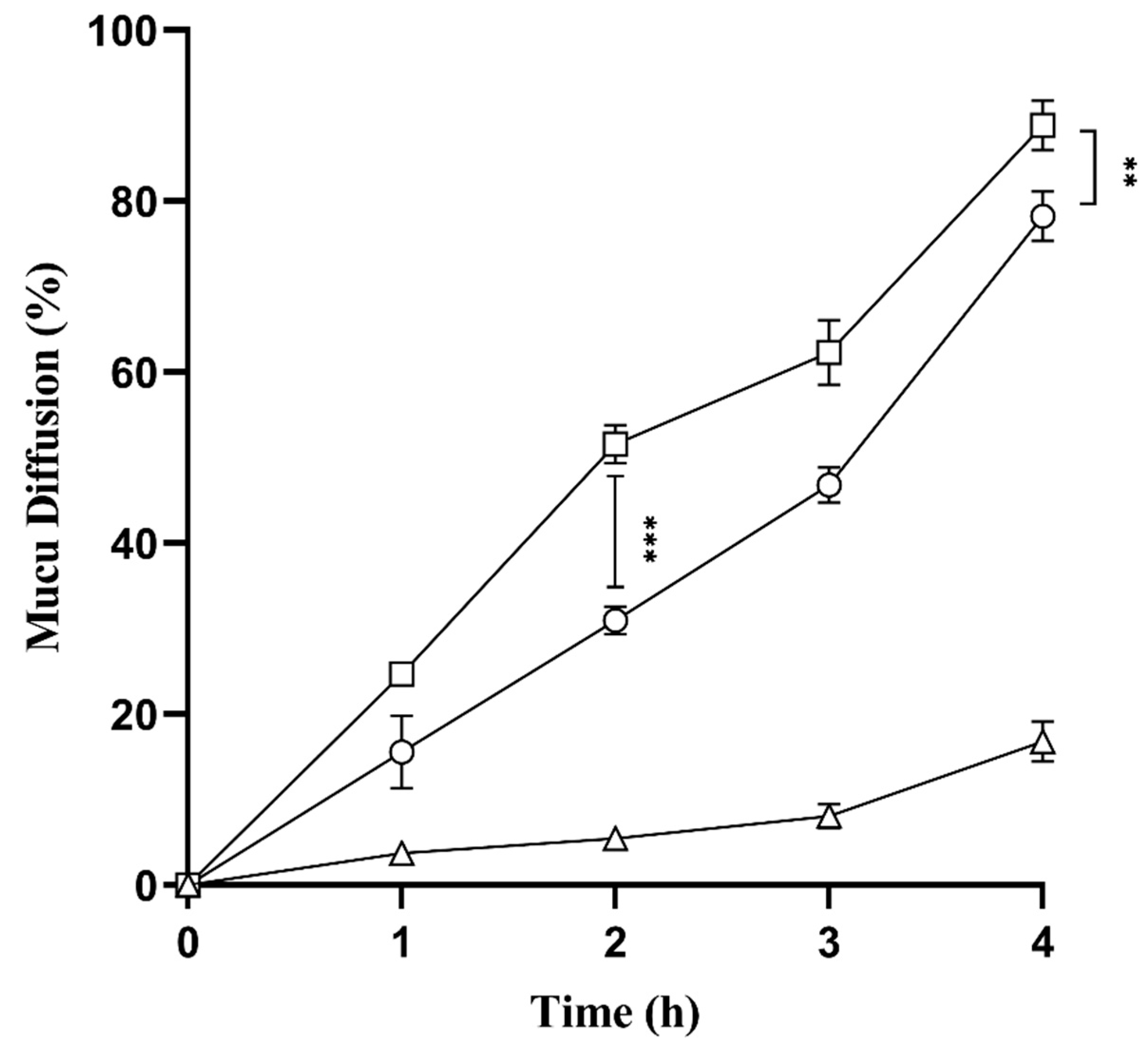
2.9. Mucus Diffusion Studies
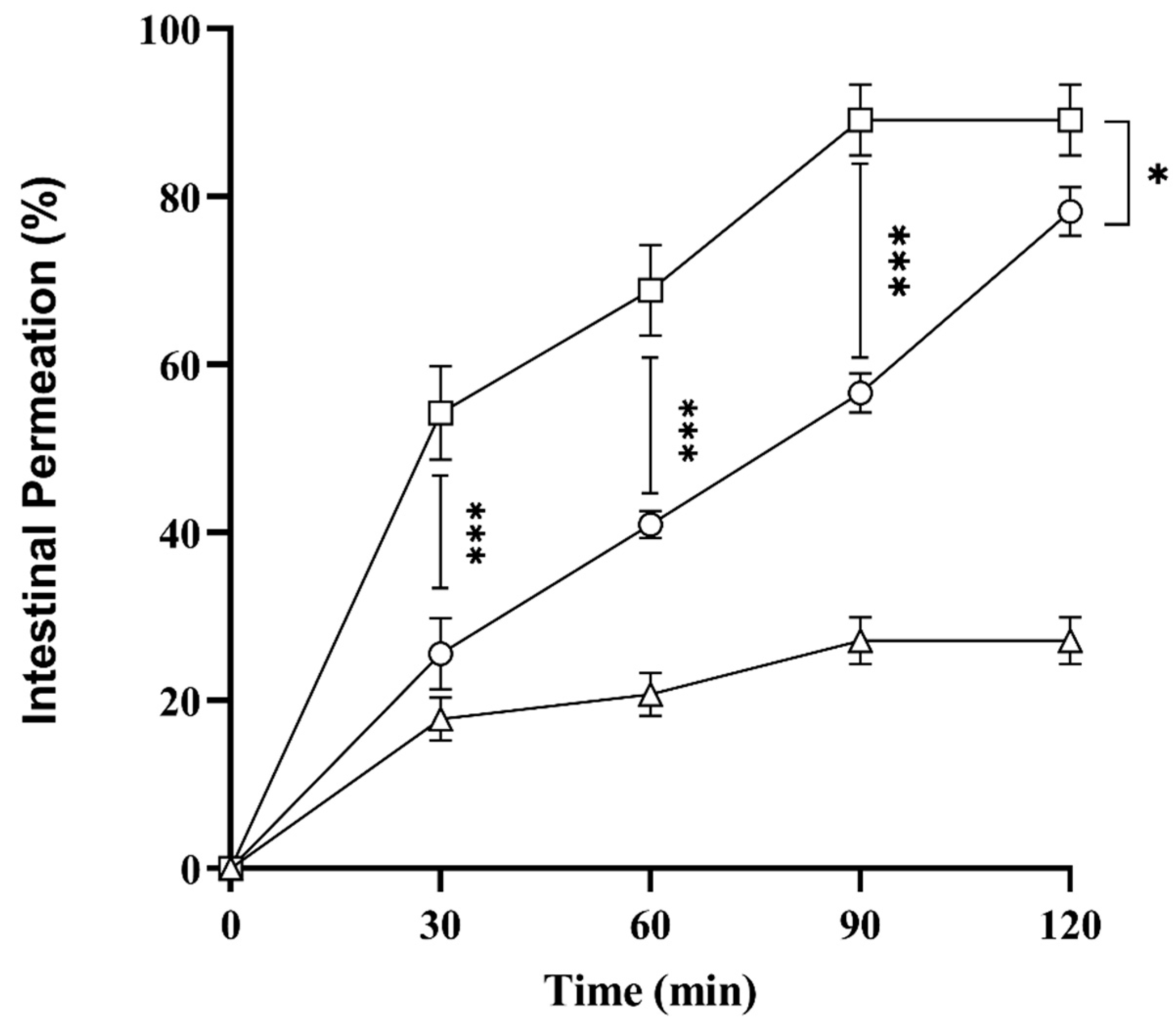
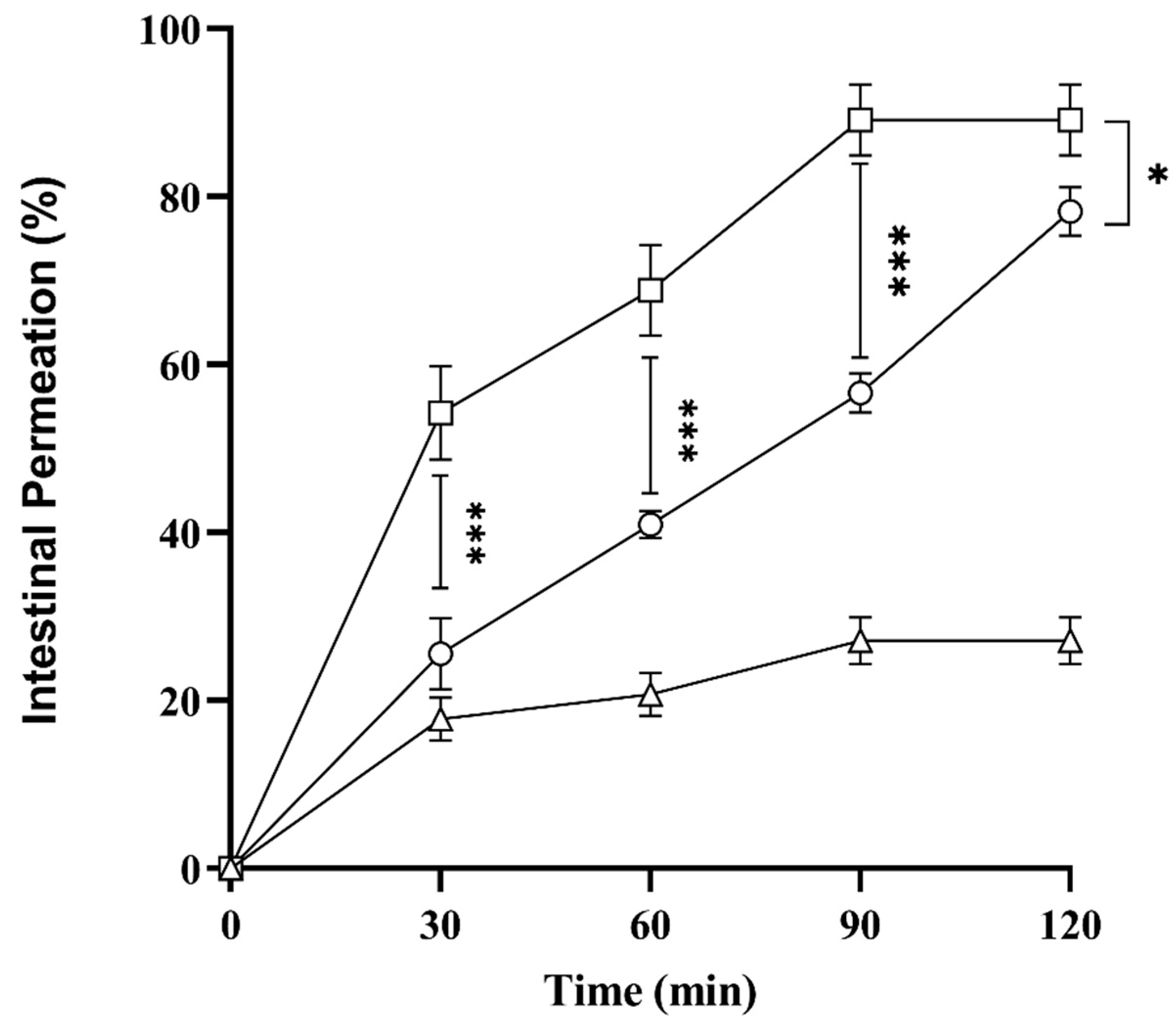
2.10. Ex Vivo Intestinal Permeation Studies
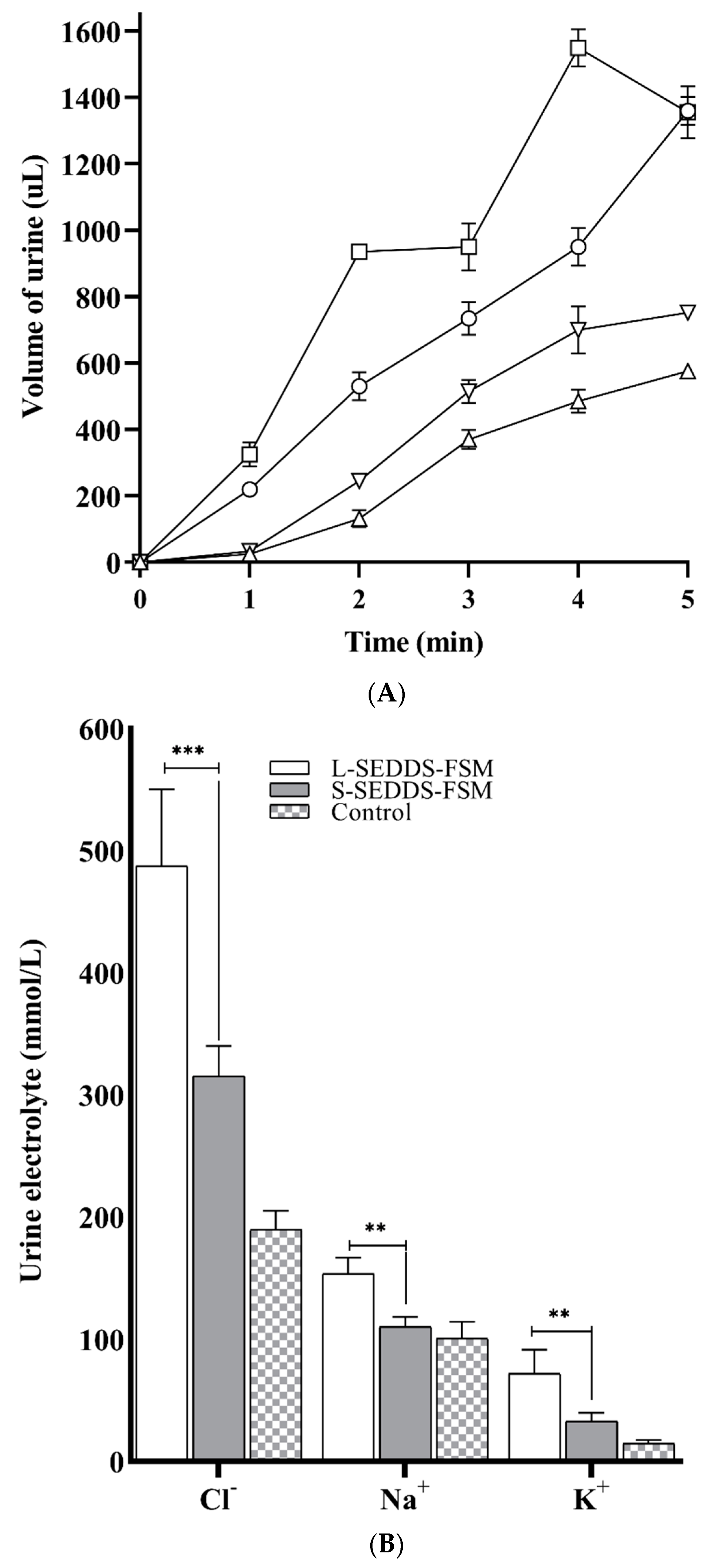
2.11. In Vivo Studies in Mice
3. Material and Methods
3.1. Materials
3.2. Methods
3.2.1. Solubility Studies
3.2.2. Investigation of Surfactant Emulsification
3.2.3. Ternary Phase Diagram
3.2.4. Preparation of SNEDDS Formulations
Preparation of Liquids SNEDDS
Preparation of Solid SNEDDS
3.2.5. SNEDDS Characterization
Droplet Size, Polydispersity Index, Zeta Potential
Thermodynamic Stability
Percent Transmittance
Stability and Robustness to Dilution
FT-IR Analysis
3.2.6. Hemolysis Assay
3.2.7. In Vitro Lipolysis Studies
3.2.8. Mucus Diffusion Studies
3.2.9. In Vitro Drug Release
3.2.10. Ex Vivo Permeation
3.2.11. In Vivo Studies Experimental Design in Mice
3.2.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Markovic, M.; Zur, M.; Ragatsky, I.; Cvijić, S.; Dahan, A. BCS Class IV oral drugs and absorption windows: Regional-dependent intestinal permeability of furosemide. Pharmaceutics 2020, 12, 1175. [Google Scholar] [CrossRef]
- van Olden, R.W.; van Meyel, J.J.; Gerlag, P.G. Acute and long-term effects of therapy with high-dose furosemide in chronic hemodialysis patients. Am. J. Nephrol. 1992, 12, 351–356. [Google Scholar] [CrossRef]
- Roopesh, S.; Reddy, K.; Chandramouli, R.; Soans, D. Enhancing the Solubility of BCS Class II and IV Drugs by Sedds Approach-A Structured Review. J. Pharm. Res. 2016, 15, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Vithani, K.; Jannin, V.; Pouton, C.W.; Boyd, B.J. Colloidal aspects of dispersion and digestion of self-dispersing lipid-based formulations for poorly water-soluble drugs. Adv. Drug Deliv. Rev. 2019, 142, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A.; Jalil, A. Do drug release studies from SEDDS make any sense? J. Control. Release 2018, 271, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Chapa, R.; Li, C.Y.; Basit, A.; Thakur, A.; Ladumor, M.K.; Sharma, S.; Singh, S.; Selen, A.; Prasad, B. Contribution of uptake and efflux transporters to oral pharmacokinetics of furosemide. ACS Omega 2020, 5, 32939–32950. [Google Scholar] [CrossRef]
- Joyce, P.; Dening, T.J.; Meola, T.R.; Schultz, H.B.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2019, 142, 102–117. [Google Scholar] [CrossRef]
- Zhao, T. Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for the Oral Delivery of Lipophilic Drugs. Ph.D. Thesis, University of Trento, Trento, Italy, 2015. [Google Scholar]
- Tran, T.H.; Guo, Y.; Song, D.; Bruno, R.S.; Lu, X. Quercetin-containing self-nanoemulsifying drug delivery system for improving oral bioavailability. J. Pharm. Sci. 2014, 103, 840–852. [Google Scholar] [CrossRef]
- McConville, C.; Friend, D. Development and characterisation of a self-microemulsifying drug delivery systems (SMEDDSs) for the vaginal administration of the antiretroviral UC-781. Eur. J. Pharm. Biopharm. 2013, 83, 322–329. [Google Scholar] [CrossRef]
- Li, P.; Ghosh, A.; Wagner, R.F.; Krill, S.; Joshi, Y.M.; Serajuddin, A.T. Effect of combined use of nonionic surfactant on formation of oil-in-water microemulsions. Int. J. Pharm. 2005, 288, 27–34. [Google Scholar] [CrossRef]
- Kommuru, T.; Gurley, B.; Khan, M.; Reddy, I. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: Formulation development and bioavailability assessment. Int. J. Pharm. 2001, 212, 233–246. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.; Shehata, E.M.; Galal, S.; Abdallah, O.Y. Self-emulsifying preconcentrates of daidzein–phospholipid complex: Design, in vitro and in vivo appraisal. Nanomedicine 2017, 12, 893–910. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Atef, E.; Belmonte, A.A. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Sci. 2008, 35, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Basalious, E.B.; Shawky, N.; Badr-Eldin, S.M. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimization. Int. J. Pharm. 2010, 391, 203–211. [Google Scholar] [CrossRef]
- Kassem, A.A.; Mohsen, A.M.; Ahmed, R.S.; Essam, T.M. Self-nanoemulsifying drug delivery system (SNEDDS) with enhanced solubilization of nystatin for treatment of oral candidiasis: Design, optimization, in vitro and in vivo evaluation. J. Mol. Liq. 2016, 218, 219–232. [Google Scholar] [CrossRef]
- Michels, R.; Foschum, F.; Kienle, A. Optical properties of fat emulsions. Opt. Express 2008, 16, 5907–5925. [Google Scholar] [CrossRef]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef]
- Kyatanwar, A.U.; Jadhav, K.R.; Kadam, V.J. Self micro-emulsifying drug delivery system (SMEDDS). J. Pharm. Res. 2010, 3, 75–83. [Google Scholar]
- Lam, H.T.; Le-Vinh, B.; Phan, T.N.Q.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems and cationic surfactants: Do they potentiate each other in cytotoxicity? J. Pharm. Pharmacol. 2019, 71, 156–166. [Google Scholar] [CrossRef]
- Rohrer, J.; Zupančič, O.; Hetenyi, G.; Kurpiers, M.; Bernkop-Schnuerch, A. Design and evaluation of SEDDS exhibiting high emulsifying properties. J. Drug Deliv. Sci. Technol. 2018, 44, 366–372. [Google Scholar] [CrossRef]
- Kollipara, S.; Gandhi, R.K. Pharmacokinetic aspects and in vitro–in vivo correlation potential for lipid-based formulations. Acta Pharm. Sin. B 2014, 4, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Dangre, P.V.; Gilhotra, R.M.; Dhole, S.N. Formulation and development of solid self micro-emulsifying drug delivery system (S-SMEDDS) containing chlorthalidone for improvement of dissolution. J. Pharm. Investig. 2016, 46, 633–644. [Google Scholar] [CrossRef]
- Trivedi, K.; Patel, P.V.; Pujara, Z. Development and characterization of liquid and solid self-emulsifying drug delivery system of fexofenadine. J. Pharm. Investig. 2013, 43, 385–394. [Google Scholar] [CrossRef]
- Abdulkarim, M.; Sharma, P.K.; Gumbleton, M. Self-emulsifying drug delivery system: Mucus permeation and innovative quantification technologies. Adv. Drug Deliv. Rev. 2019, 142, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Suchaoin, W.; de Sousa, I.P.; Netsomboon, K.; Lam, H.T.; Laffleur, F.; Bernkop-Schnürch, A. Development and in vitro evaluation of zeta potential changing self-emulsifying drug delivery systems for enhanced mucus permeation. Int. J. Pharm. 2016, 510, 255–262. [Google Scholar] [CrossRef]
- Buyukozturk, F.; Benneyan, J.C.; Carrier, R.L. Impact of emulsion-based drug delivery systems on intestinal permeability and drug release kinetics. J. Control. Release 2010, 142, 22–30. [Google Scholar] [CrossRef]
- Adin, D.; Atkins, C.; Papich, M. Pharmacodynamic assessment of diuretic efficacy and braking in a furosemide continuous infusion model. J. Vet. Cardiol. 2018, 20, 92–101. [Google Scholar] [CrossRef]
- Jassim, Z.E. Formulation and evaluation of furosemide liquisolid compact. Int. J. Appl. Pharm. 2017, 9, 39–48. [Google Scholar] [CrossRef]
- Mose, F.H.; Oczachowska-Kulik, A.E.; Fenton, R.A.; Bech, J.N. Effect of furosemide on body composition and urinary proteins that mediate tubular sodium and sodium transport—A randomized controlled trial. Physiol. Rep. 2021, 8, e14653. [Google Scholar] [CrossRef]
- Shahzadi, I.; Nazir, I.; Phan, T.N.Q.; Bernkop-Schnürch, A. About the impact of superassociation of hydrophobic ion pairs on membrane permeability. Eur. J. Pharm. Biopharm. 2020, 151, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Nagarsenker, M. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int. J. Pharm. 2007, 329, 166–172. [Google Scholar] [CrossRef] [PubMed]
- ElKasabgy, N.A. Ocular supersaturated self-nanoemulsifying drug delivery systems (S-SNEDDS) to enhance econazole nitrate bioavailability. Int. J. Pharm. 2014, 460, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Meena, A.K.; Sharma, K.; Kandaswamy, M.; Rajagopal, S.; Mullangi, R. Formulation development of an albendazole self-emulsifying drug delivery system (SEDDS) with enhanced systemic exposure. Acta Pharm. 2012, 62, 563–580. [Google Scholar] [PubMed]
- Jalil, A.; Asim, M.H.; Akkus, Z.B.; Schoenthaler, M.; Matuszczak, B.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems comprising chlorhexidine and alkyl-EDTA: A novel approach for augmented antimicrobial activity. J. Mol. Liq. 2019, 295, 111649. [Google Scholar] [CrossRef]
- Bhargavi, P.; Naha, A.; Kannan, S.; Rama, A. Development, evaluation and optimization of solid self emulsifying drug delivery system (s-sedds) of lercanidipine hydrochloride. Res. J. Pharm. Technol. 2020, 13, 4931–4940. [Google Scholar] [CrossRef]
- Jalil, A.; Asim, M.H.; Nazir, I.; Matuszczak, B.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems containing hydrophobic ion pairs of polymyxin B and agaric acid: A decisive strategy for enhanced antimicrobial activity. J. Mol. Liq. 2020, 311, 113298. [Google Scholar] [CrossRef]
- Kallakunta, V.R.; Bandari, S.; Jukanti, R.; Veerareddy, P.R. Oral self emulsifying powder of lercanidipine hydrochloride: Formulation and evaluation. Powder Technol. 2012, 221, 375–382. [Google Scholar] [CrossRef]
- Kanwal, T.; Kawish, M.; Maharjan, R.; Ghaffar, I.; Ali, H.S.; Imran, M.; Perveen, S.; Saifullah, S.; Simjee, S.U.; Shah, M.R. Design and development of permeation enhancer containing self-nanoemulsifying drug delivery system (SNEDDS) for ceftriaxone sodium improved oral pharmacokinetics. J. Mol. Liq. 2019, 289, 111098. [Google Scholar] [CrossRef]
- Alayoubi, A.; Aqueel, M.S.; Cruz, C.N.; Ashraf, M.; Zidan, A.S. Application of in vitro lipolysis for the development of oral self-emulsified delivery system of nimodipine. Int. J. Pharm. 2018, 553, 441–453. [Google Scholar] [CrossRef]
- Shahzadi, I.; Dizdarević, A.; Efiana, N.A.; Matuszczak, B.; Bernkop-Schnürch, A. Trypsin decorated self-emulsifying drug delivery systems (SEDDS): Key to enhanced mucus permeation. J. Colloid Interface Sci. 2018, 531, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Cil, O.; Haggie, P.M.; Phuan, P.-w.; Tan, J.-A.; Verkman, A.S. Small-molecule inhibitors of pendrin potentiate the diuretic action of furosemide. J. Am. Soc. Nephrol. JASN 2016, 27, 3706. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemicals | FSM (mg) | Avicel | Oleic Acid (µL) | Cremophor EL (µL) | Tween 80 (µL) | Tween 20 (µL) | PEG 400 (µL) | Zeta Size (nm) | Zeta Potential (mV) |
|---|---|---|---|---|---|---|---|---|---|
| L-SEDDS-Blk | - | 200 | 400 | 200 | 100 | 100 | 116 ± 4 | −6.3 | |
| L-SEDDS-FSM | 53 | - | 200 | 400 | 200 | 100 | 100 | 115 ± 2.6 | −5.4 |
| S-SEDDS-FSM | 53 | 1:1 | 200 | 400 | 200 | 100 | 100 | 116 ± 4.3 | −4.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gul, S.; Sridhar, S.B.; Jalil, A.; Akhlaq, M.; Arshad, M.S.; Sarwar, H.S.; Usman, F.; Shareef, J.; Thomas, S. Solid Self-Nanoemulsifying Drug Delivery Systems of Furosemide: In Vivo Proof of Concept for Enhanced Predictable Therapeutic Response. Pharmaceuticals 2024, 17, 500. https://doi.org/10.3390/ph17040500
Gul S, Sridhar SB, Jalil A, Akhlaq M, Arshad MS, Sarwar HS, Usman F, Shareef J, Thomas S. Solid Self-Nanoemulsifying Drug Delivery Systems of Furosemide: In Vivo Proof of Concept for Enhanced Predictable Therapeutic Response. Pharmaceuticals. 2024; 17(4):500. https://doi.org/10.3390/ph17040500
Chicago/Turabian StyleGul, Sania, Sathvik Belagodu Sridhar, Aamir Jalil, Muhammad Akhlaq, Muhammad Sohail Arshad, Hafiz Shoaib Sarwar, Faisal Usman, Javedh Shareef, and Sabin Thomas. 2024. "Solid Self-Nanoemulsifying Drug Delivery Systems of Furosemide: In Vivo Proof of Concept for Enhanced Predictable Therapeutic Response" Pharmaceuticals 17, no. 4: 500. https://doi.org/10.3390/ph17040500
APA StyleGul, S., Sridhar, S. B., Jalil, A., Akhlaq, M., Arshad, M. S., Sarwar, H. S., Usman, F., Shareef, J., & Thomas, S. (2024). Solid Self-Nanoemulsifying Drug Delivery Systems of Furosemide: In Vivo Proof of Concept for Enhanced Predictable Therapeutic Response. Pharmaceuticals, 17(4), 500. https://doi.org/10.3390/ph17040500