
FRET Assays for the Identification of C. albicans HSP90-Sba1 and Human HSP90α-p23 Binding Inhibitors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
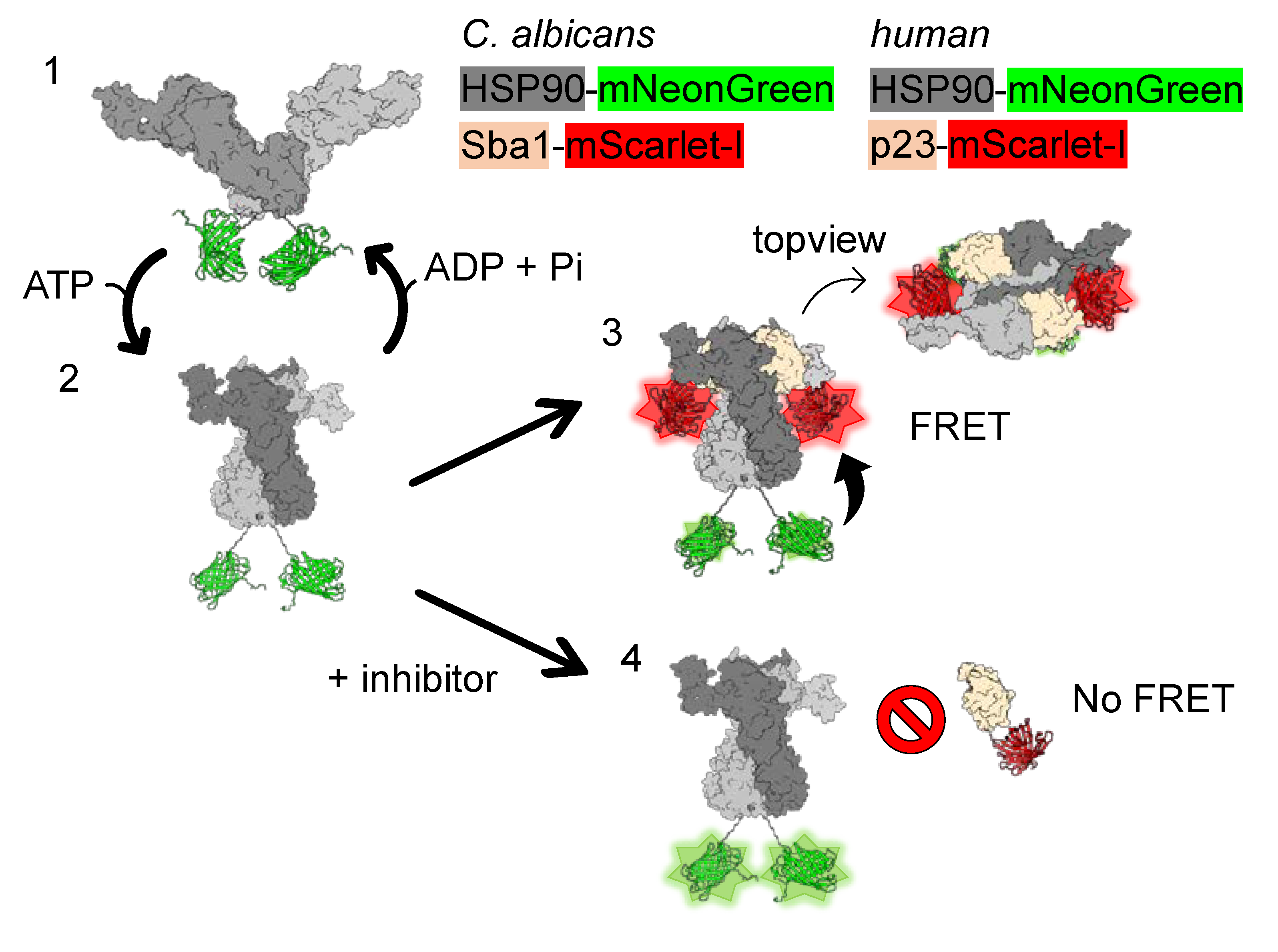
2.1. Development of FRET Assays to Characterize Fungal HSP90–Sba1 and Human HSP90α–p23 Binding
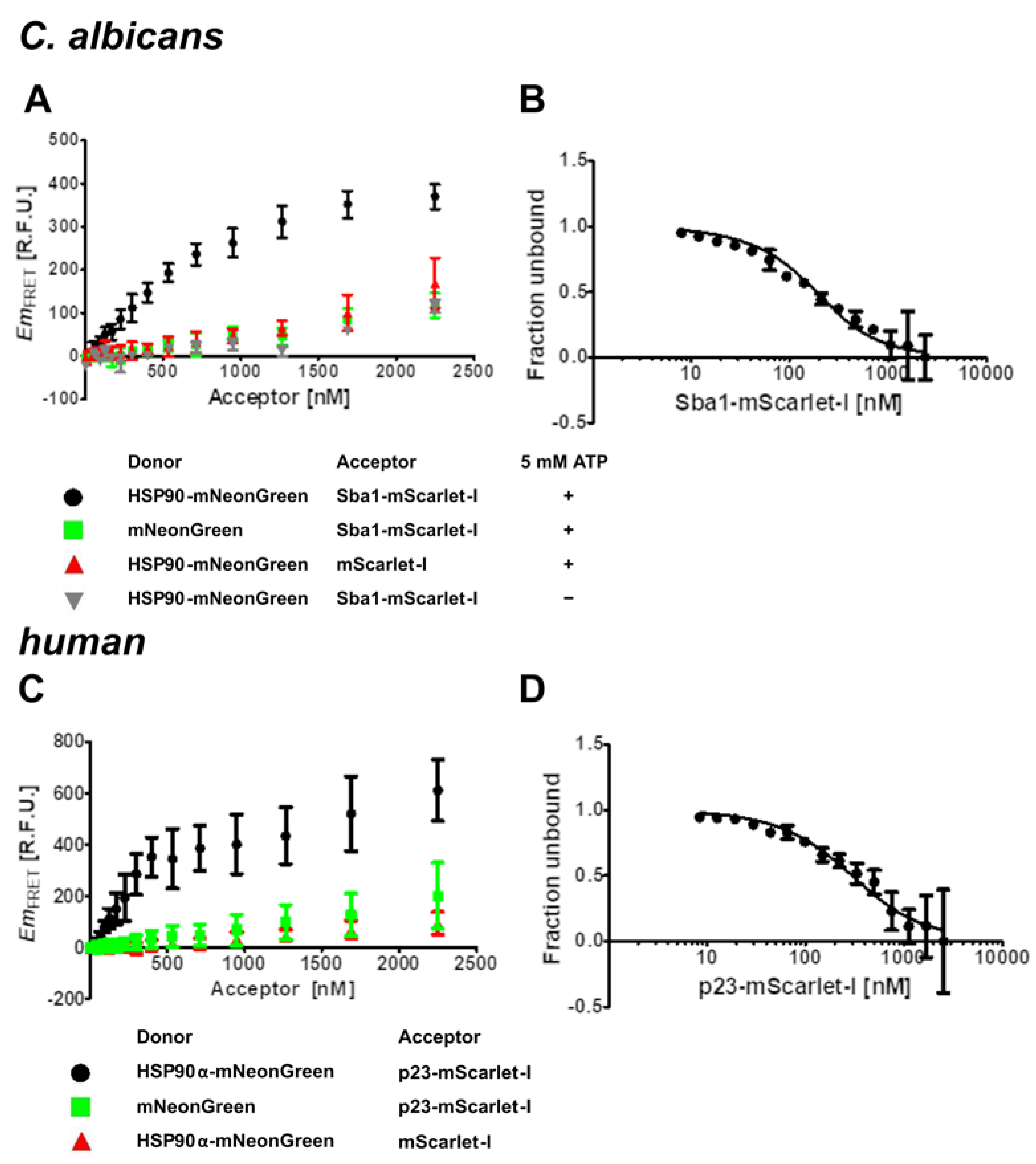
2.2. Kd Determination for C. albicans HSP90–Sba1 and Human HSP90α–p23 Binding
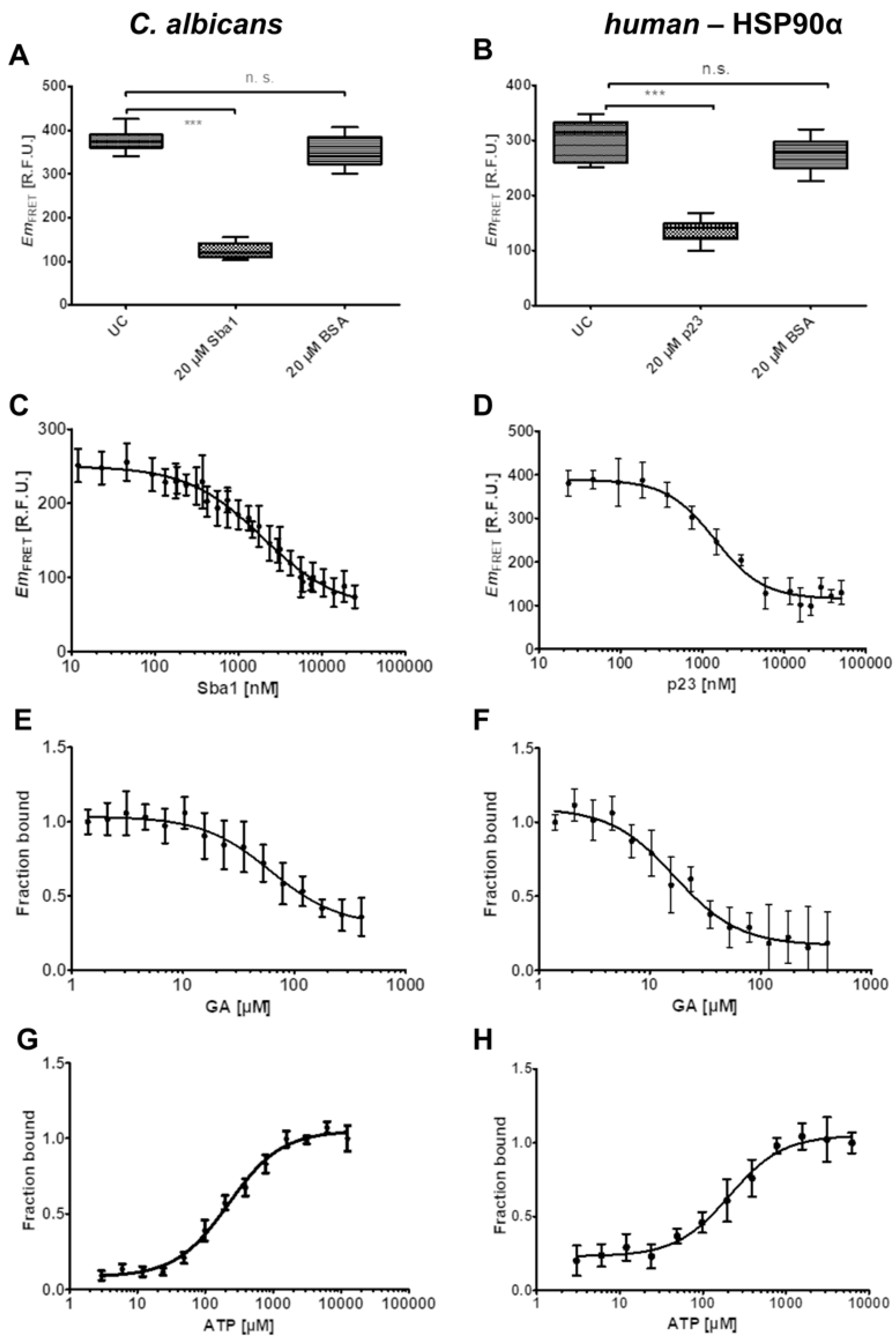
2.3. Influence of Unlabeled Sba1 on HSP90-mNeonGreen Binding to Sba1-mScarlet-I and Unlabeled p23 on Binding of HSP90α-mNeonGreen to p23-mScarlet-I
2.4. Influence of Geldanamycin on the Binding of HSP90-Sba1 and HSP90α-p23
2.5. Influence of ATP on HSP90-Sba1/p23 Binding and Optimization for Inhibitor Screening
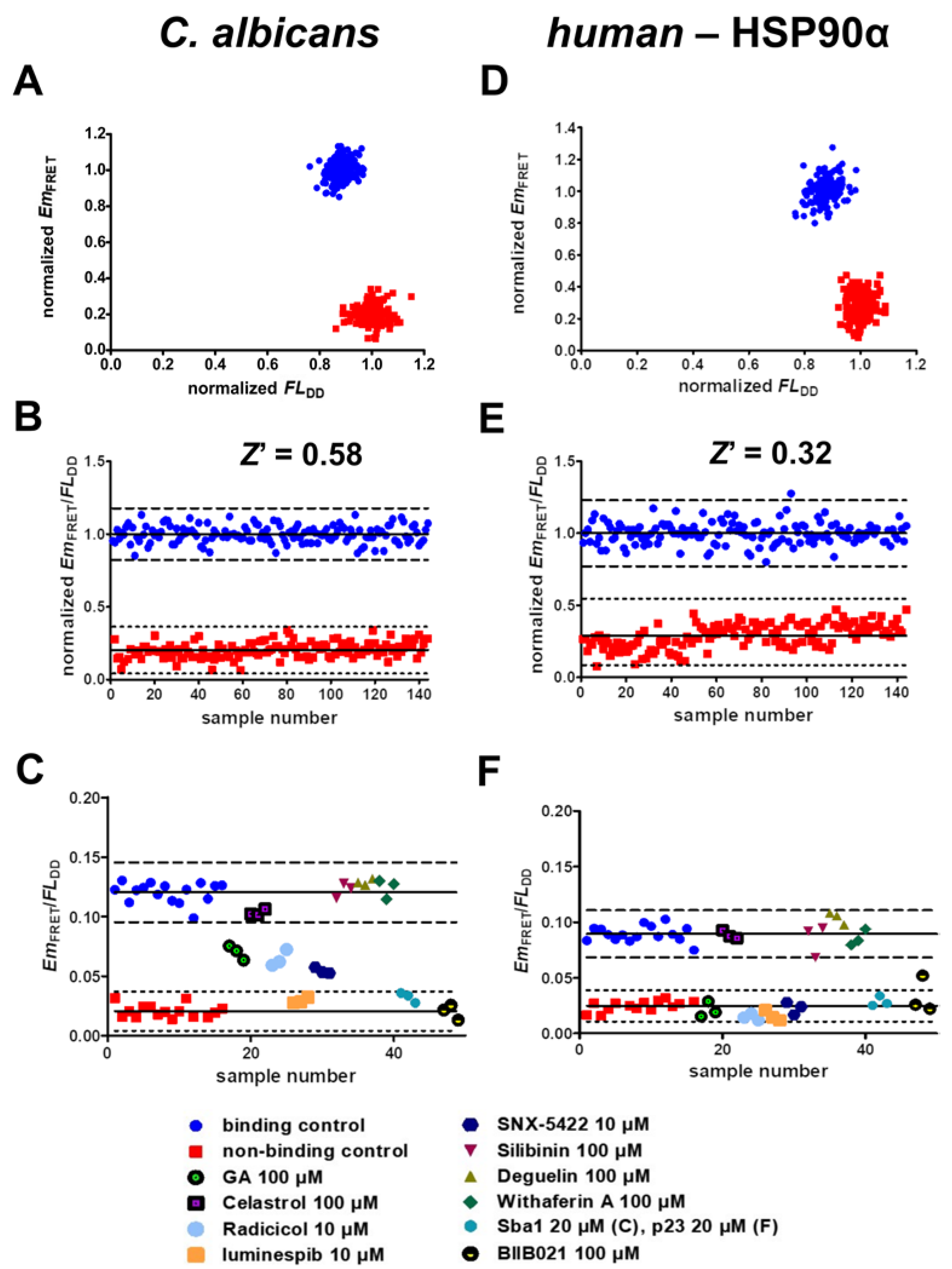
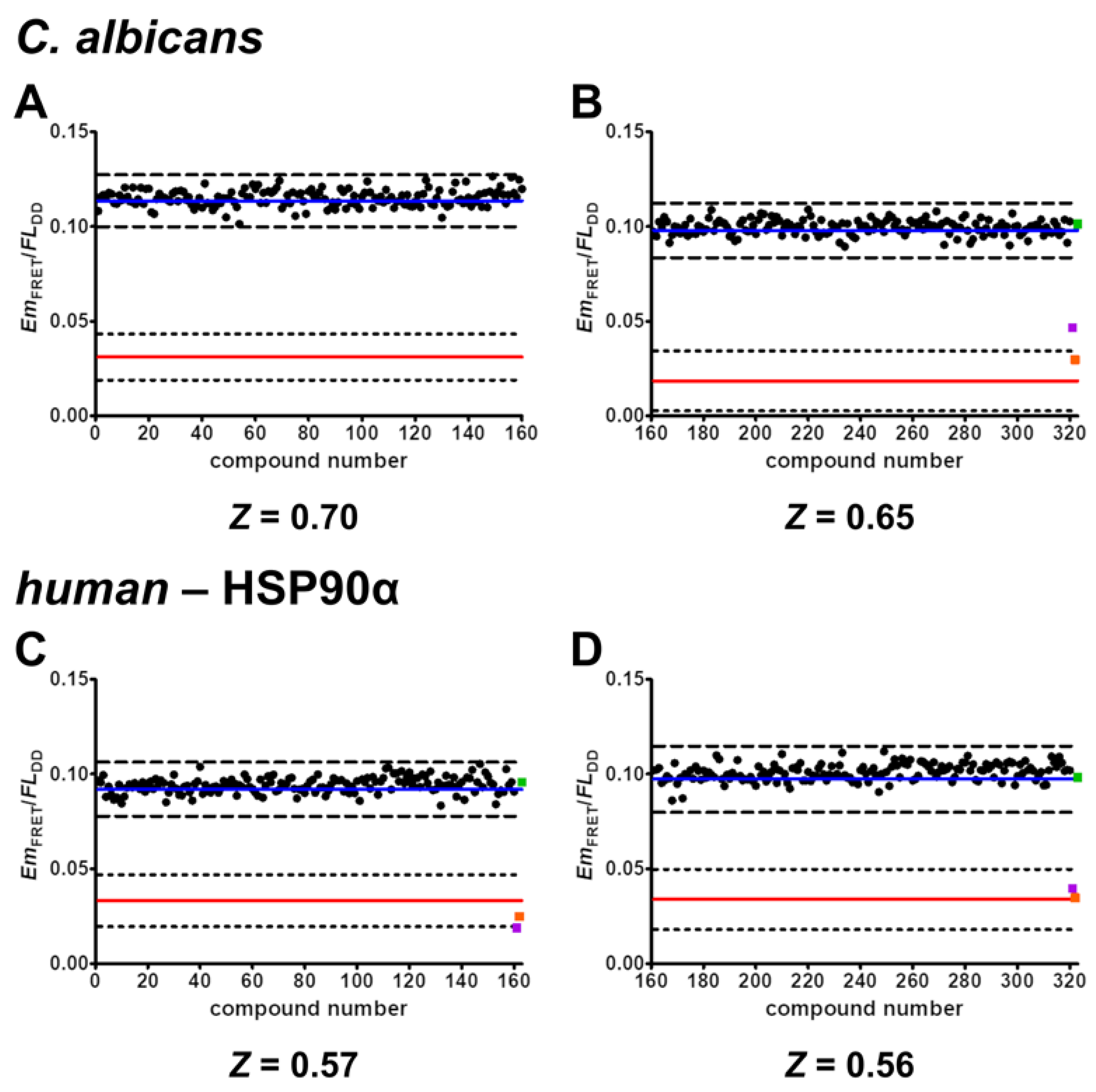
2.6. Validation of Screening Assay Conditions
2.7. Screening of Nucleoside-Mimetics Library
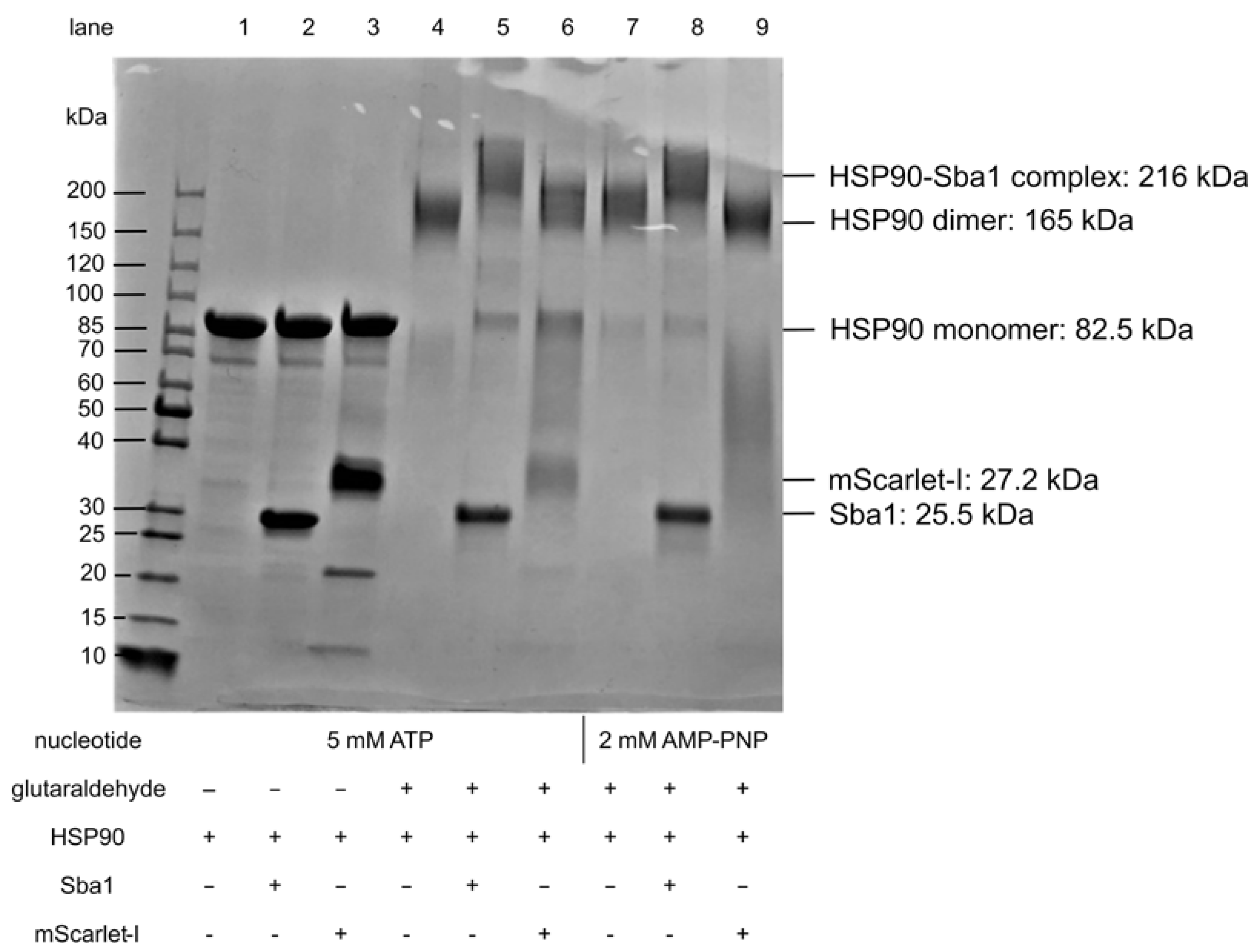
2.8. Verification of C. albicans HSP90 Homodimers and HSP90–Sba1 Complex
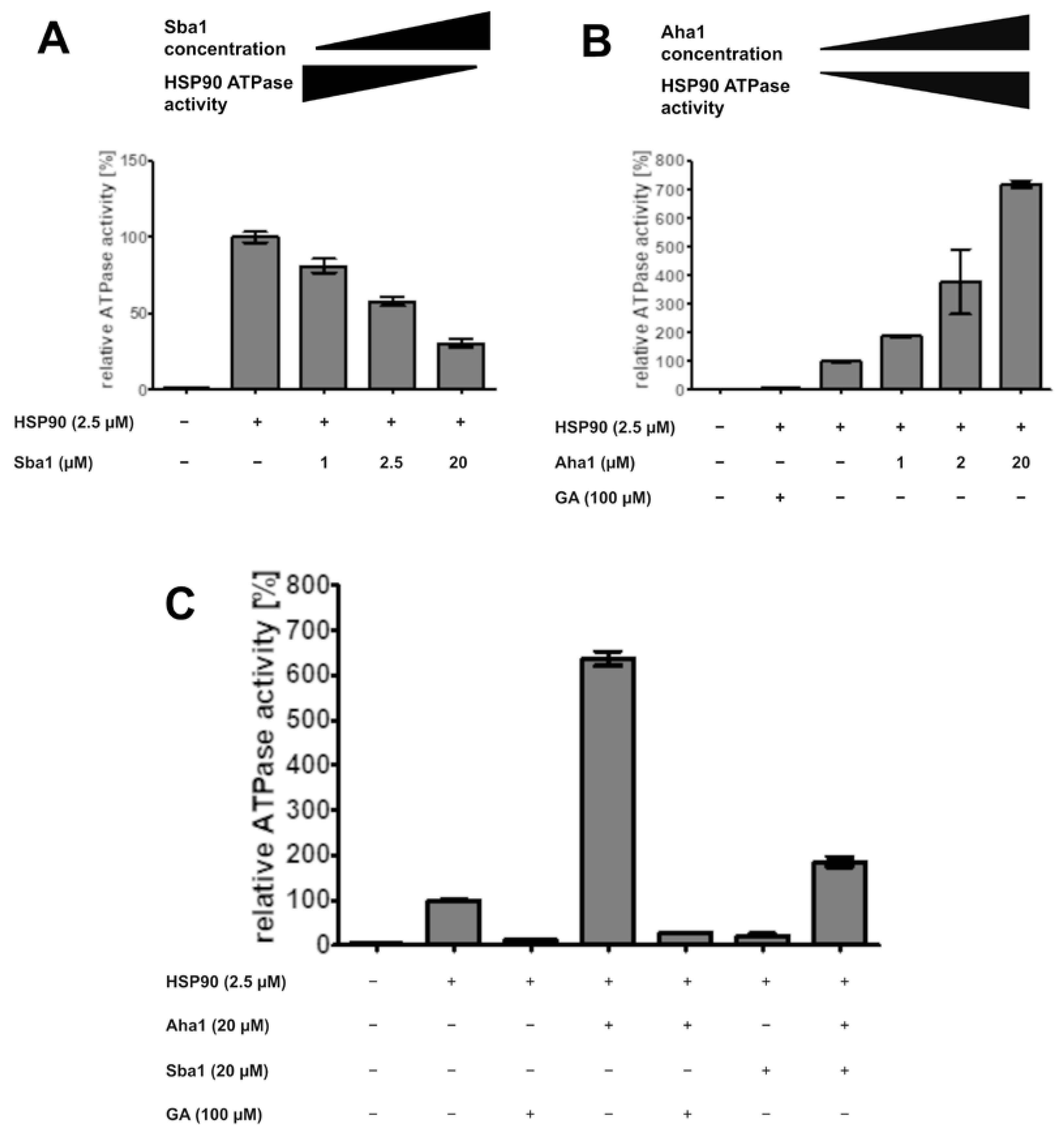
2.9. Effect of Co-Chaperones and Geldanamycin on ATPase Activity of C. albicans HSP90
3. Discussion
3.1. HSP90-Sba1 and HSP90α-p23 Binding as Drug Targets for Antifungal Drug Development or Cancer Therapy
3.2. ATP-Competitive Inhibitors
3.3. HTS in HSP90 Drug Discovery
3.4. Main Findings
- The developed FRET assays can reliably detect inhibitors of C. albicans HSP90–Sba1 and human homologous HSP90α–p23 binding as well as ATP-competitive HSP90 inhibitors that disrupt this binding. They show high robustness and low data variability with determined Z’ factors of 0.58 and 0.32. Following the definition of Zhang et al., the Z’ factor was calculated using only the control data. The Z’ factor thus represents a parameter that can be utilized for quality assessment in assay development and optimization. The Z factor is calculated including the signal of screened compounds of a screening assay [37]. For the conducted screening in duplicates, a Z factor of 0.68 and 0.57 could be achieved. The Z factors were in our case higher than Z’ because they were calculated based on the average signal of the compounds screened in duplicates. Screening in duplicates thus led to a higher robustness of the assay readout. This demonstrates the high-throughput capability of the assays. Due to the simplicity of the assays, they are suitable for automated liquid handling and screening of large compound libraries. Further miniaturization of the current 384-well to a 1536-well format could be feasible to improve throughput and reduce protein and compound consumption. However, in the current format, the assays can also be considered as HTS-capable.
- PAINs can be detected by the developed assays. If the donor emission is also reduced concomitantly with a decrease in FRET emission, these compounds can be identified as interfering with the readout signal and not disrupting the PPI per se. In this way, PAINs can be excluded from further investigation already at the stage of first screening in a simple way. The recognition of EGCG as a PAIN illustrates this capability.
- For the potential discovery of new antifungal drugs, selectivity for the pathogenic target structure is of high importance for circumventing host toxicity and reducing the risk of adverse effects. With the assays, identified hit structures can be characterized for their species selectivity by determining the IC50,rel values of both fungal and human protein interactions.
- Upon hit finding, we propose checking for ATP-competitiveness by assessing IC50,rel values by varying the ATP concentration in the presence of a fixed hit compound concentration with the FRET assays and/or evaluating the influence of the hit substance on HSP90 ATPase activity with the ADP Glo™ assay. Furthermore, the effects of identified APIs on HSP90-Sba1 complex formation as well as HSP90 homodimerization could be analyzed via cross-linking SDS-PAGE.
- With the assays, accurate Kd values of both C. albicans HSP90-Sba1 and human HSP90α-p23 binding could be determined.
4. Materials and Methods
4.1. Plasmid Construction and Mutagenesis
4.2. Protein Purification
4.3. FRET Measurements
4.3.1. FRET Binding Assays
4.3.2. Characterization of Model C. albicans HSP90–Sba1 and Human HSP90α–p23 Binding Inhibitors via FRET
4.3.3. FRET Inhibitor Screening Assays
4.4. ADP-Glo™ Assay
4.5. Cross-Linking SDS-PAGE Analysis of C. albicans HSP90 dimers and HSP90Sba1 Complex with Glutaraldehyde
4.6. Kd Determination of AMP-PNP Binding to C. albicans HSP90 via Microscale Thermophoresis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, A.; Blagg, B.S.J. Novobiocin and Additional Inhibitors of the Hsp90 C-Terminal Nucleotide- binding Pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [Google Scholar] [CrossRef] [PubMed]
- Söti, C.; Rácz, A.; Csermely, P. A Nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal binding pocket. J. Biol. Chem. 2002, 277, 7066–7075. [Google Scholar] [CrossRef] [PubMed]
- Picard, D. Table of HSP90 Interactors. Available online: https://www.picard.ch/downloads/Hsp90interactors.pdf (accessed on 21 February 2024).
- Chiosis, G.; Digwal, C.S.; Trepel, J.B.; Neckers, L. Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2023, 24, 797–815. [Google Scholar] [CrossRef]
- Prodromou, C.; Bjorklund, D.M. Advances towards Understanding the Mechanism of Action of the Hsp90 Complex. Biomolecules 2022, 12, 600. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Piel, W.H.; Gui, L.; Bruford, E.; Monteiro, A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics 2005, 86, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Siligardi, G.; Hu, B.; Panaretou, B.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 2004, 279, 51989–51998. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef]
- Gupta, A.; Bansal, A.; Hashimoto-Torii, K. HSP70 and HSP90 in neurodegenerative diseases. Neurosci. Lett. 2020, 716, 134678. [Google Scholar] [CrossRef]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in Cancer. Int. J. Mol. Sci. 2021, 22, 317. [Google Scholar] [CrossRef] [PubMed]
- Cano, L.Q.; Lavery, D.N.; Sin, S.; Spanjaard, E.; Brooke, G.N.; Tilman, J.D.; Abroaf, A.; Gaughan, L.; Robson, C.N.; Heer, R.; et al. The co-chaperone p23 promotes prostate cancer motility and metastasis. Mol. Oncol. 2015, 9, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.E.; Lambert, W.M.; Watkins, R.; Giashuddin, S.; Huang, S.J.; Oxelmark, E.; Arju, R.; Hochman, T.; Goldberg, J.D.; Schneider, R.J.; et al. High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res. 2010, 70, 8446–8456. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Cowen, L.E. Roles of Hsp90 in Candida albicans morphogenesis and virulence. Curr. Opin. Microbiol. 2023, 75, 102351. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Xue, W.; Yin, Y.; Liu, H.; Li, S.; Sun, X. The Hsp90 Co-chaperones Sti1, Aha1, and P23 Regulate Adaptive Responses to Antifungal Azoles. Front. Microbiol. 2016, 7, 1571. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, A.; Ferrara, F.; Boccellino, M.; Ponzo, A.; Cimmino, C.; Comberiati, E.; Zovi, A.; Clemente, S.; Sabbatucci, M. Antifungal Drug Resistance: An Emergent Health Threat. Biomedicines 2023, 11, 1063. [Google Scholar] [CrossRef] [PubMed]
- Aldholmi, M.; Marchand, P.; Ourliac-Garnier, I.; Le Pape, P.; Ganesan, A. A Decade of Antifungal Leads from Natural Products: 2010–2019. Pharmaceuticals 2019, 12, 182. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action. Available online: https://iris.who.int/bitstream/handle/10665/363682/9789240060241-eng.pdf?sequence=1 (accessed on 21 February 2024).
- Cosio, T.; Gaziano, R.; Zuccari, G.; Costanza, G.; Grelli, S.; Di Francesco, P.; Bianchi, L.; Campione, E. Retinoids in Fungal Infections: From Bench to Bedside. Pharmaceuticals 2021, 14, 962. [Google Scholar] [CrossRef]
- McCullock, T.W.; MacLean, D.M.; Kammermeier, P.J. Comparing the performance of mScarlet-I, mRuby3, and mCherry as FRET acceptors for mNeonGreen. PLoS ONE 2020, 15, e0219886. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R.; Raicu, V. Comparison between whole distribution- and average-based approaches to the determination of fluorescence resonance energy transfer efficiency in ensembles of proteins in living cells. Biophys. J. 2010, 98, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- James, J.R.; Oliveira, M.I.; Carmo, A.M.; Iaboni, A.; Davis, S.J. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods 2006, 3, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Ji, H.; Everton, D.; Le, A.T.H.; Krylova, S.M.; Fournier, R.; Krylov, S.N. Fundamental Determinants of the Accuracy of Equilibrium Constants for Affinity Complexes. Anal. Chem. 2023, 95, 15826–15832. [Google Scholar] [CrossRef] [PubMed]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. eLife 2020, 9, e57264. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Rukundo, J.-L.; Krylova, S.; Krylov, S. Accuracy Assessment for Equilibrium Dissociation Constant Using a Single Binding Isotherm. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Grenert, J.P.; Johnson, B.D.; Toft, D.O. The importance of ATP binding and hydrolysis by hsp90 in formation and function of protein heterocomplexes. J. Biol. Chem. 1999, 274, 17525–17533. [Google Scholar] [CrossRef] [PubMed]
- Obermann, W.M.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 1998, 143, 901–910. [Google Scholar] [CrossRef]
- Wolmarans, A.; Lee, B.; Spyracopoulos, L.; LaPointe, P. The Mechanism of Hsp90 ATPase Stimulation by Aha1. Sci. Rep. 2016, 6, 33179. [Google Scholar] [CrossRef]
- Richter, K.; Walter, S.; Buchner, J. The Co-chaperone Sba1 connects the ATPase reaction of Hsp90 to the progression of the chaperone cycle. J. Mol. Biol. 2004, 342, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.H.; Sobott, F.; Yao, Z.; Zhang, W.; Nielsen, P.R.; Grossmann, J.G.; Laue, E.D.; Robinson, C.V.; Jackson, S.E. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J. Mol. Biol. 2006, 356, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Muschler, P.; Hainzl, O.; Reinstein, J.; Buchner, J. Sti1 is a non-competitive inhibitor of the Hsp90 ATPase. Binding prevents the N-terminal dimerization reaction during the ATPase cycle. J. Biol. Chem. 2003, 278, 10328–10333. [Google Scholar] [CrossRef]
- Prodromou, C.; Panaretou, B.; Chohan, S.; Siligardi, G.; O’Brien, R.; Ladbury, J.E.; Roe, S.M.; Piper, P.W.; Pearl, L.H. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000, 19, 4383–4392. [Google Scholar] [CrossRef]
- McLaughlin, S.H.; Ventouras, L.-A.; Lobbezoo, B.; Jackson, S.E. Independent ATPase activity of Hsp90 subunits creates a flexible assembly platform. J. Mol. Biol. 2004, 344, 813–826. [Google Scholar] [CrossRef]
- Lloyd, M.D. High-Throughput Screening for the Discovery of Enzyme Inhibitors. J. Med. Chem. 2020, 63, 10742–10772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.-G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.A.; Zhan, C.G.; Sun, D. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmacol. 2010, 79, 542–551. [Google Scholar] [CrossRef]
- Yin, Z.; Henry, E.C.; Gasiewicz, T.A. (−)-Epigallocatechin-3-gallate Is a Novel Hsp90 Inhibitor. Biochemistry 2009, 48, 336–345. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Jiang, Y.; Lee, H.-F.; Schwartz, S.J.; Sun, D. (−)-Epigallocatechin-3-gallate Inhibits Hsp90 Function by Impairing Hsp90 Association with Cochaperones in Pancreatic Cancer Cell Line Mia Paca-2. Mol. Pharm. 2009, 6, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, M.G.; Newbatt, Y.M.; Prodromou, C.; Pearl, L.H.; Workman, P.; Aherne, W. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal. Biochem. 2004, 327, 176–183. [Google Scholar] [CrossRef]
- Richter, K.; Muschler, P.; Hainzl, O.; Buchner, J. Coordinated ATP hydrolysis by the Hsp90 dimer. J. Biol. Chem. 2001, 276, 33689–33696. [Google Scholar] [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [CrossRef]
- Halpin, J.C.; Street, T.O. Hsp90 Sensitivity to ADP Reveals Hidden Regulation Mechanisms. J. Mol. Biol. 2017, 429, 2918–2930. [Google Scholar] [CrossRef]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef]
- Li, J.; Richter, K.; Reinstein, J.; Buchner, J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol. 2013, 20, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 2010, 37, 344–354. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Harst, A.; Lin, H.; Obermann, W.M.J. Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 2005, 387, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.K.; Hutt, D.M.; Tait, B.; Guy, N.C.; Sivils, J.C.; Ortiz, N.R.; Payan, A.N.; Komaragiri, S.K.; Owens, J.J.; Culbertson, D.; et al. Management of Hsp90-Dependent Protein Folding by Small Molecules Targeting the Aha1 Co-Chaperone. Cell Chem. Biol. 2020, 27, 292–305.e6. [Google Scholar] [CrossRef] [PubMed]
- Biebl, M.M.; Lopez, A.; Rehn, A.; Freiburger, L.; Lawatscheck, J.; Blank, B.; Sattler, M.; Buchner, J. Structural elements in the flexible tail of the co-chaperone p23 coordinate client binding and progression of the Hsp90 chaperone cycle. Nat. Commun. 2021, 12, 828. [Google Scholar] [CrossRef] [PubMed]
- Oxelmark, E.; Knoblauch, R.; Arnal, S.; Su, L.F.; Schapira, M.; Garabedian, M.J. Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J. Biol. Chem. 2003, 278, 36547–36555. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, C.A.; Fauq, A.; Peterson, L.B.; Miller, C.; Blagg, B.S.J.; Chadli, A. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J. Biol. Chem. 2013, 288, 7313–7325. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Peng, S.; Wang, J.; Chen, H.; Cong, X.; Chen, A.; Hu, M.; Qin, M.; Wu, H.; Gao, S.; et al. Ailanthone targets p23 to overcome MDV3100 resistance in castration-resistant prostate cancer. Nat. Commun. 2016, 7, 13122. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-N.; Luo, Y. HSP90 inhibitors and cancer: Prospects for use in targeted therapies (Review). Oncol. Rep. 2022, 49, 6. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Pimitespib: First Approval. Drugs 2022, 82, 1413–1418. [Google Scholar] [CrossRef]
- Mishra, S.J.; Liu, W.; Beebe, K.; Banerjee, M.; Kent, C.N.; Munthali, V.; Koren, J.; Taylor, J.A.; Neckers, L.M.; Holzbeierlein, J.; et al. The Development of Hsp90β-Selective Inhibitors to Overcome Detriments Associated with pan-Hsp90 Inhibition. J. Med. Chem. 2021, 64, 1545–1557. [Google Scholar] [CrossRef]
- Ernst, J.T.; Liu, M.; Zuccola, H.; Neubert, T.; Beaumont, K.; Turnbull, A.; Kallel, A.; Vought, B.; Stamos, D. Correlation between chemotype-dependent binding conformations of HSP90α/β and isoform selectivity-Implications for the structure-based design of HSP90α/β selective inhibitors for treating neurodegenerative diseases. Bioorg. Med. Chem. Lett. 2014, 24, 204–208. [Google Scholar] [CrossRef]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.H.; Smith, H.W.; Jackson, S.E. Stimulation of the weak ATPase activity of human hsp90 by a client protein. J. Mol. Biol. 2002, 315, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Soroka, J.; Skalniak, L.; Leskovar, A.; Hessling, M.; Reinstein, J.; Buchner, J. Conserved conformational changes in the ATPase cycle of human Hsp90. J. Biol. Chem. 2008, 283, 17757–17765. [Google Scholar] [CrossRef] [PubMed]
- Southworth, D.R.; Agard, D.A. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol. Cell 2008, 32, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Rouges, C.; Asad, M.; Laurent, A.D.; Marchand, P.; Le Pape, P. Is the C-Terminal Domain an Effective and Selective Target for the Design of Hsp90 Inhibitors against Candida Yeast? Microorganisms 2023, 11, 2837. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Robbins, N.; Huang, D.S.; McLellan, C.A.; Shekhar-Guturja, T.; LeBlanc, E.V.; Nation, C.S.; Hui, R.; Hutchinson, A.; Collins, C.; et al. Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun. 2019, 10, 402. [Google Scholar] [CrossRef]
- Huang, D.S.; LeBlanc, E.V.; Shekhar-Guturja, T.; Robbins, N.; Krysan, D.J.; Pizarro, J.; Whitesell, L.; Cowen, L.E.; Brown, L.E. Design and Synthesis of Fungal-Selective Resorcylate Aminopyrazole Hsp90 Inhibitors. J. Med. Chem. 2020, 63, 2139–2180. [Google Scholar] [CrossRef] [PubMed]
- Marcyk, P.T.; LeBlanc, E.V.; Kuntz, D.A.; Xue, A.; Ortiz, F.; Trilles, R.; Bengtson, S.; Kenney, T.M.G.; Huang, D.S.; Robbins, N.; et al. Fungal-Selective Resorcylate Aminopyrazole Hsp90 Inhibitors: Optimization of Whole-Cell Anticryptococcal Activity and Insights into the Structural Origins of Cryptococcal Selectivity. J. Med. Chem. 2021, 64, 1139–1169. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.J.; Khandelwal, A.; Banerjee, M.; Balch, M.; Peng, S.; Davis, R.E.; Merfeld, T.; Munthali, V.; Deng, J.; Matts, R.L.; et al. Selective Inhibition of the Hsp90α Isoform. Angew. Chem. Int. Ed. Engl. 2021, 60, 10547–10551. [Google Scholar] [CrossRef]
- Mishra, S.J.; Ghosh, S.; Stothert, A.R.; Dickey, C.A.; Blagg, B.S.J. Transformation of the Non-Selective Aminocyclohexanol-Based Hsp90 Inhibitor into a Grp94-Seletive Scaffold. ACS Chem. Biol. 2017, 12, 244–253. [Google Scholar] [CrossRef]
- Lee, C.; Park, H.-K.; Jeong, H.; Lim, J.; Lee, A.-J.; Cheon, K.Y.; Kim, C.-S.; Thomas, A.P.; Bae, B.; Kim, N.D.; et al. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J. Am. Chem. Soc. 2015, 137, 4358–4367. [Google Scholar] [CrossRef]
- Patel, P.D.; Yan, P.; Seidler, P.M.; Patel, H.J.; Sun, W.; Yang, C.; Que, N.S.; Taldone, T.; Finotti, P.; Stephani, R.A.; et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol. 2013, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jin, X.; Xie, Z.; Zhang, H.; Liu, T.; Zheng, H.; Luan, X.; Sun, Y.; Fang, W.; Chang, W.; et al. Benzamidine Conjugation Converts Expelled Potential Active Agents into Antifungals against Drug-Resistant Fungi. J. Med. Chem. 2023, 66, 13684–13704. [Google Scholar] [CrossRef]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; De Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef]
- Cheung, K.-M.J.; Matthews, T.P.; James, K.; Rowlands, M.G.; Boxall, K.J.; Sharp, S.Y.; Maloney, A.; Roe, S.M.; Prodromou, C.; Pearl, L.H.; et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3338–3343. [Google Scholar] [CrossRef]
- Fadden, P.; Huang, K.H.; Veal, J.M.; Steed, P.M.; Barabasz, A.F.; Foley, B.; Hu, M.; Partridge, J.M.; Rice, J.; Scott, A.; et al. Application of chemoproteomics to drug discovery: Identification of a clinical candidate targeting hsp90. Chem. Biol. 2010, 17, 686–694. [Google Scholar] [CrossRef]
- Patwardhan, C.A.; Alfa, E.; Lu, S.; Chadli, A. Progesterone receptor chaperone complex-based high-throughput screening assay: Identification of capsaicin as an inhibitor of the Hsp90 machine. J. Biomol. Screen. 2015, 20, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Schoepfer, J.; Radimerski, T.; Massey, A.; Guy, C.T.; Brueggen, J.; Quadt, C.; Buckler, A.; Cozens, R.; Drysdale, M.J.; et al. NVP-AUY922: A small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008, 10, R33. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, K.; Zhang, H.; Brekken, J.; Huser, N.; Powell, R.E.; Timple, N.; Busch, D.J.; Neely, L.; Sensintaffar, J.L.; Yang, Y.C.; et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol. Cancer Ther. 2009, 8, 921–929. [Google Scholar] [CrossRef]
- Zhao, H.; Brandt, G.E.; Galam, L.; Matts, R.L.; Blagg, B.S. Identification and initial SAR of silybin: An Hsp90 inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 2659–2664. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Woo, J.K.; Yazici, Y.D.; Myers, J.N.; Kim, W.Y.; Jin, Q.; Hong, S.S.; Park, H.J.; Suh, Y.G.; Kim, K.W.; et al. Structural basis for depletion of heat shock protein 90 client proteins by deguelin. J. Natl. Cancer Inst. 2007, 99, 949–961. [Google Scholar] [CrossRef]
- Chang, D.J.; An, H.; Kim, K.S.; Kim, H.H.; Jung, J.; Lee, J.M.; Kim, N.J.; Han, Y.T.; Yun, H.; Lee, S.; et al. Design, synthesis, and biological evaluation of novel deguelin-based heat shock protein 90 (HSP90) inhibitors targeting proliferation and angiogenesis. J. Med. Chem. 2012, 55, 10863–10884. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohlmann, P.; Krylov, S.N.; Marchand, P.; Jose, J. FRET Assays for the Identification of C. albicans HSP90-Sba1 and Human HSP90α-p23 Binding Inhibitors. Pharmaceuticals 2024, 17, 516. https://doi.org/10.3390/ph17040516
Kohlmann P, Krylov SN, Marchand P, Jose J. FRET Assays for the Identification of C. albicans HSP90-Sba1 and Human HSP90α-p23 Binding Inhibitors. Pharmaceuticals. 2024; 17(4):516. https://doi.org/10.3390/ph17040516
Chicago/Turabian StyleKohlmann, Philip, Sergey N. Krylov, Pascal Marchand, and Joachim Jose. 2024. "FRET Assays for the Identification of C. albicans HSP90-Sba1 and Human HSP90α-p23 Binding Inhibitors" Pharmaceuticals 17, no. 4: 516. https://doi.org/10.3390/ph17040516
APA StyleKohlmann, P., Krylov, S. N., Marchand, P., & Jose, J. (2024). FRET Assays for the Identification of C. albicans HSP90-Sba1 and Human HSP90α-p23 Binding Inhibitors. Pharmaceuticals, 17(4), 516. https://doi.org/10.3390/ph17040516