

Investigation on the Combined Effect of Hydroxypropyl Beta-Cyclodextrin (HPβCD) and Polysorbate in Monoclonal Antibody Formulation

 ,
,
Abstract
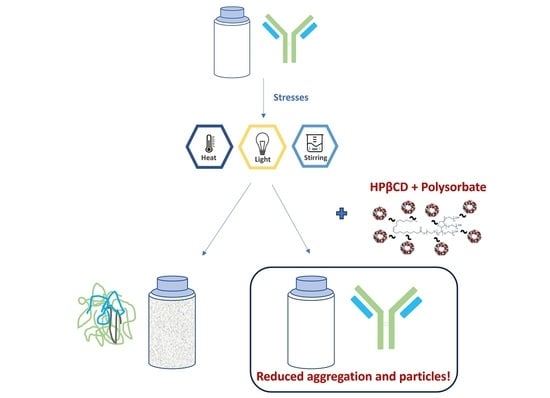
1. Introduction
2. Results
2.1. Stabilizing Effects of KLEPTOSE® HPB on Monoclonal Antibody
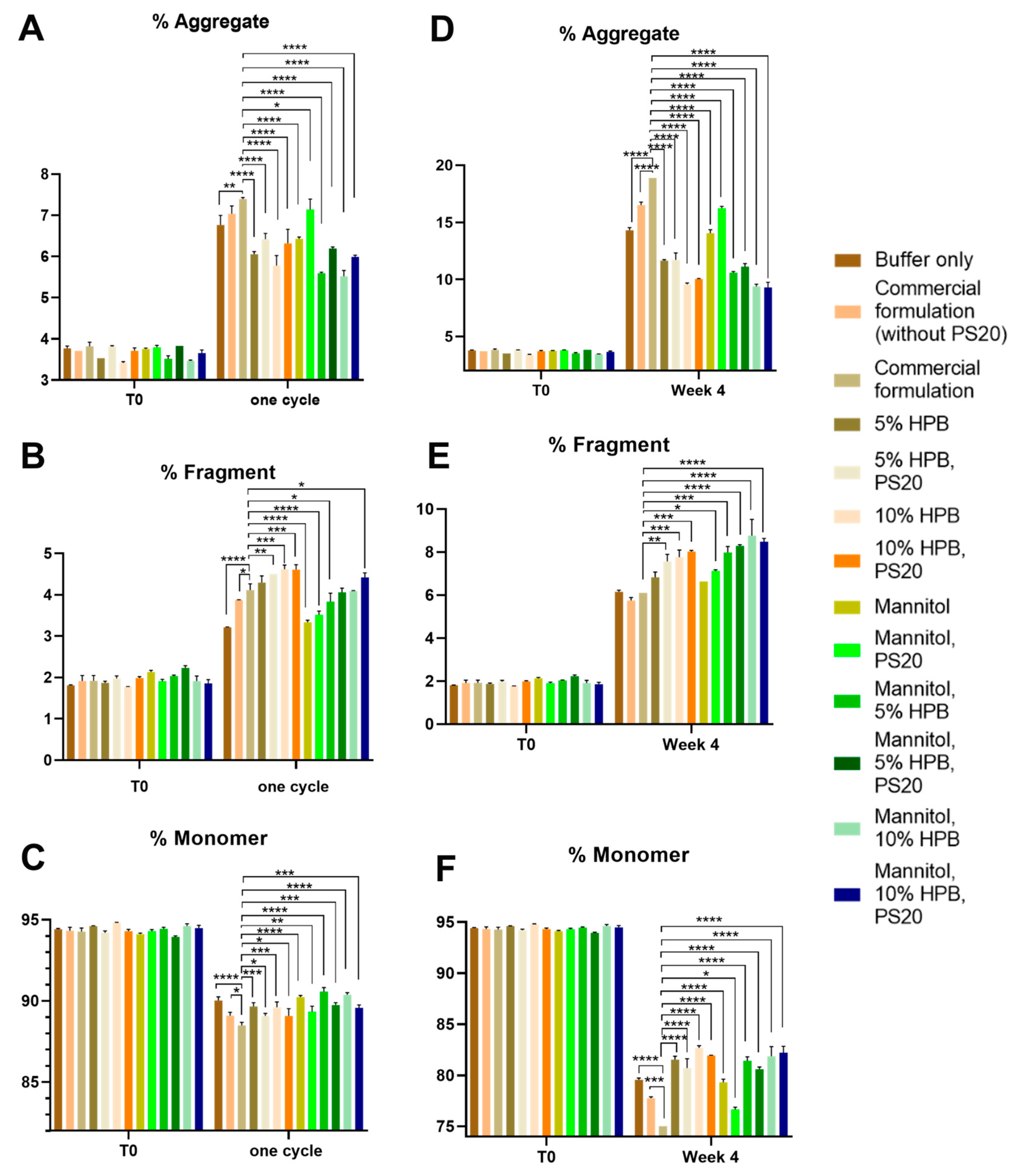
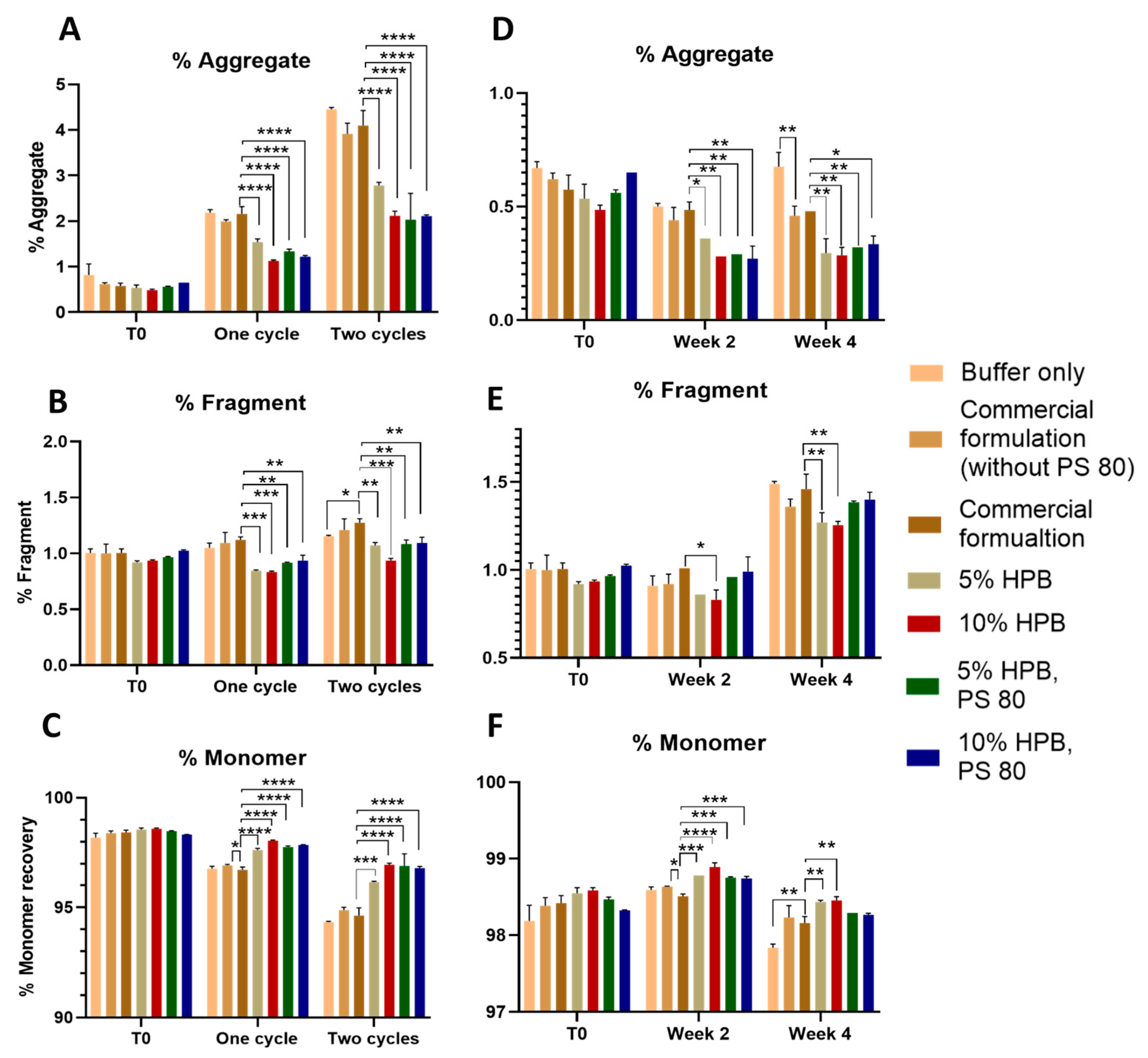
2.1.1. Bevacizumab Stability under Various Stresses
Bevacizumab Aggregation and Fragmentation Profiles under Light and Heat Stresses
Bevacizumab Charge Variant Profiles under Light Stress
Bevacizumab Aggregation and Fragmentation Profiles under Agitation Stress
2.1.2. Ipilimumab Stability under Various Stresses
2.2. Mechanistic Studies
2.2.1. Effect of KLEPTOSE® HPB on Protein–Protein Interaction
2.2.2. Surface Activity of KLEPTOSE® HPB
2.3. Enhancing Monoclonal Antibody Stability in Subvisible Particle Formation by the Combination of KLEPTOSE® HPB and Polysorbate
2.4. Interactions of KLEPTOSE® HPB and Polysorbate
2.4.1. Weak Binding Interactions between KLEPTOSE® HPB and Polysorbate
2.4.2. Enhancement of Polysorbate Surface Activity by KLEPTOSE® HPB
3. Discussion
3.1. Mechanistic Understanding of KLEPTOSE® HPB Stabilization Mechanism
3.2. Enhanced Effects in Protein Formulation with the Combination of KLEPTOSE® HPB and Polysorbate
3.3. Interactions between Polysorbates and KLEPTOSE® HPB
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Excipient Preparation
4.2.2. Antibody Formulation and Stability Studies
4.2.3. Size Exclusion Chromatography (SEC)–HPLC
4.2.4. Cation-Exchange Chromatography (CEX) for Charge Heterogeneity Profiling of Bevacizumab
4.2.5. Subvisible Particles Analysis by Micro-Flow Imaging (MFI)
4.2.6. Surface Activity Evaluation
4.2.7. Binding Affinity by Isothermal Titration Calorimetry (ITC) Analysis
4.2.8. Determination of Diffusion Interaction Parameter (KD) by Dynamic Light Scattering (DLS)
4.2.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef] [PubMed]
- Bajracharya, R.; Song, J.G.; Back, S.Y.; Han, H.-K. Recent advancements in non-invasive formulations for protein drug delivery. Comput. Struct. Biotechnol. J. 2019, 17, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Wang, W.; Arakawa, T.; Ohtake, S. Developments and Challenges for mAb-Based Therapeutics. Antibodies 2013, 2, 452–500. [Google Scholar] [CrossRef]
- Rahban, M.; Ahmad, F.; Piatyszek, M.A.; Haertlé, T.; Saso, L.; Saboury, A.A. Stabilization challenges and aggregation in protein-based therapeutics in the pharmaceutical industry. RSC Adv. 2023, 13, 35947–35963. [Google Scholar] [CrossRef] [PubMed]
- Rathore, N.; Rajan, R.S. Current perspectives on stability of protein drug products during formulation, fill and finish operations. Biotechnol. Prog. 2008, 24, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.S.; Capela, E.V.; Loureiro, A.M.; Tavares, A.P.; Freire, M.G. An overview on the recent advances in alternative solvents as stabilizers of proteins and enzymes. ChemEngineering 2022, 6, 51. [Google Scholar] [CrossRef]
- Agarkhed, M.; O’Dell, C.; Hsieh, M.C.; Zhang, J.; Goldstein, J.; Srivastava, A. Effect of polysorbate 80 concentration on thermal and photostability of a monoclonal antibody. AAPS PharmSciTech 2013, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Matthew Fesinmeyer, R.; Pierini, C.J.; Siska, C.C.; Litowski, J.R.; Brych, S.; Wen, Z.-Q.; Kleemann, G.R. Free Fatty Acid Particles in Protein Formulations, Part 1: Microspectroscopic Identification. J. Pharm. Sci. 2015, 104, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Roy, I.; Patel, A.; Kumar, V.; Nanda, T.; Assenberg, R.; Wuchner, K.; Amin, K. Polysorbate Degradation and Particle Formation in a High Concentration mAb: Formulation Strategies to Minimize Effect of Enzymatic Polysorbate Degradation. J. Pharm. Sci. 2021, 110, 3313–3323. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, A.; Demeule, B.; Lin, B.; Yadav, S. Polysorbate 20 Degradation in Biopharmaceutical Formulations: Quantification of Free Fatty Acids, Characterization of Particulates, and Insights into the Degradation Mechanism. Mol. Pharm. 2015, 12, 3805–3815. [Google Scholar] [CrossRef] [PubMed]
- Pegues, M.A.; Szczepanek, K.; Sheikh, F.; Thacker, S.G.; Aryal, B.; Ghorab, M.K.; Wolfgang, S.; Donnelly, R.P.; Verthelyi, D.; Rao, V.A. Effect of fatty acid composition in polysorbate 80 on the stability of therapeutic protein formulations. Pharm. Res. 2021, 38, 1961–1975. [Google Scholar] [CrossRef] [PubMed]
- Castañeda Ruiz, A.J.; Shetab Boushehri, M.A.; Phan, T.; Carle, S.; Garidel, P.; Buske, J.; Lamprecht, A. Alternative Excipients for Protein Stabilization in Protein Therapeutics: Overcoming the Limitations of Polysorbates. Pharmaceutics 2022, 14, 2575. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, R.; Razzaq, F.A.; Asghar, S.; Irfan, M.; Khan, I.U.; Khalid, S.H. Cyclodextrins: An overview of fundamentals, types, and applications. In Cyclodextrins—Core Concepts and New Frontiers; IntechOpen: London, UK, 2022; Available online: https://www.intechopen.com/chapters/84117 (accessed on 14 April 2024).
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hong, S.; Tan, S.S.K.; Peng, T.; Goh, L.Y.H.; Lam, K.H.; Chow, K.T.; Gokhale, R. Polysorbates versus Hydroxypropyl Beta-Cyclodextrin (HPβCD): Comparative Study on Excipient Stability and Stabilization Benefits on Monoclonal Antibodies. Molecules 2022, 27, 6497. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Garidel, P.; Michaela, B. HP-β-CD for the formulation of IgG and Ig-based biotherapeutics. Int. J. Pharm. 2021, 601, 120531. [Google Scholar] [CrossRef] [PubMed]
- Braga, S.S. Cyclodextrins: Emerging medicines of the new millennium. Biomolecules 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed]
- Rospiccio, M.; Arsiccio, A.; Winter, G.; Pisano, R. The role of cyclodextrins against interface-induced denaturation in pharmaceutical formulations: A molecular dynamics approach. Mol. Pharm. 2021, 18, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Kozarewicz, P.; Loftsson, T. Novel excipients—Regulatory challenges and perspectives—The EU insight. Int. J. Pharm. 2018, 546, 176–179. [Google Scholar] [CrossRef]
- Bhatia, V.; Dhingra, A.; Chopra, B.; Guarve, K. Co-processed excipients: Recent advances and future perspective. J. Drug Deliv. Sci. Technol. 2022, 71, 103316. [Google Scholar] [CrossRef]
- Bin, L.K.; Hui, H.S.; Uddin, A.H.; Sarker, Z.I.; Ling, C.Y. Co-processed Excipients: A Revisit of Its Development in the Past Two Decades; A Review. J. Pharm. Negat. Results 2022, 13, 96–103. [Google Scholar]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Excipients for Room Temperature Stable Freeze-Dried Monoclonal Antibody Formulations. J. Pharm. Sci. 2020, 109, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Saluja, A.; Fesinmeyer, R.M.; Hogan, S.; Brems, D.N.; Gokarn, Y.R. Diffusion and sedimentation interaction parameters for measuring the second virial coefficient and their utility as predictors of protein aggregation. Biophys. J. 2010, 99, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Sorret, L.L.; DeWinter, M.A.; Schwartz, D.K.; Randolph, T.W. Challenges in Predicting Protein-Protein Interactions from Measurements of Molecular Diffusivity. Biophys. J. 2016, 111, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Hartl, E.; Dixit, N.; Besheer, A.; Kalonia, D.; Winter, G. Weak antibody-cyclodextrin interactions determined by quartz crystal microbalance and dynamic/static light scattering. Eur. J. Pharm. Biopharm. 2013, 85, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Stolzke, T.; Krieg, F.; Peng, T.; Zhang, H.; Hausler, O.; Brandenbusch, C. Hydroxylpropyl-beta-cyclodextrin as Potential Excipient to Prevent Stress-Induced Aggregation in Liquid Protein Formulations. Molecules 2022, 27, 5094. [Google Scholar] [CrossRef] [PubMed]
- Serno, T.; Härtl, E.; Besheer, A.; Miller, R.; Winter, G. The role of polysorbate 80 and HPβCD at the air-water interface of IgG solutions. Pharm. Res. 2013, 30, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Kishore, R.S.; Kiese, S.; Fischer, S.; Pappenberger, A.; Grauschopf, U.; Mahler, H.-C. The degradation of polysorbates 20 and 80 and its potential impact on the stability of biotherapeutics. Pharm. Res. 2011, 28, 1194–1210. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, M.; Blech, M.; Presser, I.; Garidel, P. Polysorbate degradation in biotherapeutic formulations: Identification and discussion of current root causes. Int. J. Pharm. 2018, 552, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Vaclaw, C.; Merritt, K.; Griffin, V.P.; Whitaker, N.; Gokhale, M.; Volkin, D.B.; Ogunyankin, M.O.; Dhar, P. Comparison of Protein Particle Formation in IgG1 mAbs Formulated with PS20 vs. PS80 When Subjected to Interfacial Dilatational Stress. AAPS PharmSciTech 2023, 24, 104. [Google Scholar] [CrossRef] [PubMed]
- Mitigating Particle Formation During Ultrafiltration_Diafiltration of Biologics with KLEPTOSE® HPβCD (Hydroxypropyl Beta-Cyclodextrin). Available online: https://www.roquette.com/innovation-hub/pharma/expert-opinion/mitigating-particle-formation-during-ultrafiltration-diafiltration-of-biologics-with-kleptose (accessed on 13 June 2023).
- Härtl, E.; Winter, G.; Besheer, A. Influence of hydroxypropyl-beta-cyclodextrin on the stability of dilute and highly concentrated immunoglobulin G formulations. J. Pharm. Sci. 2013, 102, 4121–4131. [Google Scholar] [CrossRef] [PubMed]
- Yuk, I.H.; Koulis, T.; Doshi, N.; Gregoritza, K.; Hediger, C.; Lebouc-Haefliger, V.; Giddings, J.; Khan, T.A. Formulation mitigations for particle formation induced by enzymatic hydrolysis of polysorbate 20 in protein-based drug products: Insights from a full-factorial longitudinal study. AAPS Open 2022, 8, 18. [Google Scholar] [CrossRef]
- Shalaby, K.S.; Ismail, M.I.; Lamprecht, A. Cyclodextrin Complex Formation with Water-Soluble Drugs: Conclusions from Isothermal Titration Calorimetry and Molecular Modeling. AAPS PharmSciTech 2021, 22, 232. [Google Scholar] [CrossRef] [PubMed]
- DRG ICH. Stability testing of new drug substances and products Q1A (R2). In Proceedings of the International Conference on Harmonization, Geneva, Switzerland, 6 February 2003. [Google Scholar]
- ICH Harmonised Tripartite Guideline. Stability Testing: Photostability Testing of New Drug Substances and Products. Q1B Current Step 4 Version. 1996, pp. 1–8. Available online: https://gxp-academy.org/upload/iblock/994/9944e3302b7e2bea9541bc18361a1a03.pdf (accessed on 14 April 2024).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bevacizumab | Buffer | Excipients | ||||
|---|---|---|---|---|---|---|
| Trehalose | PS 20 | Mannitol | KLEPTOSE® HPB | |||
| Condition 1 (buffer only) | 5 mg/mL | 50 mM sodium phosphate, pH 6.2 | ||||
| Condition 2 | 5 mg/mL | 60 mg/mL | ||||
| Condition 3 (commercial formulation) | 5 mg/mL | 60 mg/mL | 0.04% w/v | |||
| Condition 4 | 5 mg/mL | 60 mg/mL | 5% w/v | |||
| Condition 5 | 5 mg/mL | 60 mg/mL | 0.04% w/v | 5% w/v | ||
| Condition 6 | 5 mg/mL | 60 mg/mL | 10% w/v | |||
| Condition 7 | 5 mg/mL | 60 mg/mL | 0.04% w/v | 10% w/v | ||
| Condition 8 | 5 mg/mL | 10 mg/mL | ||||
| Condition 9 | 5 mg/mL | 0.04% w/v | 10 mg/mL | |||
| Condition 10 | 5 mg/mL | 10 mg/mL | 5% w/v | |||
| Condition 11 | 5 mg/mL | 0.04% w/v | 10 mg/mL | 5% w/v | ||
| Condition 12 | 5 mg/mL | 10 mg/mL | 10% w/v | |||
| Condition 13 | 5 mg/mL | 0.04% w/v | 10 mg/mL | 10% w/v | ||
| Ipilimumab | Buffer | Excipients | |||
|---|---|---|---|---|---|
| PS 80 | Mannitol | KLEPTOSE® HPB | |||
| Condition 1 (buffer only) | 5 mg/mL | 5.85 mg/mL sodium chloride, 3.15 mg/mL tris hydrochloride, 0.04 mg/mL DTPA, pH 7.0 | |||
| Condition 2 | 5 mg/mL | 10 mg/mL | |||
| Condition 3 (commercial formulation) | 5 mg/mL | 0.1% w/v | 10 mg/mL | ||
| Condition 4 | 5 mg/mL | 5% w/v | |||
| Condition 5 | 5 mg/mL | 0.1% w/v | 5% w/v | ||
| Condition 6 | 5 mg/mL | 10% w/v | |||
| Condition 7 | 5 mg/mL | 0.1% w/v | 10% w/v | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Hong, S.; Goh, L.Y.H.; Zhang, H.; Peng, T.; Chow, K.T.; Gokhale, R.; Tuliani, V. Investigation on the Combined Effect of Hydroxypropyl Beta-Cyclodextrin (HPβCD) and Polysorbate in Monoclonal Antibody Formulation. Pharmaceuticals 2024, 17, 528. https://doi.org/10.3390/ph17040528
Huang J, Hong S, Goh LYH, Zhang H, Peng T, Chow KT, Gokhale R, Tuliani V. Investigation on the Combined Effect of Hydroxypropyl Beta-Cyclodextrin (HPβCD) and Polysorbate in Monoclonal Antibody Formulation. Pharmaceuticals. 2024; 17(4):528. https://doi.org/10.3390/ph17040528
Chicago/Turabian StyleHuang, Jiayi, Shiqi Hong, Lucas Yuan Hao Goh, Hailong Zhang, Tao Peng, Keat Theng Chow, Rajeev Gokhale, and Vinod Tuliani. 2024. "Investigation on the Combined Effect of Hydroxypropyl Beta-Cyclodextrin (HPβCD) and Polysorbate in Monoclonal Antibody Formulation" Pharmaceuticals 17, no. 4: 528. https://doi.org/10.3390/ph17040528
APA StyleHuang, J., Hong, S., Goh, L. Y. H., Zhang, H., Peng, T., Chow, K. T., Gokhale, R., & Tuliani, V. (2024). Investigation on the Combined Effect of Hydroxypropyl Beta-Cyclodextrin (HPβCD) and Polysorbate in Monoclonal Antibody Formulation. Pharmaceuticals, 17(4), 528. https://doi.org/10.3390/ph17040528