
Nature-Inspired 1-Phenylpyrrolo[2,1-a]isoquinoline Scaffold for Novel Antiproliferative Agents Circumventing P-Glycoprotein-Dependent Multidrug Resistance

 ,
,  ,
,  , ,
, ,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
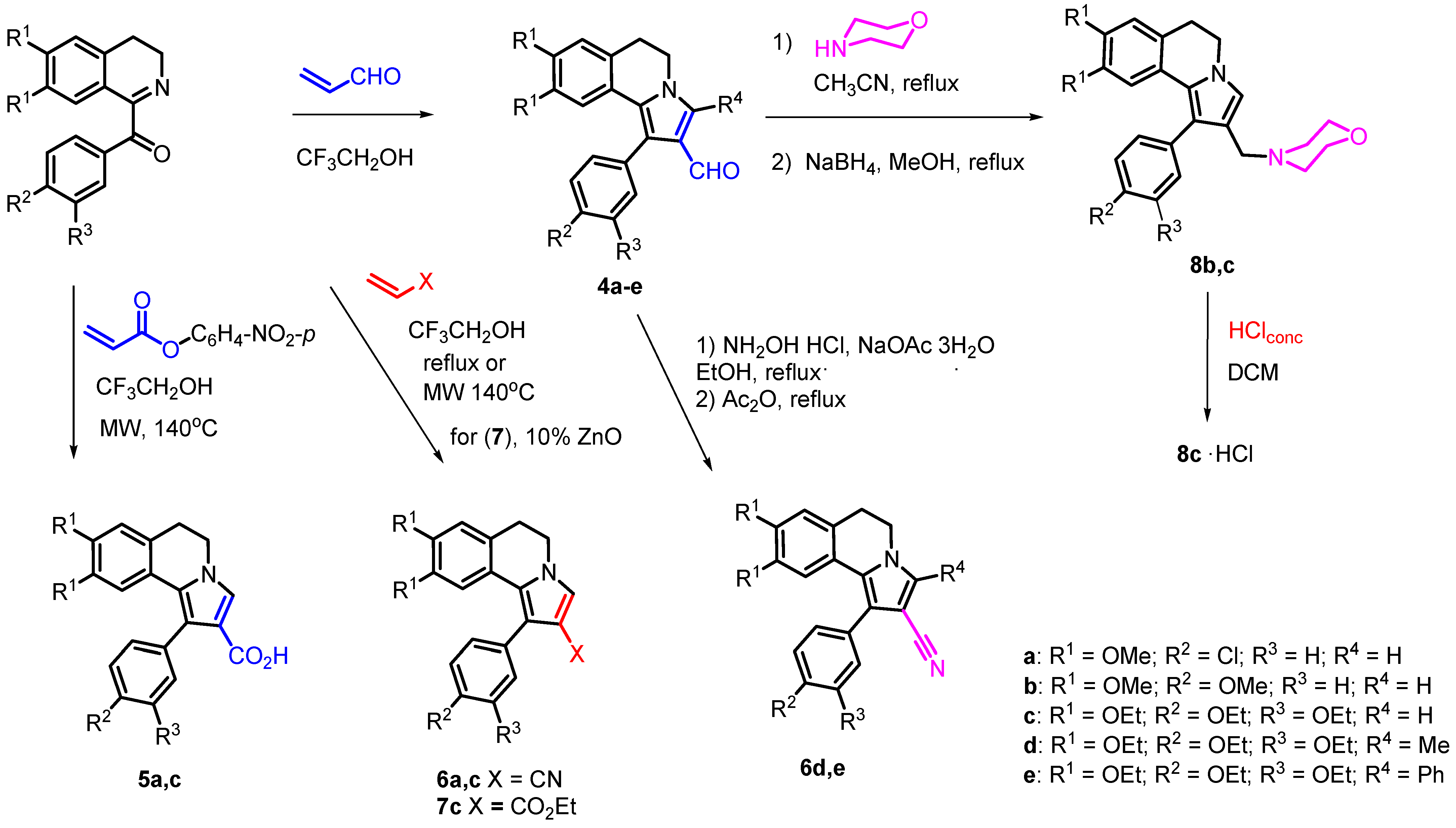
2.1. Synthesis
2.2. Biological Evaluation
2.2.1. In Vitro Cytotoxicity Screening
2.2.2. P-gp and MRP1 Inhibitory Potency
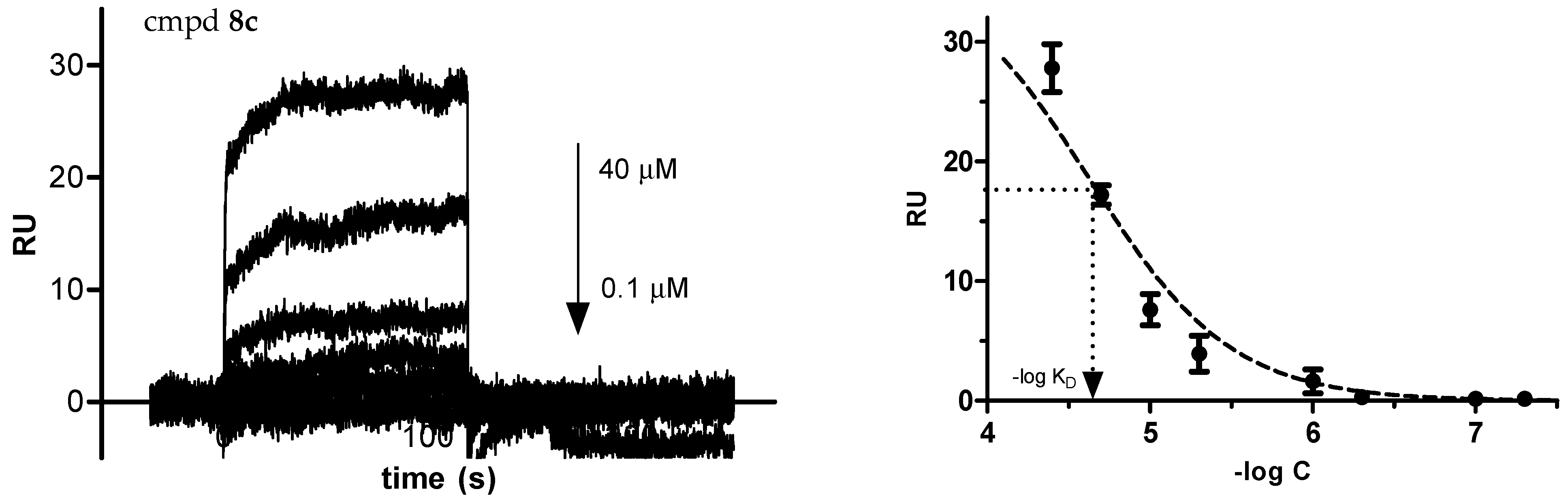
2.2.3. Binding Affinity to Human Serum Albumin (HSA)
2.3. Solvation-Related Parameters
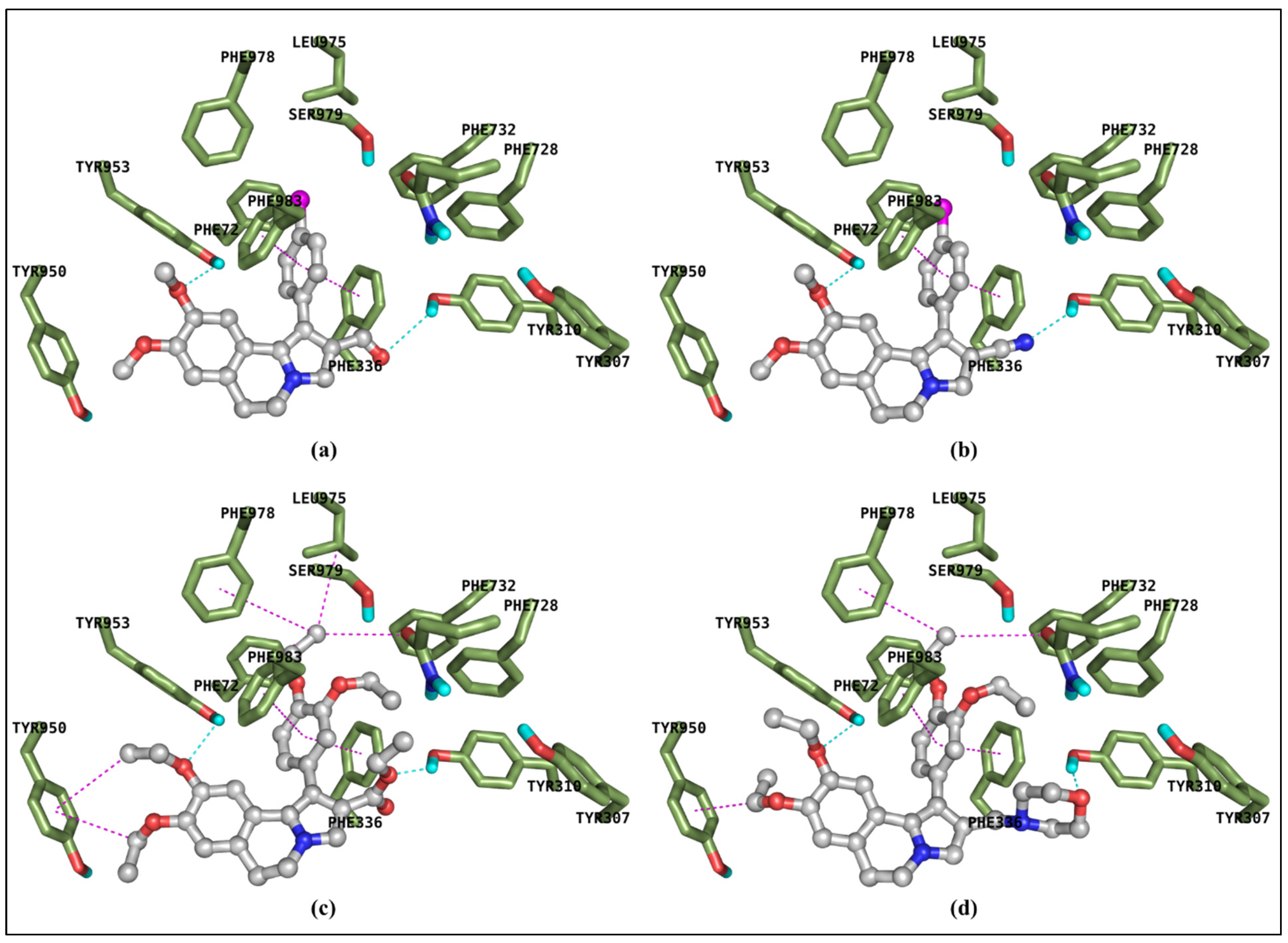
2.4. Molecular Docking Calculation
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of (1-(4-Methoxyphenyl)-8,9-dimethoxy-5,6-dihydropyrrolo[2,1-a] Isoquinoline-2-carbaldehyde (4b)
3.1.2. Synthesis of 1-Aryl-5,6-dihydropyrrolo[2,1-a]isoquinoline-2-carboxylic acids 5a,c
3.1.3. Synthesis of Carbonitriles 6d,e
3.1.4. Synthesis of Ethyl 1-(3,4-Diethoxyphenyl)-8,9-diethoxy-5,6-dihydro pyrrolo[2,1-a]isoquinoline-2-carboxylate (7c)
3.1.5. Synthesis of 2-(Morpholin-4-yl-methyl)-5,6-dihydropyrrolo[2,1-a]isoquinolines 8b,c
3.1.6. Synthesis of 4-{[1-(3,4-Diethoxyphenyl)-8,9-diethoxy-5,6-dihydropyrrolo[2,1-a]isoquinolin-2-yl]methyl}morpholin-4-ium chloride (8c·HCl)
3.2. Biological Evaluation
3.2.1. Cell Cultures
3.2.2. Cytotoxicity Assay
3.2.3. Inhibition Assays of P-Glycoprotein (P-gp) and Multidrug-Resistance-Associated Protein-1 (MRP1)
3.2.4. Affinity to Human Serum Albumin (HSA) by Surface Plasmon Resonance (SPR)
3.3. Aqueous Solubility and Lipophilicity
3.3.1. Determination of Kinetic Solubility in PBS
3.3.2. Determination of Lipophilicity by RP-HPLC
3.4. Molecular Dooking Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukuda, T.; Ishibashi, F.; Iwao, M. Synthesis and biological activity of lamellarin alkaloids: An overview. Heterocycles 2011, 83, 491–529. [Google Scholar] [CrossRef]
- Matveeva, M.D.; Purgatorio, R.; Voskressensky, L.G.; Altomare, C.D. Pyrrolo[2,1-a]isoquinoline scaffold in drug discovery: Advances in synthesis and medicinal chemistry. Future Med. Chem. 2019, 11, 2735–2755. [Google Scholar] [CrossRef] [PubMed]
- Kakhki, S.; Shahosseini, S.; Zarghi, A. Design, synthesis and cytotoxicity evaluation of new 2-aryl-5, 6-dihydropyrrolo[2,1-a]isoquinoline derivatives as topoisomerase inhibitors. Iran. J. Pharm. Res. 2014, 13, 71–77. [Google Scholar] [PubMed]
- Kakhki, S.; Shahosseini, S.; Zarghi, A. Design and synthesis of pyrrolo [2,1-a] isoquinoline-based derivatives as new cytotoxic agents. Iran. J. Pharm. Res. 2016, 15, 743–751. [Google Scholar] [PubMed]
- Nevskaya, A.A.; Miftyakhova, A.R.; Anikina, L.V.; Borisova, T.N.; Varlamov, A.V.; Voskressensky, L.G. Synthesis and Cytotoxicity of Novel 1-Arylindolizines and 1-Arylpyrrolo[2,1-a]isoquinolines. Tetrahedron Lett. 2021, 87, 153552. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- .Dehner, C.A.; Rudzinski, E.R.; Davis, J.L. Rhabdomyosarcoma: Updates on classification and the necessity of molecular testing beyond immunohistochemistry. Hum. Pathol. 2023, 21, S0046-8177(23)00251-4. [Google Scholar] [CrossRef] [PubMed]
- Nevskaya, A.A.; Anikina, L.V.; Purgatorio, R.; Catto, M.; Nicolotti, O.; de Candia, M.; Pisani, L.; Borisova, T.N.; Miftyakhova, A.R.; Varlamov, A.V.; et al. Homobivalent Lamellarin-like Schiff Bases: In Vitro Evaluation of Their Cancer Cell Cytotoxicity and Multitargeting Anti-Alzheimer’s Disease Potential. Molecules 2021, 26, 359. [Google Scholar] [CrossRef] [PubMed]
- Nevskaya, A.A.; Matveeva, M.D.; Borisova, T.N.; Niso, M.; Colabufo, N.A.; Boccarelli, A.; Purgatorio, R.; de Candia, M.; Cellamare, S.; Voskressensky, L.G.; et al. A New Class of 1-Aryl-5,6-dihydropyrrolo[2,1-a]isoquinoline derivatives as reversers of P-glycoprotein-mediated multidrug resistance in tumor cells. ChemMedChem 2018, 13, 1588–1596. [Google Scholar] [CrossRef]
- Dufour, E.; Storer, A.C.; Menard, R. Peptide aldehydes and nitriles as transition state analog inhibitors of cysteine proteases. Biochemistry 1995, 34, 9136–9143. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T. Molecular Mechanisms of Aldehyde Toxicity: A Chemical Perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 80, 743–816. [Google Scholar] [CrossRef]
- Matveeva, M.; Borisova, T.; Titov, A.; Anikina, L.; Dyachenko, S.; Astakhov, G.; Varlamov, A.; Voskressensky, L. Domino Reactions of 1-Aroyl-3,4-Dihydroisoquinolines with α,β-Unsaturated Aldehydes. Synthesis 2017, 49, 5251–5257. [Google Scholar] [CrossRef]
- Astakhov, G.S.; Shigaev, R.R.; Borisova, T.N.; Ershova, A.A.; Titov, A.A.; Varlamov, A.V.; Voskressensky, L.G.; Matveeva, M.D. Facile Synthesis of Pyrrolo[2,1-a]Isoquinolines by Domino Reaction of 1-Aroyl-3,4-Dihydroisoquinolines with Conjugated Ketones, Nitroalkenes and Nitriles. Mol. Divers. 2021, 25, 2441–2446. [Google Scholar] [CrossRef]
- Trompier, D.; Chang, X.-B.; Barattin, R.; d’Hardemare, A.D.M.; Di Pietro, A.; Baubichon-Cortay, H. Verapamil and Its Derivative Trigger Apoptosis through Glutathione Extrusion by Multidrug Resistance Protein MRP1. Cancer Res. 2004, 64, 4950–4956. [Google Scholar] [CrossRef]
- Fabini, E.; Danielson, U.H. Monitoring Drug–Serum Protein Interactions for Early ADME Prediction through Surface Plasmon Resonance Technology. J. Pharm. Biomed. Anal. 2017, 144, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Frostell-Karlsson, Å.; Remaeus, A.; Roos, H.; Andersson, K.; Borg, P.; Hämäläinen, M.; Karlsson, R. Biosensor Analysis of the Interaction between Immobilized Human Serum Albumin and Drug Compounds for Prediction of Human Serum Albumin Binding Levels. J. Med. Chem. 2000, 43, 1986–1992. [Google Scholar] [CrossRef]
- Eur Pharmacopeia. Free Access to Supportive Pharmacopoeial Texts in the Field of Vaccines for Human Use during the Coronavirus Disease (COVID-19) Pandemic Updated Package—October 2020.
- Purgatorio, R.; de Candia, M.; De Palma, A.; De Santis, F.; Pisani, L.; Campagna, F.; Cellamare, S.; Altomare, C.; Catto, M. Insights into Structure-Activity Relationships of 3-Arylhydrazonoindolin-2-One Derivatives for Their Multitarget Activity on β-Amyloid Aggregation and Neurotoxicity. Molecules 2018, 23, 1544. [Google Scholar] [CrossRef]
- Purgatorio, R.; Kulikova, L.N.; Pisani, L.; Catto, M.; De Candia, M.; Carrieri, A.; Cellamare, S.; De Palma, A.; Beloglazkin, A.A.; Reza Raesi, G.; et al. Scouting around 1,2,3,4-Tetrahydrochromeno[3,2-c]Pyridin-10-ones for Single- and Multitarget Ligands Directed towards Relevant Alzheimer’s Targets. ChemMedChem 2020, 15, 1947–1955. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM Structures Reveal Distinct Mechanisms of Inhibition of the Human Multidrug Transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Schrödinger Release 2024-1: MacroModel; Schrödinger, LLC: New York, NY, USA, 2024. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second-Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Molecular Modeling Software|OpenEye Scientific. Available online: https://www.eyesopen.com (accessed on 20 September 2023).
- Forli, S.; Olson, A.J. A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- El Khoury, L.; Santos-Martins, D.; Sasmal, S.; Eberhardt, J.; Bianco, G.; Ambrosio, F.A.; Solis-Vasquez, L.; Koch, A.; Forli, S.; Mobley, D.L. Comparison of Affinity Ranking Using AutoDock-GPU and MM-GBSA Scores for BACE-1 Inhibitors in the D3R Grand Challenge 4. J. Comput.-Aided Mol. Des. 2019, 33, 1011–1020. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| N | X | R1 | R2 | R3 | R4 | RD | HCT116 | HeLa | A549 |
| 4a b | CHO | OMe | Cl | H | H | 17.6 (3.2) | 22.0 (4.0) | 33.0 (4.5) | 38.6 (3.2) |
| 4b | CHO | OMe | OMe | H | H | 95.2 (7.1) | >100 | >100 | >100 |
| 4c c | CHO | OEt | OEt | OEt | H | 21.3 (1.2) | 11.8 (0.2) | 44.5 (2.0) | 19.7 (0.3) |
| 5a | CO2H | OMe | Cl | H | H | >100 | >100 | >100 | >100 |
| 5c | CO2H | OEt | OEt | OEt | H | n.a. | n.a. | n.a. | n.a. |
| 6a | CN | OMe | Cl | H | H | >100 | >100 | >100 | >100 |
| 6c | CN | OEt | OEt | OEt | H | >100 | >100 | >100 | >100 |
| 6d | CN | OEt | OEt | OEt | Me | >100 | >100 | >100 | >100 |
| 6e | CN | OEt | OEt | OEt | Ph | >100 | n.a. | n.a. | n.a. |
| 7c | CO2Et | OEt | OEt | OEt | H | n.a. | n.a. | n.a. | n.a. |
| 8b | MM d | OMe | OMe | H | H | 37.7 (1.6) | 56.4 (1.7) | 65.4 (4.5) | 66.0 (1.6) |
| 8c | MM·HCl | OEt | OEt | OEt | H | 18.6 (2.7) | 15.7 (0.7) | 17.5 (1.4) | 20.8 (3.6) |
| Camptothecin | 16.0 (0.2) | 12.3 (0.5) | 0.33 (0.07) | 3.32 (0.02) | |||||
| Doxorubicin | 0.29 (0.02) | 0.14 (0.01) | 0.89 (0.01) | 0.38 (0.02) | |||||
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | X | R1 | R2 | R3 | P-gp, IC50 (μM) a | MRP1, IC50 (μM) a | HSA, KD (μM) b | S (μM) c | CLog P d | Log k′w e |
| 4a | CHO | OMe | Cl | H | 25.3 (1.9) | 21.9 (1.5) | 1.60 (0.04) | 6.66 (0.09) | 5.62 | 4.32 |
| 4b | CHO | OMe | OMe | H | 4.48 (0.17) | 6.42 (0.27) | 1.95 (0.06) | 19.8 (0.1) | 4.86 | 3.39 |
| 5c | CO2H | OEt | OEt | OEt | 0.35 (0.04) | >100 | 5.20 (0.20) | 14.6 (0.6) | 4.70 | 6.91 |
| 6a | CN | OMe | Cl | H | 5.89 (0.42) | 16.6 (0.5) | 4.60 (0.10) | 1.42 (0.01) | 5.75 | 4.42 |
| 6c | CN | OEt | OEt | OEt | 0.39 (0.06) | >100 | 1.80 (0.20) | 1.06 (0.05) | 6.79 | 5.09 |
| 7c | CO2Et | OEt | OEt | OEt | 0.32 (0.08) | 3.23 (0.29) | 12.1 (0.1) | 41.7 (1.1) g | 7.75 | 5.49 |
| 8b | MM f | OMe | OMe | H | 0.36 (0.02) | 1.80 (0.31) | 19.3 (0.5) | 112 (4) g | 4.73 | 3.86 |
| 8c | MM·HCl | OEt | OEt | OEt | 0.45 (0.03) | 12.1 (2.1) | 26.9 (0.5) | 41.7 (1.2) g | 6.53 | 4.31 |
| MC18 | 1.20 (0.3) | |||||||||
| Verapamil | 4.53 (0.50) | |||||||||
| Warfarin | 5.30 (0.35) | |||||||||
| N | FEB (a) | ΔE (b) | LE (c) | TAN (d) | POP (e) |
|---|---|---|---|---|---|
| 4a | −8.18 | 0.65 | 0.315 | 0.254 | 251/1000 |
| 6a | −7.96 | 1.04 | 0.306 | 0.268 | 17/1000 |
| 7c | −11.0 | 0.08 | 0.307 | 0.307 | 265/1000 |
| 8c | −10.6 | 0.21 | 0.279 | 0.276 | 121/1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevskaya, A.A.; Purgatorio, R.; Borisova, T.N.; Varlamov, A.V.; Anikina, L.V.; Obydennik, A.Y.; Nevskaya, E.Y.; Niso, M.; Colabufo, N.A.; Carrieri, A.; et al. Nature-Inspired 1-Phenylpyrrolo[2,1-a]isoquinoline Scaffold for Novel Antiproliferative Agents Circumventing P-Glycoprotein-Dependent Multidrug Resistance. Pharmaceuticals 2024, 17, 539. https://doi.org/10.3390/ph17040539
Nevskaya AA, Purgatorio R, Borisova TN, Varlamov AV, Anikina LV, Obydennik AY, Nevskaya EY, Niso M, Colabufo NA, Carrieri A, et al. Nature-Inspired 1-Phenylpyrrolo[2,1-a]isoquinoline Scaffold for Novel Antiproliferative Agents Circumventing P-Glycoprotein-Dependent Multidrug Resistance. Pharmaceuticals. 2024; 17(4):539. https://doi.org/10.3390/ph17040539
Chicago/Turabian StyleNevskaya, Alisa A., Rosa Purgatorio, Tatiana N. Borisova, Alexey V. Varlamov, Lada V. Anikina, Arina Yu. Obydennik, Elena Yu. Nevskaya, Mauro Niso, Nicola A. Colabufo, Antonio Carrieri, and et al. 2024. "Nature-Inspired 1-Phenylpyrrolo[2,1-a]isoquinoline Scaffold for Novel Antiproliferative Agents Circumventing P-Glycoprotein-Dependent Multidrug Resistance" Pharmaceuticals 17, no. 4: 539. https://doi.org/10.3390/ph17040539
APA StyleNevskaya, A. A., Purgatorio, R., Borisova, T. N., Varlamov, A. V., Anikina, L. V., Obydennik, A. Y., Nevskaya, E. Y., Niso, M., Colabufo, N. A., Carrieri, A., Catto, M., de Candia, M., Voskressensky, L. G., & Altomare, C. D. (2024). Nature-Inspired 1-Phenylpyrrolo[2,1-a]isoquinoline Scaffold for Novel Antiproliferative Agents Circumventing P-Glycoprotein-Dependent Multidrug Resistance. Pharmaceuticals, 17(4), 539. https://doi.org/10.3390/ph17040539