



In Silico Design of Potential Small-Molecule Antibiotic Adjuvants against Salmonella typhimurium Ortho Acetyl Sulphydrylase Synthase to Address Antimicrobial Resistance

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Pharmacophore Modeling
2.2. Molecular Docking
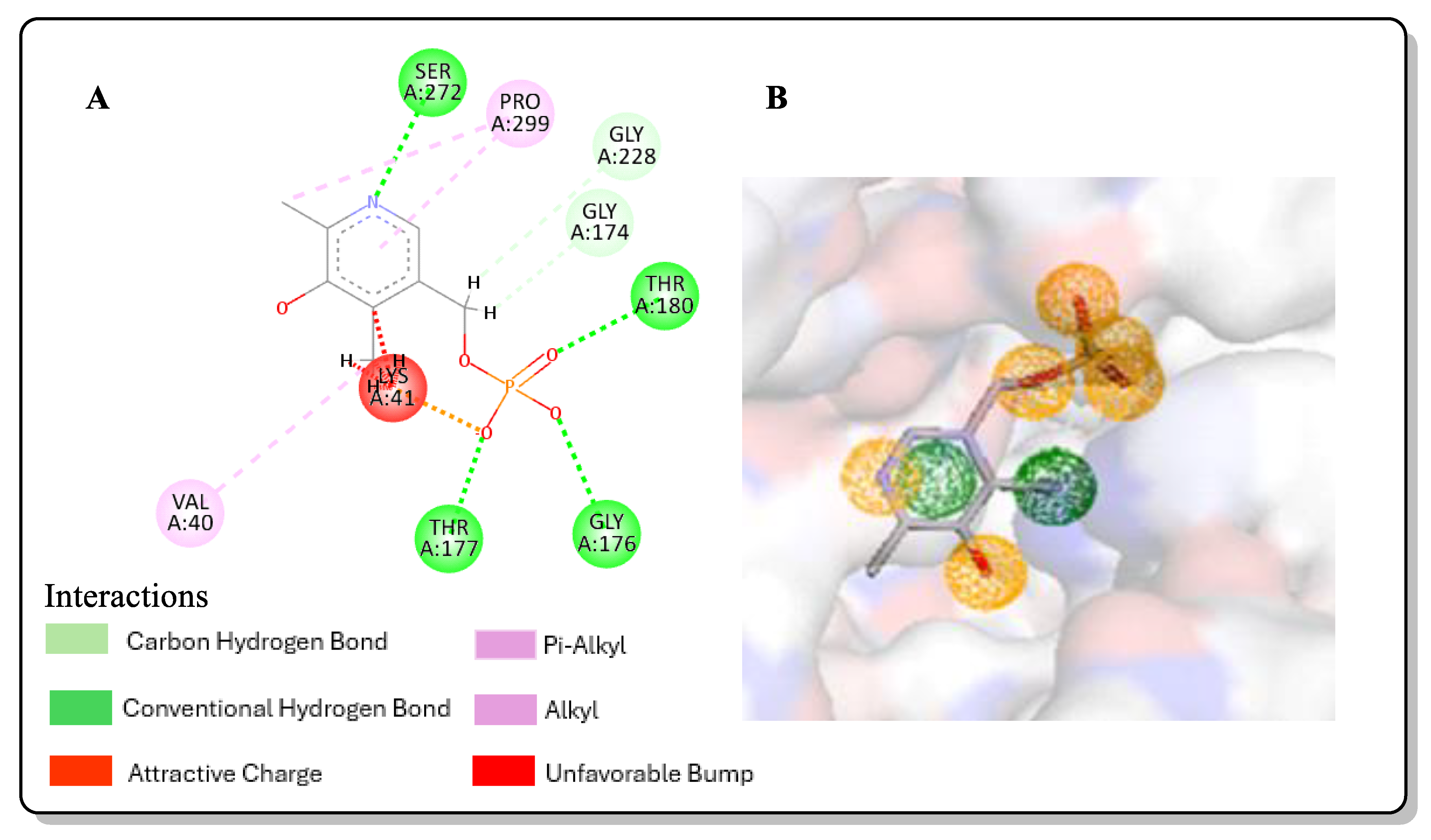
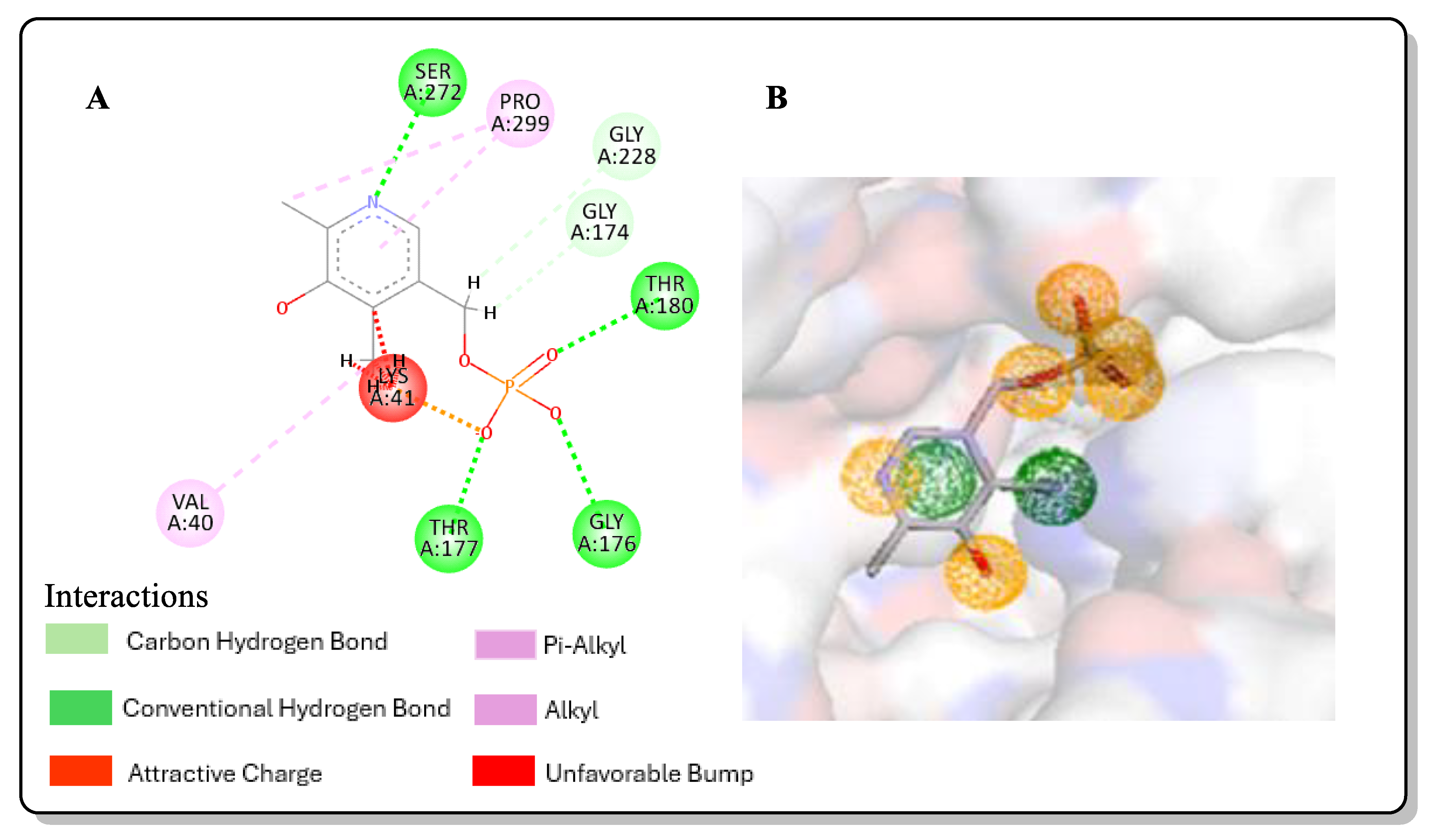
2.3. Post-Screening Analysis
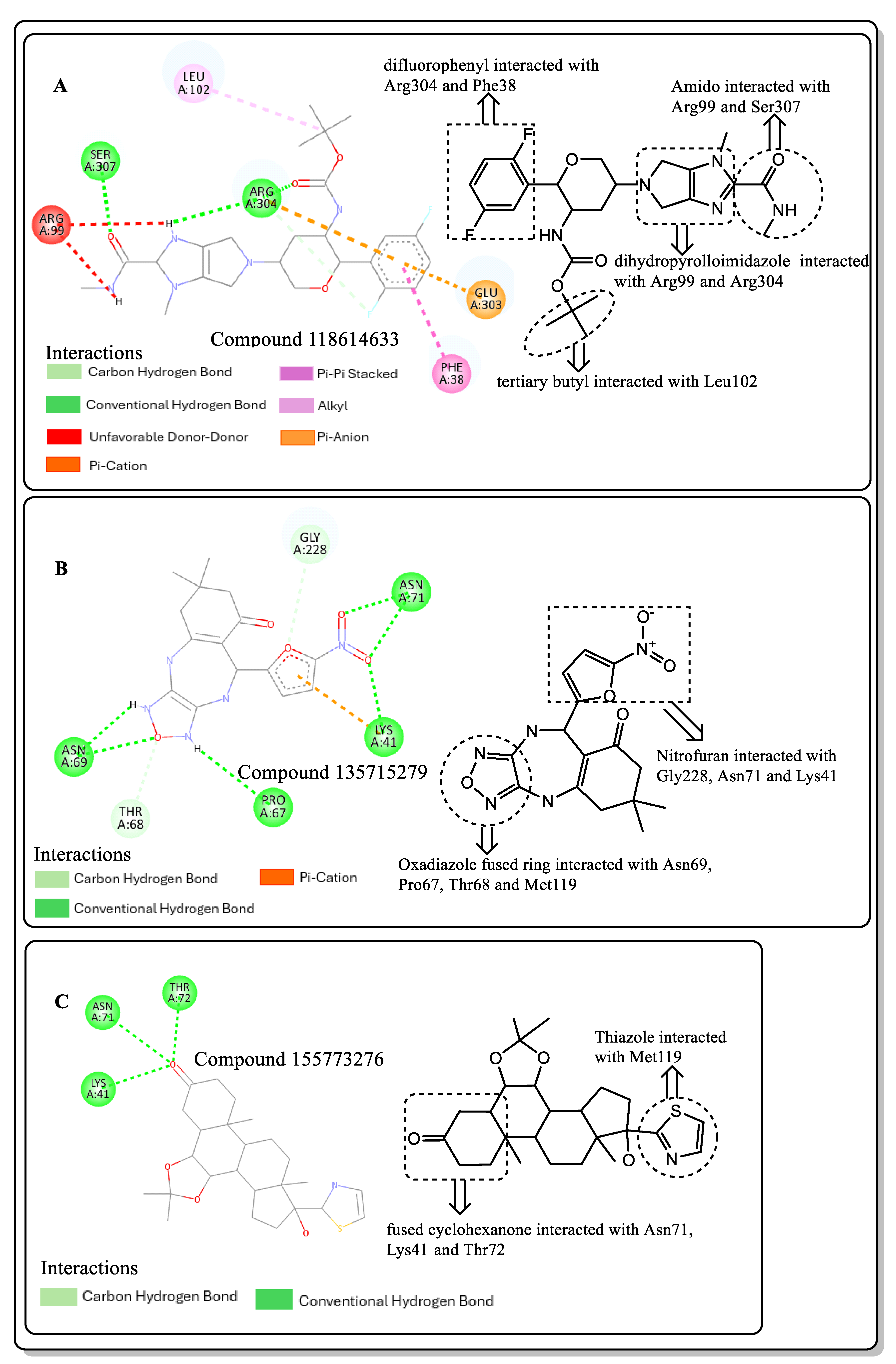
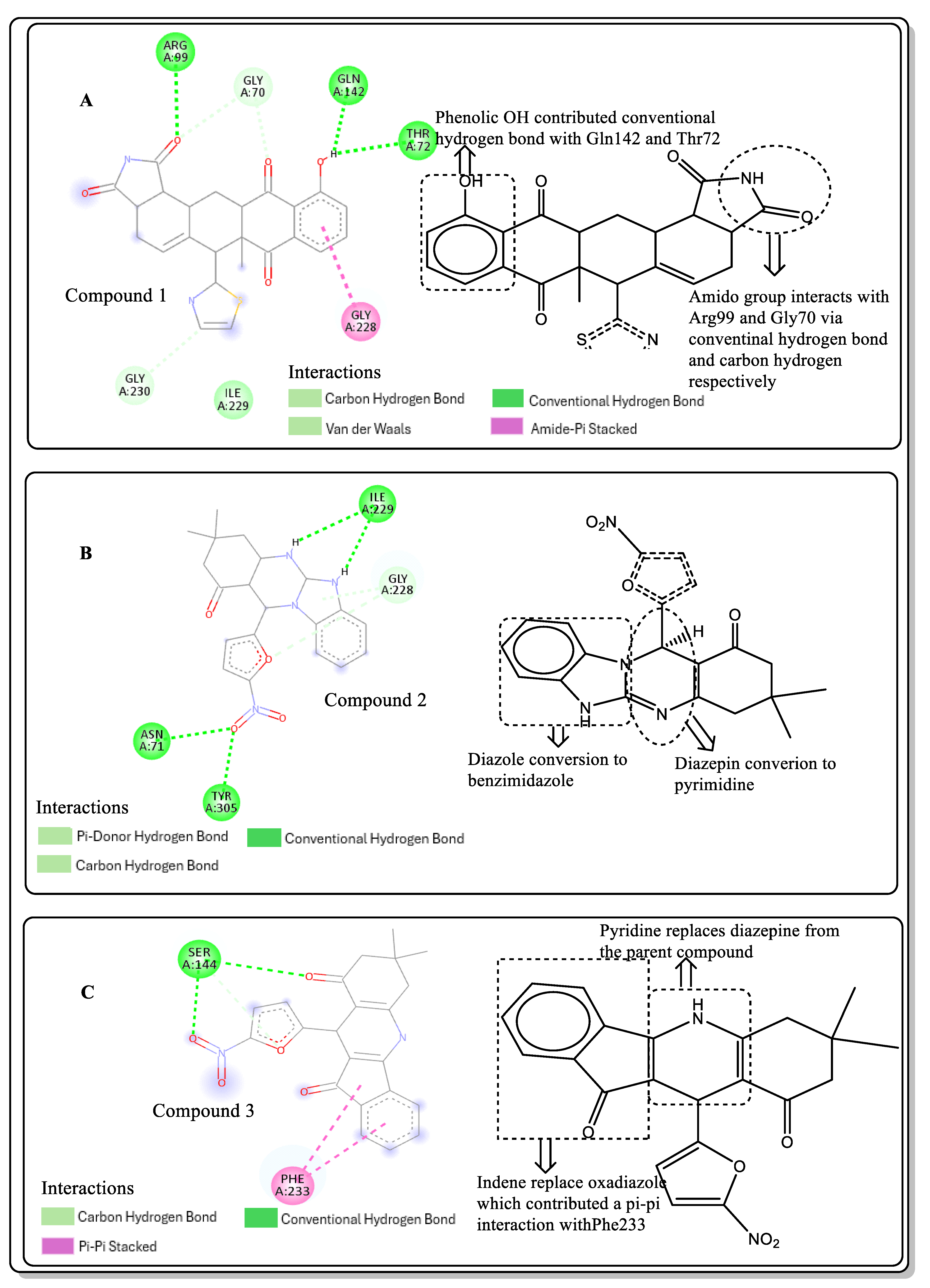
2.4. Structural Activity Relationship (SAR) of Best Hits from Docking Study
2.5. In Silico Toxicity and Druglikeness Prediction
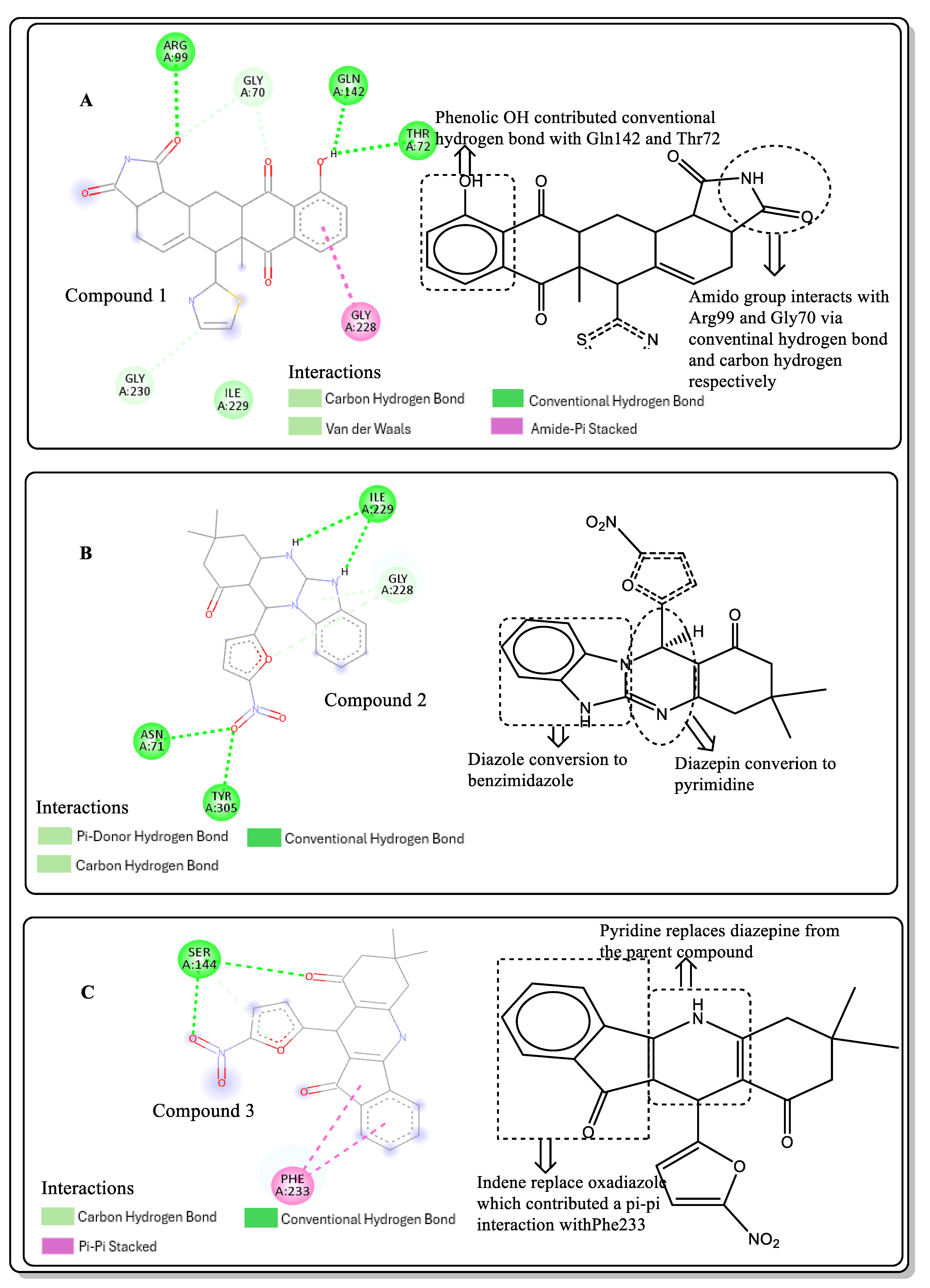
2.6. Lead Optimisation via Scaffold Hopping
Druglikeness and Toxicity Profiling of Optimized Compounds
2.7. Molecular Dynamics Simulation
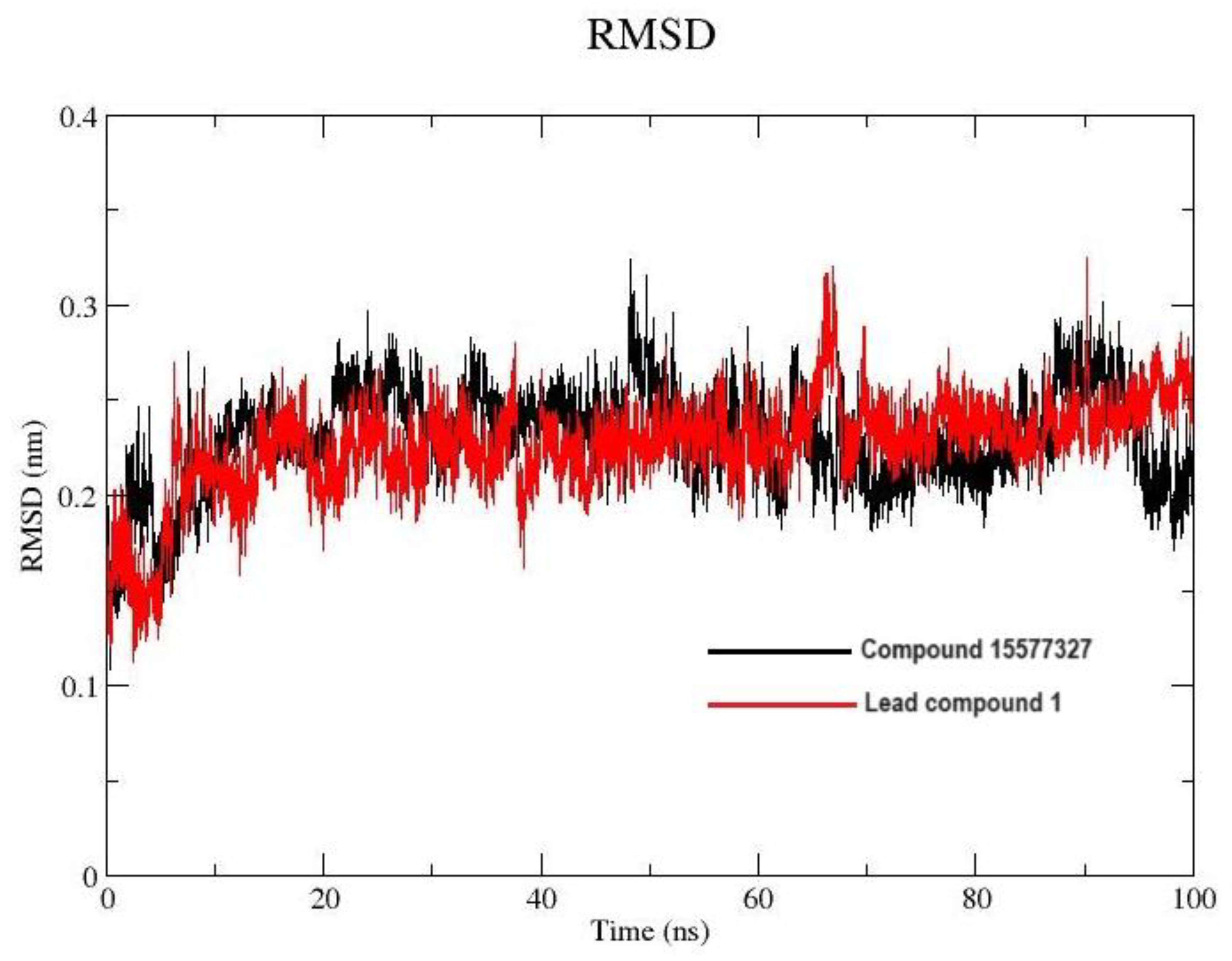
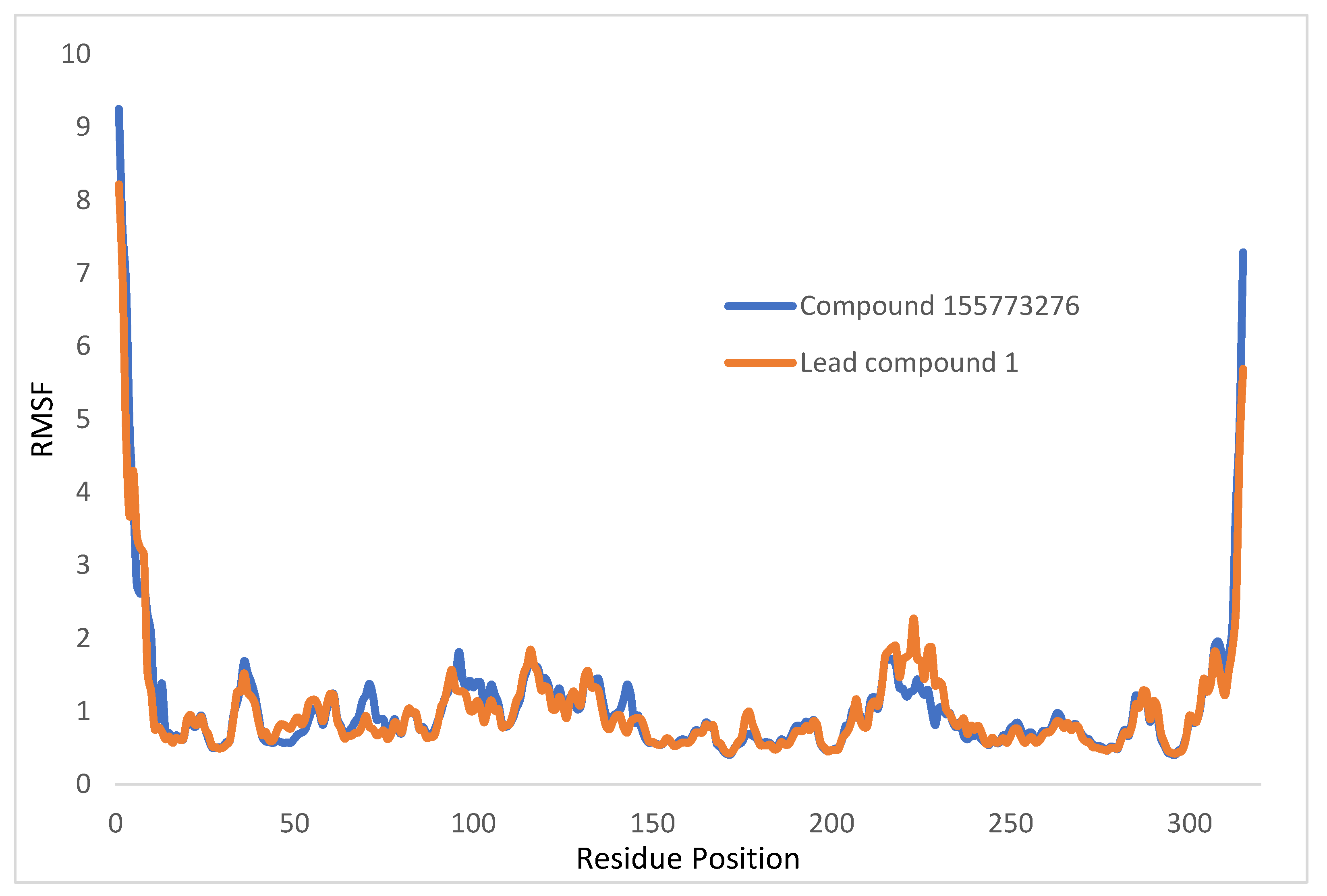
2.7.1. Root Mean Square Deviation and Root Mean Square Fluctuation
2.7.2. Principal Component Analysis
2.7.3. Radius of Gyration
2.7.4. Solvent-Accessible Surface Area
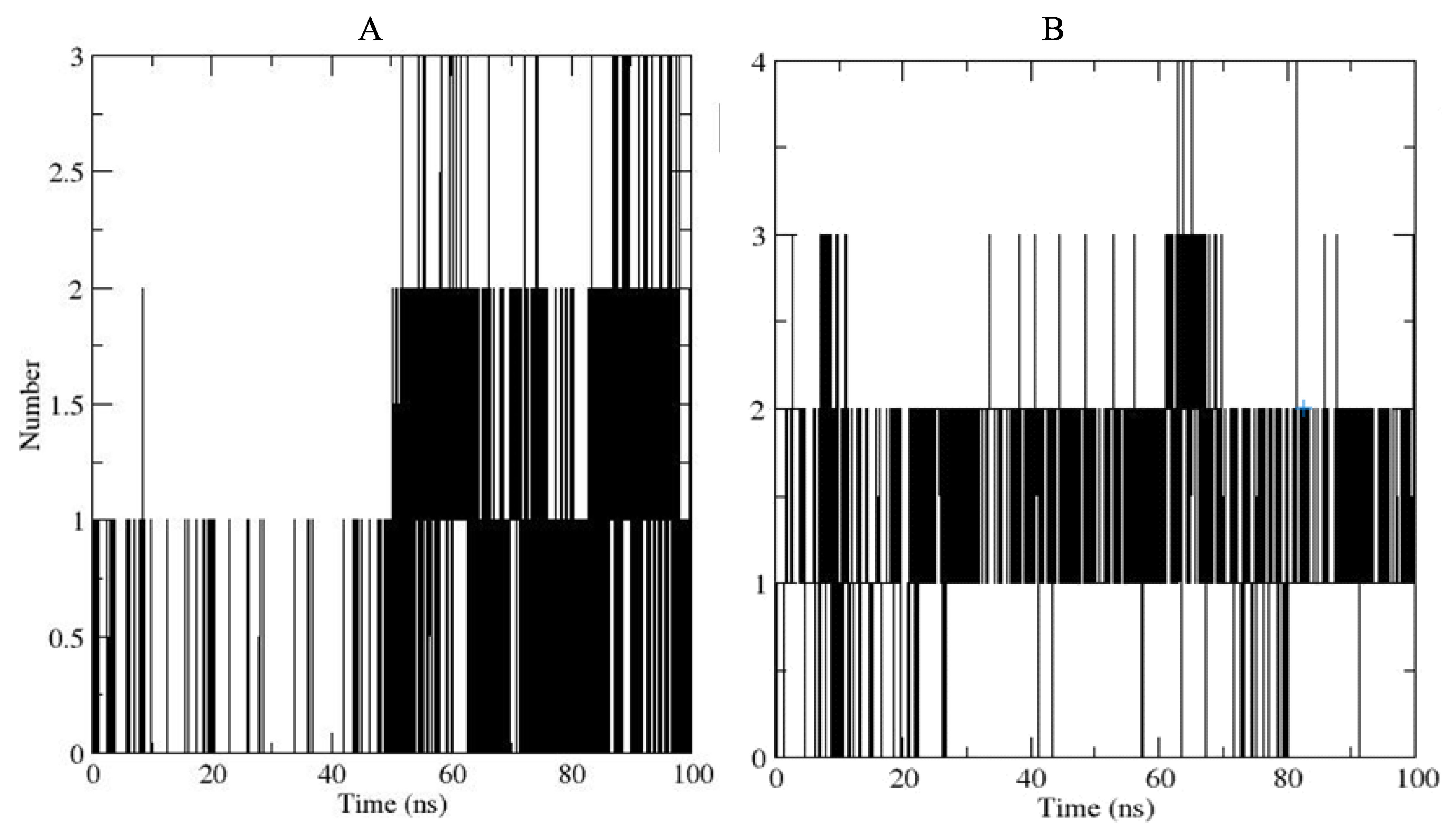
2.7.5. Hydrogen Bond Analysis
3. Discussion
4. Materials and Methods
4.1. Protein Structure Preparation
4.2. Pharmacophore Modeling
4.3. Ligand Library Preparation
4.4. Virtual Screening and Post-Screening Analyses
4.5. Hit-to-Lead: Best Hit Optimization
4.6. In Silico Druglikeness Prediction
4.7. Molecular Dynamics Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- WHO. Global Action Plan on Antimicrobial Resistance; WHO: Geneva, Switzerland, 2017; pp. 1–28. [Google Scholar]
- Annunziato, G. Strategies to overcome antimicrobial resistance (AMR) making use of non-essential target inhibitors: A review. Int. J. Mol. Sci. 2019, 20, 5844. [Google Scholar] [CrossRef]
- Fereshteh, S.; Goodarzi, N.N.; Kalhor, H.; Rahimi, H.; Barzi, S.M.; Badmasti, F. Identification of Putative Drug Targets in Highly Resistant Gram-Negative Bacteria; and Drug Discovery Against Glycyl-tRNA Synthetase as a New Target. Bioinform. Biol. Insights 2023, 17, 11779322231152980. [Google Scholar] [CrossRef]
- Murugaiyan, J.; Anand Kumar, P.; Rao, G.S.; Iskandar, K.; Hawser, S.; Hays, J.P.; Mohsen, Y.; Adukkadukkam, S.; Awuah, W.A.; Jose RA, M.; et al. Progress in Alternative Strategies to Combat Antimicrobial Resistance: Focus on Antibiotics. Antibiotics 2022, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.L.; Oldham KE, A.; McGarvie, J.; Walker, E.J. Combatting antimicrobial resistance via the cysteine biosynthesis pathway in bacterial pathogens. Biosci. Rep. 2022, 42, BSR20220368. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, G.; Spadini, C.; Marchetti, M.; Franko, N.; Pavone, M.; Iannarelli, M.; Bruno, A.; Pieroni, M.; Bettati, S.; Cabassi, C.S.; et al. Inhibitors of O-Acetylserine Sulfhydrylase with a Cyclopropane-Carboxylic Acid Scaffold Are Effective Colistin Adjuvants in Gram Negative Bacteria. Pharmaceuticals 2022, 15, 766. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Chiba, Y.; Hirai, M.Y. Metabolism and Regulatory Functions of O-Acetylserine, S-Adenosylmethionine, Homocysteine, and Serine in Plant Development and Environmental Responses. Front. Plant Sci. 2021, 12, 643403. [Google Scholar] [CrossRef] [PubMed]
- Tumbull, A.L.; Surette, M.G. L-Cysteine is required for induced antibiotic resistance in actively swarming Salmonella enterica serovar Typhimurium. Microbiology 2008, 154, 3410–3419. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Nlebemuo, M.T.; Adekoya, J.A.; Ogunniran, K.O.; Siyanbola, T.O.; Ajanaku, C.O. Chemistry and pharmacological diversity of quinoxaline motifs as anticancer agents. Acta Pharm. 2019, 69, 177–196. [Google Scholar] [CrossRef]
- Adedeji, E.O.; Oduselu, G.O.; Ogunlana, O.O.; Fatumo, S.; Koenig, R.; Adebiyi, E. Anopheles gambiae Trehalase Inhibitors for Malaria Vector Control: A Molecular Docking and Molecular Dynamics Study. Insects 2022, 13, 1070. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19 ed B Alatas. Biomed Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Jukič, M.; Bren, U. Machine Learning in Antibacterial Drug Design. Front. Pharmacol. 2022, 13, 864412. [Google Scholar] [CrossRef]
- Fantacuzzi, M.; De Filippis, B.; Gallorini, M.; Ammazzalorso, A.; Giampietro, L.; Maccallini, C.; Aturki, Z.; Donati, E.; Ibrahim, R.S.; Shawky, E. Synthesis, biological evaluation, and docking study of indole aryl sulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 2020, 185, 111815. [Google Scholar] [CrossRef]
- Zell, L.; Lainer, C.; Kollár, J.; Temml, V.; Schuster, D. Identification of Novel Dopamine D2 Receptor Ligands—A Combined In Silico/In Vitro Approach. Molecules 2022, 27, 4435. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.Y.; Kwon, M.; Choi, H.E.; Kim, K.S. Recent advances in transdermal drug delivery systems: A review. Biomater. Res. 2021, 25, 24. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Ahmad, C.; Khan, H.; Ullahkhan, I.; Tariq, M.; Ullah, N. Synthesis, spectroscopic characterization, biological screening and POM analysis of potentially bioactive copper(II) carboxylate complexes. Bull. Chem. Soc. Ethiop. 2022, 36, 57–71. [Google Scholar] [CrossRef]
- O’Donovan, D.H.; De Fusco, C.; Kuhnke, L.; Reichel, A. Trends in Molecular Properties, Bioavailability, and Permeability across the Bayer Compound Collection: Miniperspective. J. Med. Chem. 2023, 66, 2347–2360. [Google Scholar] [CrossRef]
- Girase, R.; Ahmad, I.; Pawara, R.; Patel, H. Optimizing cardio, hepato and phospholipidosis toxicity of the Bedaquiline by chemoinformatics and molecular modelling approach. SAR QSAR Environ. Res. 2022, 33, 215–235. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Proença-Assunção, J.; Constantino, E.; Farias-de-França, A.P.; Nogueira, F.A.R.; Consonni, S.R.; Chaud, M.V.; dos Santos, C.A.; Oshima-Franco, Y. Mutagenicity of silver nanoparticles synthesized with curcumin (Cur-AgNPs). J. Saudi Chem. Soc. 2021, 25, 101321. [Google Scholar] [CrossRef]
- Ouyang, Y.; Huang, J.; Wang, Y.-L.; Zhong, H.; Song, B.-A.; Hao, G. In Silico resources of drug-likeness as a mirror: What are we lacking in pesticide-likeness? J. Agric. Food Chem. 2021, 69, 10761–10773. [Google Scholar] [CrossRef]
- Bilodeau, C.; Jin, W.; Jaakkola, T.; Barzilay, R.; Jensen, K.F. Generative models for molecular discovery: Recent advances and challenges. WIREs Comput. Mol. Sci. 2022, 12, e1608. [Google Scholar] [CrossRef]
- Ahmed, A.; Aziz, M.; Ejaz, S.A.; Channar, P.A.; Saeed, A.; Zargar, S.; Wani, T.A.; Hamad, A.; Abbas, Q.; Raza, H.; et al. Design, Synthesis, Kinetic Analysis and Pharmacophore-Directed Discovery of 3-Ethylaniline Hybrid Imino-Thiazolidinone as Potential Inhibitor of Carbonic Anhydrase II: An Emerging Biological Target for Treatment of Cancer. Biomolecules 2022, 12, 1696. [Google Scholar] [CrossRef]
- Verma, A.; Kumar, A.; Debnath, M. Molecular docking and simulation studies to give insight of surfactin amyloid interaction for destabilizing Alzheimer ’s A b 42 protofibrils. Med. Chem. Res. 2016, 25, 1616–1622. [Google Scholar] [CrossRef]
- Ghahremanian, S.; Rashidi, M.M.; Raeisi, K.; Toghraie, D. Molecular dynamics simulation approach for discovering potential inhibitors against SARS-CoV-2: A structural review. J. Mol. Liq. 2022, 354, 118901. [Google Scholar] [CrossRef] [PubMed]
- Shahbaaz, M.; Nkaule, A.; Christoffels, A. Designing novel possible kinase inhibitor derivatives as therapeutics against Mycobacterium tuberculosis: An in silico study. Sci. Rep. 2019, 9, 4405. [Google Scholar] [CrossRef] [PubMed]
- Oluyori, A.P.; Olanipekun, B.E.; Adeyemi, O.S.; Egharevba, G.O.; Adegboyega, A.E.; Oladeji, O.S. Molecular docking, pharmacophore modelling, MD simulation and in silico ADMET study reveals bitter cola constituents as potential inhibitors of SARS-CoV-2 main protease and RNA dependent-RNA polymerase. J. Biomol. Struct. Dyn. 2022, 41, 1510–1525. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Amori, L.; Costantino, G. Computational insights into the mechanism of inhibition of OASS-A by a small molecule inhibitor. Mol. Inform. 2013, 32, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho BM, D.C.; et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2019, 47, 1070–1081. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef]
- da Silva CP, M.; das Neves, G.M.; Poser GL von Eifler-Lima, V.L.; Rates SM, K. In Silico Prediction of ADMET/Drug-likeness Properties of Bioactive Phloroglucinols from Hypericum genus. Med. Chem. 2023, 19, 1002–1017. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel. J. Cheminform. 2011, 3, 1–14. [Google Scholar]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Vijayan, T.; Kim, J.; Azam, M.; Al-Resayes, S.I.; Stalin, A.; Kannan, B.S.; Jayamani, A.; Ayyakannu, A.; Nallathambi, S. Influence of co-ligand on the biological properties of Schiff base metal complexes: Synthesis, characterization, cytotoxicity, and antimicrobial studies. Appl. Organomet. Chem. 2022, 36, e6542. [Google Scholar] [CrossRef]
- Ding, J.; Tang, S.; Mei, Z.; Wang, L.; Huang, Q.; Hu, H.; Ling, M.; Wu, J. Vina-GPU 2.0: Further Accelerating AutoDock Vina and Its Derivatives with Graphics Processing Units. J. Chem. Inf. Model. 2023, 63, 1982–1998. [Google Scholar] [CrossRef] [PubMed]
- Muslikh, F.A.; Samudra, R.R.; Ma’arif, B.; Ulhaq, Z.S.; Hardjono, S.; Agil, M. In Silico Molecular Docking and ADMET Analysis for Drug Development of Phytoestrogens Compound with Its Evaluation of Neurodegenerative Diseases. Borneo J. Pharm. 2022, 5, 357–366. [Google Scholar] [CrossRef]
- Perkin, V.O.; Antonyan, G.V.; Radchenko, E.V.; Palyulin, V.A. Web Services for the Prediction of ADMET Parameters Relevant to the Design of Neuroprotective Drugs. In Computational Modeling of Drugs against Alzheimer’s Disease; Roy, K., Ed.; Springer: New York, NY, USA, 2023; pp. 465–485. [Google Scholar]
- Sander, T.; Freyss, J.; von Korff, M.; Reich, J.; Rufener, C. OSIRIS, an Entirely in-House Developed Drug Discovery Informatics System. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Oduselu, G.O.; Afolabi, R.; Ademuwagun, I.; Vaughan, A.; Adebiyi, E. Structure-based pharmacophore modeling, virtual screening, and molecular dynamics simulation studies for identification of Plasmodium falciparum 5-aminolevulinate synthase inhibitors. Front. Med. 2023, 9, 1022429. [Google Scholar] [CrossRef] [PubMed]
- Nisha, C.M.; Kumar, A.; Nair, P.; Gupta, N.; Silakari, C.; Tripathi, T.; Kumar, A. Molecular docking and in silico ADMET study reveals acylguanidine 7a as a potential inhibitor of β-secretase. Adv. Bioinform. 2016, 2016, 9258578. [Google Scholar] [CrossRef] [PubMed]
- Ajay Kumar, T.V.; Kabilan, S.; Parthasarathy, V. Screening and Toxicity Risk Assessment of Selected Compounds to Target Cancer using QSAR and Pharmacophore Modelling. Int. J. PharmTech Res. 2017, 10, 219–224. [Google Scholar] [CrossRef]
- Pinto, E.C.; de Souza, L.G.; Velozo, C.T.; Viana, G.M.; Cabral, L.M.; de Sousa, V.P. Macitentan: An overview of its degradation products, process-related impurities, and in silico toxicity. Comput. Toxicol. 2022, 25, 100255. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Turabi, K.; Sannakki, J.A.; Garse, S.; Iyer, D. Computational screening of phytochemicals for anti-bacterial drug discovery. In Phytochemistry, Computational Tools and Databases in Drug Discovery; Elsevier: Amsterdam, The Netherlands, 2023; pp. 213–243. [Google Scholar]
- Vaiwala, R.; Sharma, P.; Ganapathy Ayappa, K. Differentiating interactions of antimicrobials with Gram-negative and Gram-positive bacterial cell walls using molecular dynamics simulations. Biointerphases 2022, 17, 061008. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PubChem ID | 3D Structure | Docking Score (kcal/mol) | nHBA | nHBD |
|---|---|---|---|---|
| 118614633 | 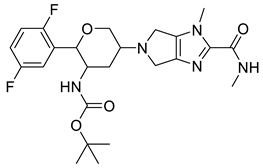 | −9.1 | 9 | 2 |
| 135715279 | 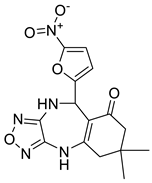 | −8.9 | 10 | 2 |
| 155773276 | 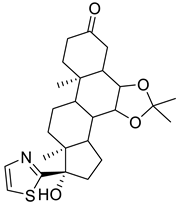 | −8.8 | 5 | 1 |
| 118505490 |  | −8.8 | 9 | 5 |
| 123531073 | 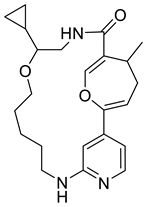 | −8.8 | 6 | 2 |
| 132083481 | 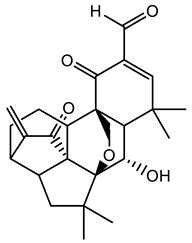 | −8.8 | 5 | 1 |
| 153409783 |  | −8.6 | 8 | 2 |
| 136030136 | 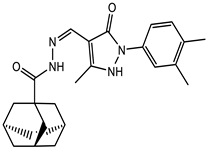 | −8.5 | 6 | 2 |
| 153368440 |  | −8.5 | 9 | 3 |
| 156238864 | 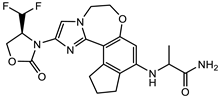 | −8.5 | 9 | 3 |
| Plp |  | −5.6 | 7 | 3 |
| Gentamicin | 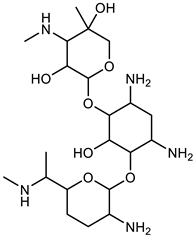 | −7.8 | 12 | 11 |
| PubChem ID |
MW (g/mol) | Clogp |
Tpsa (Å2) | logS | Drug Score | Mutagen | Tumorigenic | Irritant | Reproductive Effective |
|---|---|---|---|---|---|---|---|---|---|
| 118614633 | 491 | 2.29 | 102.5 | −3.04 | 0.36 | Low | Low | Low | Low |
| 135715279 | 345 | 1.27 | 139.3 | −3.49 | 0.43 | Low | Low | Low | Low |
| 118505490 | 453 | 1.99 | 134.0 | −3.73 | 0.32 | Low | Low | Low | Low |
| 123531073 | 397 | 2.37 | 72.48 | −3.60 | 0.57 | Low | Low | Low | Low |
| 155773276 | 445 | 3.48 | 96.89 | −4.37 | 0.30 | Low | Low | Low | High |
| 153409783 | 431 | −0.59 | 99.08 | −1.93 | 0.27 | Medium | High | Low | Medium |
| 136030136 | 406 | 2.62 | 73.8 | −6.18 | 0.35 | Low | High | Low | Low |
| 153368440 | 461 | 1.23 | 111.7 | −5.08 | 0.19 | High | Low | Low | Low |
| 156238864 | 447 | 1.9 | 111.7 | −5.40 | 0.29 | Low | Low | low | Low |
| PLP | 247 | −3.2 | 126.7 | −1.20 | 0.29 | High | Low | low | Low |
| Gentamicin | 477 | −4.21 | 199.7 | −0.59 | 0.77 | Low | Low | low | Low |
| Compound ID | Medicinal Chemistry Score | Toxicity Profile | |||||||
|---|---|---|---|---|---|---|---|---|---|
| QED | Synthetic Accessibility | MCE | GSK | Pfizer | HHT | AMES | hERG Blocker | BBB | |
| PubChem 155773276 Compound 1 | 0.68 | 4.92 | 142.09 | Rejected | Rejected | - | --- | +++ | |
| 0.51 | 4.56 | 121.97 | Rejected | Accepted | -- | -- | - | -- | |
| PubChem 135715279 Compound 2 Compound 3 | 0.62 | 3.95 | 81.14 | Accepted | Accepted | + | +++ | --- | +++ |
| 0.62 | 3.49 | 95.92 | Accepted | Accepted | ++ | +++ | --- | -- | |
| 0.61 | 0.35 | 98.57 | Accepted | Accepted | - | +++ | --- | --- | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elebiju, O.F.; Oduselu, G.O.; Ogunnupebi, T.A.; Ajani, O.O.; Adebiyi, E. In Silico Design of Potential Small-Molecule Antibiotic Adjuvants against Salmonella typhimurium Ortho Acetyl Sulphydrylase Synthase to Address Antimicrobial Resistance. Pharmaceuticals 2024, 17, 543. https://doi.org/10.3390/ph17050543
Elebiju OF, Oduselu GO, Ogunnupebi TA, Ajani OO, Adebiyi E. In Silico Design of Potential Small-Molecule Antibiotic Adjuvants against Salmonella typhimurium Ortho Acetyl Sulphydrylase Synthase to Address Antimicrobial Resistance. Pharmaceuticals. 2024; 17(5):543. https://doi.org/10.3390/ph17050543
Chicago/Turabian StyleElebiju, Oluwadunni F., Gbolahan O. Oduselu, Temitope A. Ogunnupebi, Olayinka O. Ajani, and Ezekiel Adebiyi. 2024. "In Silico Design of Potential Small-Molecule Antibiotic Adjuvants against Salmonella typhimurium Ortho Acetyl Sulphydrylase Synthase to Address Antimicrobial Resistance" Pharmaceuticals 17, no. 5: 543. https://doi.org/10.3390/ph17050543
APA StyleElebiju, O. F., Oduselu, G. O., Ogunnupebi, T. A., Ajani, O. O., & Adebiyi, E. (2024). In Silico Design of Potential Small-Molecule Antibiotic Adjuvants against Salmonella typhimurium Ortho Acetyl Sulphydrylase Synthase to Address Antimicrobial Resistance. Pharmaceuticals, 17(5), 543. https://doi.org/10.3390/ph17050543