


Evaluating Drug Interactions between Ritonavir and Opioid Analgesics: Implications from Physiologically Based Pharmacokinetic Simulation

 ,
,
Abstract
1. Introduction
2. Results
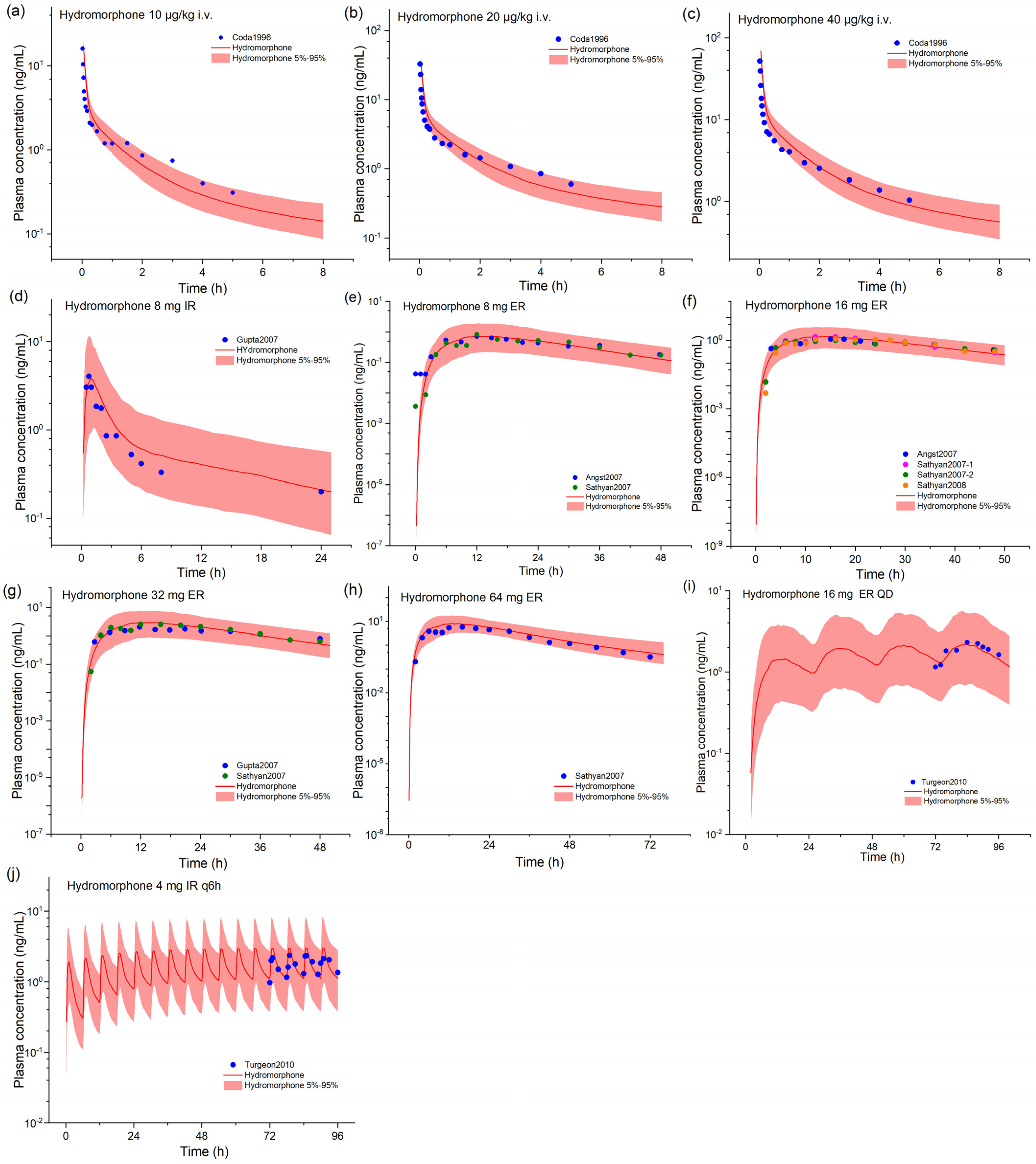
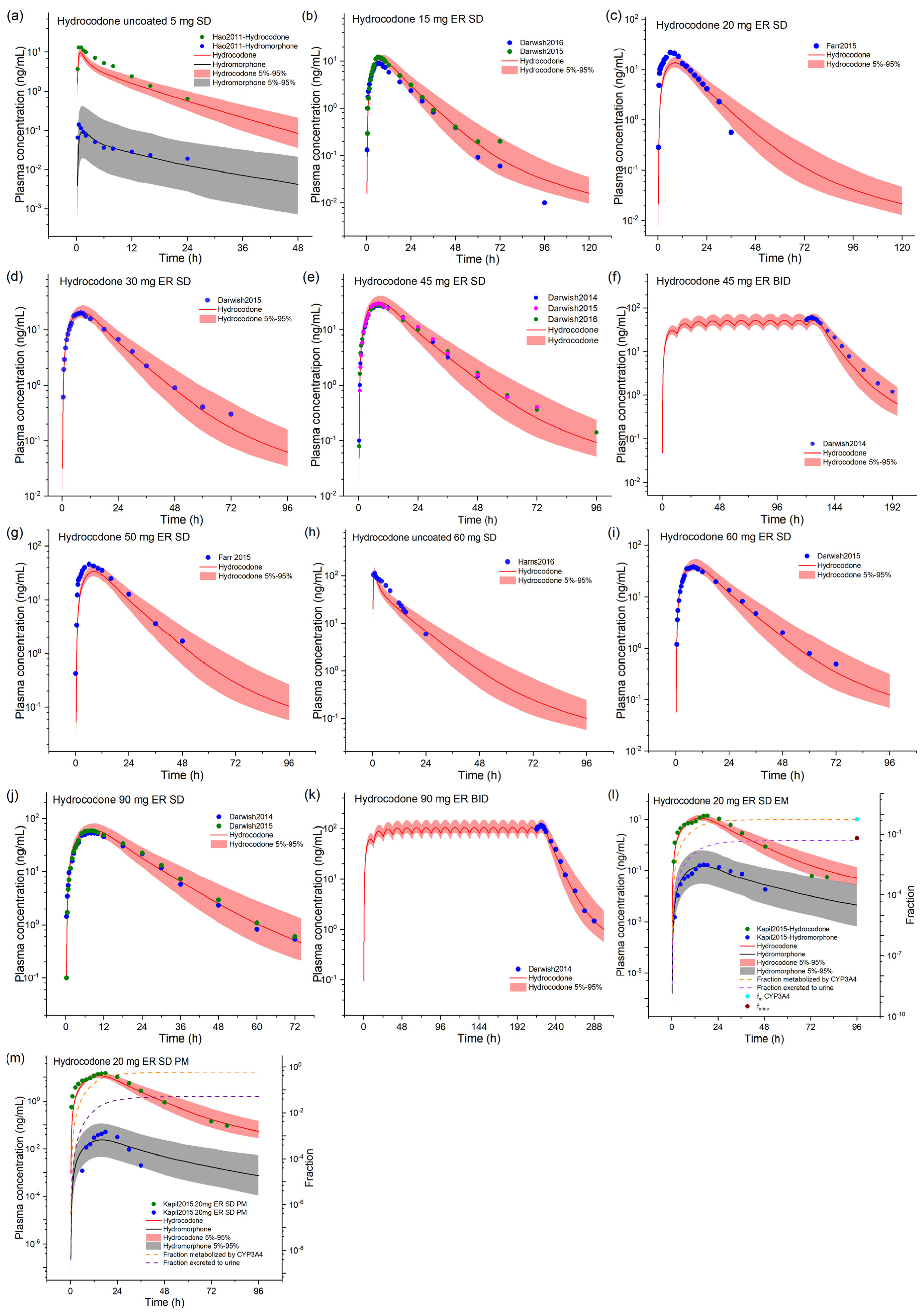
2.1. Hydrocodone and Hydromorphone Model Development and Evaluation
2.2. Model-Based DDI Prediction between Ritonavir and Fentanyl Analogs
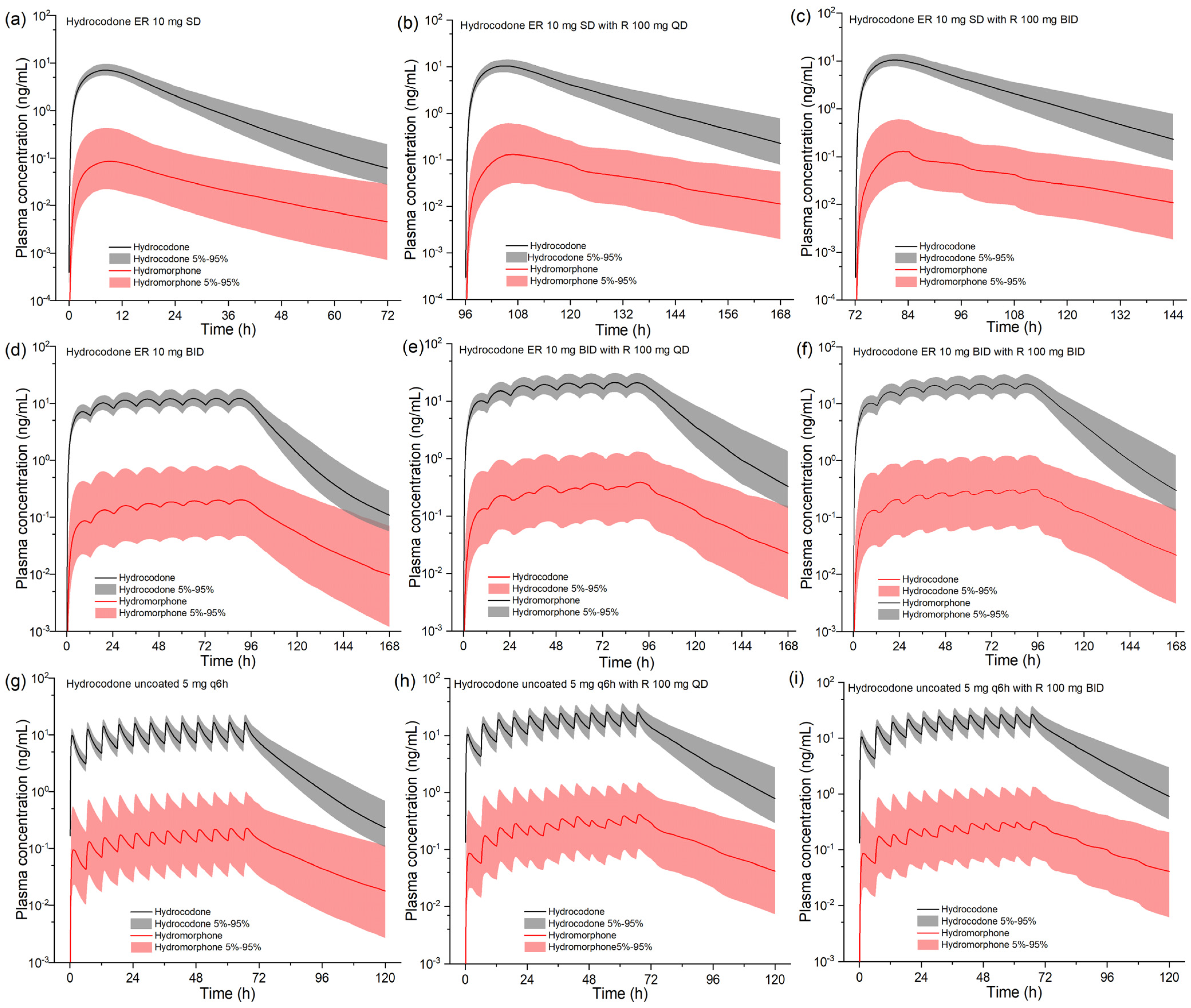
2.3. Model-Based DDI Prediction between Ritonavir and Hydrocodone
3. Discussion
4. Materials and Methods
4.1. PBPK Modeling Platform and Related Software
4.2. Drug Model Preparation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Hydromorphone | Reference/Source | Hydrocodone | Reference/Source |
|---|---|---|---|---|
| Lipophilicity | 1.8 | [36] | 2.0 | [37] |
| Plasma fraction unbound | 0.86 | Drugbank | 0.64 | Drug label |
| MW | 285.30 g/mol | Drugbank | 299.40 g/mol | Drugbank |
| pKa | 8.5 | PubChem | 8.23 | PubChem |
| Solubility | 0.149 mg/mL | PubChem | 0.797 mg/mL | PubChem |
| Specific intestinal permeability | 3.27 × 10−6 cm/min | Optimized | 2.00 × 10−4 cm/min | Optimized |
| Partition coefficients calculation | Diverse | PK-Sim standard | Diverse | Schmitt |
| Cellular permeability | 3.37 × 10−3 cm/min | PK-Sim standard | 4.00 × 10−3 cm/min | Charge-dependent Schmitt normalized to PK-Sim |
| UGT2B7 specific clearance | 6.11 1/min | Optimized | NA | |
| Formulation | IR dissolved, ER Weibull | Weibull | ||
| 50% dissolution time | ER 8.48 h | Optimized | Uncoated 0.80 h, ER 6.40 h | Optimized |
| Dissolution shape | ER 2.94 | Optimized | Uncoated 0.79 ER 1.79 | Optimized |
| GFR fraction | 1.0 | Assumed | 1.0 | Assumed |
| Km,CYP3A4 | NA | 2.60 mmol/L | [19] | |
| Kcat,CYP3A4 | NA | 361 1/min | Optimized | |
| Km,CYP2D6 | NA | 54 μmol/L | [19] | |
| Kcat,CYP2D6 (EM) | NA | 7.12 1/min | Optimized | |
| Kcat,CYP2D6 (PM) | NA | 1.07 1/min | Optimized | |
| UGT2B7-specific clearance | 6.11 1/min | Optimized | NA | |
| Hepatic clearance (non-CYP) | NA | 0.36 1/min | Optimized | |
| Ritonavir | ||||
| Ki,CYP3A4 | 0.25 μM | [38] | ||
| Kinact,CYP3A4 | 0.40 1/min | [39] | ||
| Kinact_half,CYP3A4 | 0.20 μM | [39] | ||
| Ki,CYP2D6 | 0.04 μM | [40] | ||
| EC50CYP3A4 | 0.17 μM | [25] | ||
| EmaxCYP3A4 | 7.47 | [25] | ||
| Ki,P-gp | 0.20 μM | [41] |
4.3. PBPK Modeling-Based Simulation for Ritonavir–Opioid DDIs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mercadante, S. Opioid metabolism and clinical aspects. Eur. J. Pharmacol. 2015, 769, 71–78. [Google Scholar] [CrossRef]
- Lv, J.; Liu, F.; Feng, N.; Sun, X.; Tang, J.; Xie, L.; Wang, Y. CYP3A4 gene polymorphism is correlated with individual consumption of sufentanil. Acta Anaesthesiol. Scand. 2018, 62, 1367–1373. [Google Scholar] [CrossRef]
- Olkkola, K.T.; Palkama, V.J.; Neuvonen, P.J. Ritonavir’s role in reducing fentanyl clearance and prolonging its half-life. Anesthesiology 1999, 91, 681–685. [Google Scholar] [CrossRef]
- Nieminen, T.H.; Hagelberg, N.M.; Saari, T.I.; Neuvonen, M.; Neuvonen, P.J.; Laine, K.; Olkkola, K.T. Oxycodone concentrations are greatly increased by the concomitant use of ritonavir or lopinavir/ritonavir. Eur. J. Clin. Pharmacol. 2010, 66, 977–985. [Google Scholar] [CrossRef]
- Svedmyr, A.; Hack, H.; Anderson, B.J. Interactions of the protease inhibitor, ritonavir, with common anesthesia drugs. Paediatr. Anaesth. 2022, 32, 1091–1099. [Google Scholar] [CrossRef]
- Feng, X.Q.; Zhu, L.L.; Zhou, Q. Opioid analgesics-related pharmacokinetic drug interactions: From the perspectives of evidence based on randomized controlled trials and clinical risk management. J. Pain Res. 2017, 10, 1225–1239. [Google Scholar] [CrossRef]
- Tseng, A.; Szadkowski, L.; Walmsley, S.; Salit, I.; Raboud, J. Association of age with polypharmacy and risk of drug interactions with antiretroviral medications in HIV-positive patients. Ann. Pharmacother. 2013, 47, 1429–1439. [Google Scholar] [CrossRef]
- Gerhart, J.; Draica, F.; Benigno, M.; Atkinson, J.; Reimbaeva, M.; Francis, D.; Baillon-Plot, N.; Sidhu, G.S.; Damle, B.D. Real-World Evidence of the Top 100 Prescribed Drugs in the USA and Their Potential for Drug Interactions with Nirmatrelvir; Ritonavir. AAPS J. 2023, 25, 73. [Google Scholar] [CrossRef]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef]
- Lea, A.P.; Faulds, D. Ritonavir. Drugs 1996, 52, 541–546, discussion 547–548. [Google Scholar] [CrossRef]
- Vadivelu, N.; Kai, A.M.; Kodumudi, V.; Sramcik, J.; Kaye, A.D. The Opioid Crisis: A Comprehensive Overview. Curr. Pain Headache Rep. 2018, 22, 16. [Google Scholar] [CrossRef]
- World Health Organization. WHO Guidelines for the Pharmacological and Radiotherapeutic Management of Cancer Pain in Adults and Adolescents; WHO Guidelines Approved by the Guidelines Review Committee; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Feierman, D.E.; Lasker, J.M. Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4. Drug Metab. Dispos. 1996, 24, 932–939. [Google Scholar]
- Kharasch, E.D.; Walker, A.; Isoherranen, N.; Hoffer, C.; Sheffels, P.; Thummel, K.; Whittington, D.; Ensign, D. Influence of CYP3A5 genotype on the pharmacokinetics and pharmacodynamics of the cytochrome P4503A probes alfentanil and midazolam. Clin. Pharmacol. Ther. 2007, 82, 410–426. [Google Scholar] [CrossRef]
- Tateishi, T.; Krivoruk, Y.; Ueng, Y.F.; Wood, A.J.; Guengerich, F.P.; Wood, M. Identification of human liver cytochrome P-450 3A4 as the enzyme responsible for fentanyl and sufentanil N-dealkylation. Anesth. Analg. 1996, 82, 167–172. [Google Scholar] [CrossRef]
- Shafi, A.; Berry, A.J.; Sumnall, H.; Wood, D.M.; Tracy, D.K. Synthetic opioids: A review and clinical update. Ther. Adv. Psychopharmacol. 2022, 12, 20451253221139616. [Google Scholar] [CrossRef]
- Dhillon, S. Hydrocodone Bitartrate ER (Hysingla((R)) ER): A Review in Chronic Pain. Clin. Drug Investig. 2016, 36, 969–980. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Menelaou, A.; Foster, D.J.; Coller, J.K.; Somogyi, A.A. CYP2D6 and CYP3A4 involvement in the primary oxidative metabolism of hydrocodone by human liver microsomes. Br. J. Clin. Pharmacol. 2004, 57, 287–297. [Google Scholar] [CrossRef]
- Smith, H.S. The metabolism of opioid agents and the clinical impact of their active metabolites. Clin. J. Pain 2011, 27, 824–838. [Google Scholar] [CrossRef]
- Polepally, A.R.; King, J.R.; Ding, B.; Shuster, D.L.; Dumas, E.O.; Khatri, A.; Chiu, Y.L.; Podsadecki, T.J.; Menon, R.M. Drug-Drug Interactions Between the Anti-Hepatitis C Virus 3D Regimen of Ombitasvir, Paritaprevir/Ritonavir, and Dasabuvir and Eight Commonly Used Medications in Healthy Volunteers. Clin. Pharmacokinet. 2016, 55, 1003–1014. [Google Scholar] [CrossRef]
- Jones, H.M.; Gardner, I.B.; Watson, K.J. Modelling and PBPK simulation in drug discovery. AAPS J. 2009, 11, 155–166. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.; Unadkat, J.D.; Zhang, X.; Wu, D.; Heimbach, T. Applications, Challenges, and Outlook for PBPK Modeling and Simulation: A Regulatory, Industrial and Academic Perspective. Pharm. Res. 2022, 39, 1701–1731. [Google Scholar] [CrossRef]
- Moj, D.; Hanke, N.; Britz, H.; Frechen, S.; Kanacher, T.; Wendl, T.; Haefeli, W.E.; Lehr, T. Clarithromycin, Midazolam, and Digoxin: Application of PBPK Modeling to Gain New Insights into Drug-Drug Interactions and Co-medication Regimens. AAPS J. 2017, 19, 298–312. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, W.; Olkkola, K.T.; Dallmann, A.; Ni, L.; Zhao, Y.; Wang, L.; Zhang, Q.; Hu, W. Physiologically Based Pharmacokinetic Modeling of Ritonavir-Oxycodone Drug Interactions and Its Implication for Dosing Strategy. Eur. J. Pharm. Sci. 2024, 194, 106697. [Google Scholar] [CrossRef]
- Kovar, L.; Weber, A.; Zemlin, M.; Kohl, Y.; Bals, R.; Meibohm, B.; Selzer, D.; Lehr, T. Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients. Pharmaceutics 2020, 12, 908. [Google Scholar] [CrossRef]
- Ziesenitz, V.C.; Konig, S.K.; Mahlke, N.S.; Skopp, G.; Haefeli, W.E.; Mikus, G. Pharmacokinetic interaction of intravenous fentanyl with ketoconazole. J. Clin. Pharmacol. 2015, 55, 708–717. [Google Scholar] [CrossRef]
- Palkama, V.J.; Neuvonen, P.J.; Olkkola, K.T. The CYP 3A4 inhibitor itraconazole has no effect on the pharmacokinetics of i.v. fentanyl. Br. J. Anaesth. 1998, 81, 598–600. [Google Scholar] [CrossRef]
- Crews, K.R.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Klein, T.E.; Caudle, K.E.; Haidar, C.E.; Shen, D.D.; Callaghan, J.T.; Sadhasivam, S.; et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin. Pharmacol. Ther. 2014, 95, 376–382. [Google Scholar] [CrossRef]
- Glass, P.S.A.; Gan, T.J.; Howell, S. A review of the pharmacokinetics and pharmacodynamics of remifentanil. Anesth. Analg. 1999, 89, 7. [Google Scholar] [CrossRef]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef]
- Coates, S.; Lazarus, P. Hydrocodone, Oxycodone, and Morphine Metabolism and Drug–Drug Interactions. J. Pharmacol. Exp. Ther. 2023, 387, 150–169. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. In Vitro 2008, 22, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Kapil, R.P.; Friedman, K.; Cipriano, A.; Michels, G.; Shet, M.; Mondal, S.A.; Harris, S.C. Effects of paroxetine, a CYP2D6 inhibitor, on the pharmacokinetic properties of hydrocodone after coadministration with a single-entity, once-daily, extended--release hydrocodone tablet. Clin. Ther. 2015, 37, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Yang, H.; Dallmann, A.; Jiang, X.; Wang, L.; Hu, W. Physiologically Based Pharmacokinetic Modeling in Pregnant Women Suggests Minor Decrease in Maternal Exposure to Olanzapine. Front. Pharmacol. 2021, 12, 793346. [Google Scholar] [CrossRef] [PubMed]
- Landolf, K.M.; Rivosecchi, R.M.; Gomez, H.; Sciortino, C.M.; Murray, H.N.; Padmanabhan, R.R.; Sanchez, P.G.; Harano, T.; Sappington, P.L. Comparison of Hydromorphone versus Fentanyl-based Sedation in Extracorporeal Membrane Oxygenation: A Propensity-Matched Analysis. Pharmacotherapy 2020, 40, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Concheiro, M.; Chesser, R.; Pardi, J.; Cooper, G. Postmortem Toxicology of New Synthetic Opioids. Front. Pharmacol. 2018, 9, 1210. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.J.; Collier, A.C.; Kharasch, E.D.; Whittington, D.; Thummel, K.E.; Unadkat, J.D. Complex drug interactions of HIV protease inhibitors 1: Inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab. Dispos. 2011, 39, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Stresser, D.M.; Mao, J.; Kenny, J.R.; Jones, B.C.; Grime, K. Exploring concepts of in vitro time-dependent CYP inhibition assays. Expert Opin. Drug Metab. Toxicol. 2014, 10, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Pansari, A.; Kilford, P.J.; Jamei, M.; Turner, D.B.; Gardner, I. A Mechanistic Absorption and Disposition Model of Ritonavir to Predict Exposure and Drug-Drug Interaction Potential of CYP3A4/5 and CYP2D6 Substrates. Eur. J. Drug Metab. Pharmacokinet. 2022, 47, 483–495. [Google Scholar] [CrossRef]
- Alsmadi, M.M. The investigation of the complex population-drug-drug interaction between ritonavir-boosted lopinavir and chloroquine or ivermectin using physiologically-based pharmacokinetic modeling. Drug Metab. Pers. Ther. 2023, 38, 87–105. [Google Scholar] [CrossRef]
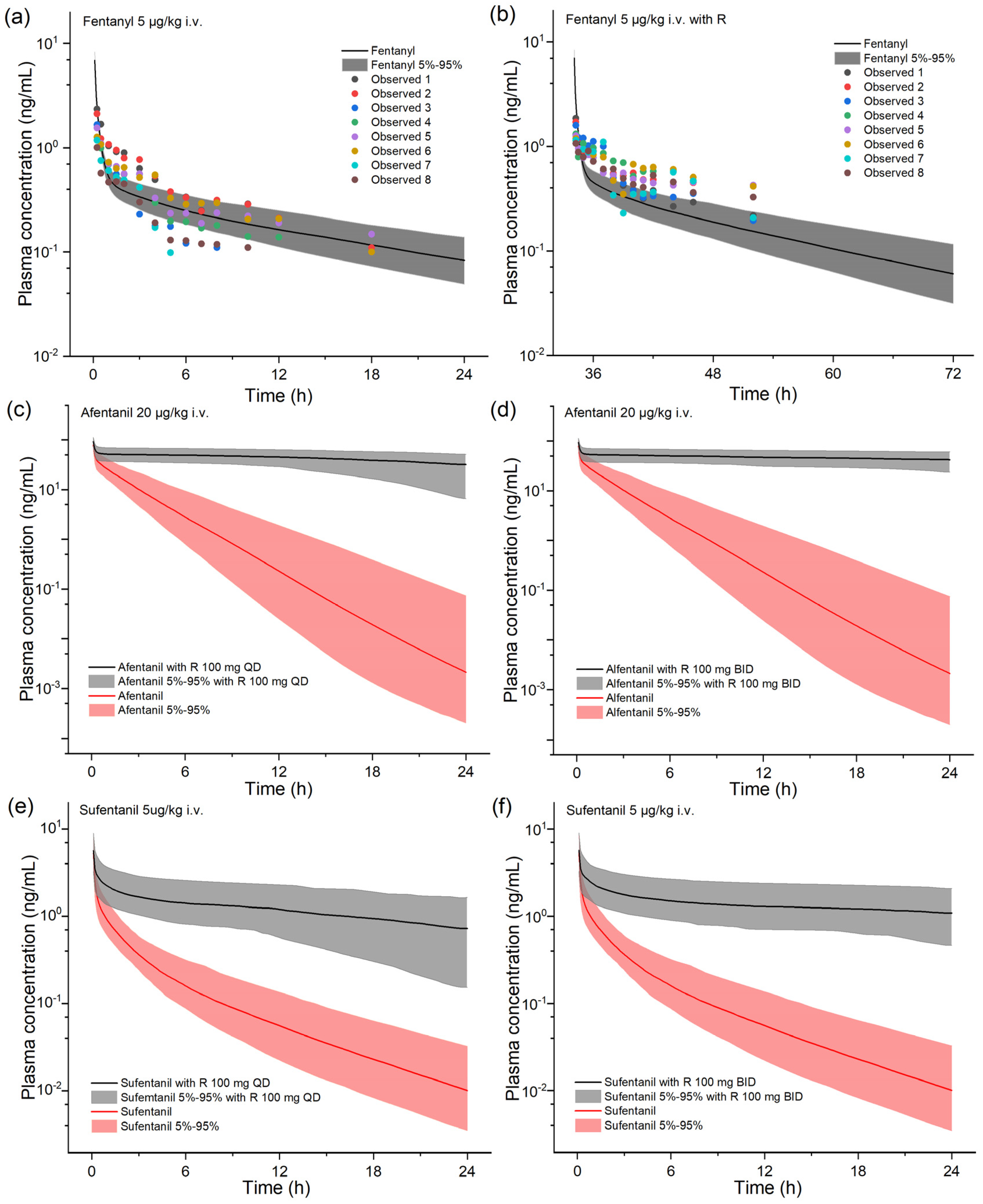
| Drugs | CmaxR | AUCR | ||
|---|---|---|---|---|
| Predicted | Observed | Predicted | Observed | |
| Fentanyl | NA | NA | 1.28 | 2.70 |
| Hydrocodone | 1.24 | 1.27 | 1.81 | 1.90 |
| Drugs | Protocols | AUC0~∞ (ng × h/mL) | T1/2 (h) |
|---|---|---|---|
| Alfentanil | Alfentanil 20 μg/kg i.v. | 94.9 | 2.29 |
| Alfentanil 20 μg/kg i.v. + R 100 mg QD | 2717 (+2766%) | 17.4 (+660%) | |
| Alfentanil 20 μg/kg i.v. + R 100 mg BID | 5581 (+5787%) | 63.7 (+2681%) | |
| Sufentanil | Sufentanil 5 μg/kg i.v. | 4.58 | 5.42 |
| Sufentanil 5 μg/kg i.v. + R 100 mg QD | 59.1 (+1192%) | 11.4 (+110%) | |
| Sufentanil 5 μg/kg i.v. 84 h after R 100 mg QD withdrawal | 5.54 | 5.38 | |
| Sufentanil 5 μg/kg i.v. + R 100 mg BID | 117 (+2448%) | 39.8 (+634%) | |
| Sufentanil 5 μg/kg i.v. 96 h after R 100 mg BID withdrawal | 5.57 | 6.46 |
| Protocols | Cmax/Cmax-ss (ng/mL) | AUC/AUCss (ng×h/mL) | T1/2 (h) | |||
|---|---|---|---|---|---|---|
| HYD | HYM | HYD | HYM | HYD | HYM | |
| HYD ER 10 mg SD | 7.11 | 0.09 | 135 | 2.35 | 13.1 | 19.7 |
| HYD ER 10 mg SD + R 100 mg QD | 10.4 (46.3%) | 0.13 (44.4%) | 241 (78.5%) | 4.22 (79.6%) | 12.3 (−6.1%) | 18.9 (−4.1%) |
| HYD ER 10 mg SD + R 100 mg BID | 10.5 (46.3%) | 0.13 (44.4%) | 248 (83.7%) | 3.87 (64.7%) | 15.7 (19.8%) | 20.8 (5.6%) |
| HYD ER 10 mg BID | 12.24 | 0.20 | 135 | 2.24 | 10.2 | 13.9 |
| HYD ER 10 mg BID + R 100 mg QD | 21.2 (73.2%) | 0.39 (95.0%) | 235 (74.1%) | 3.78 (68.8%) | 13.6 (33.3%) | 24.1 (73.4%) |
| HYD ER 10 mg BID + R 100 mg BID | 22.1 (80.6%) | 0.31 (55.0%) | 245 (81.5%) | 3.40 (51.8%) | 13.6 (33.3%) | 24.1 (73.4%) |
| HYD uncoated 5 mg q6h | 17.1 | 0.23 | 65.1 | 1.07 | 5.73 | 5.22 |
| HYD uncoated 5 mg q6h + R 100 mg QD | 26.4 (54.4%) | 0.42 (82.6%) | 116 (78.2%) | 2.01 (87.9%) | 7.11 (24.1%) | 8.89 (70.3%) |
| HYD uncoated 5 mg q6h + R 100 mg BID | 27.3 (59.6%) | 0.32 (39.1%) | 122 (87.4%) | 1.60 (49.5%) | 7.11 (24.1%) | 8.89 (70.3%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, L.; Cao, Z.; Jiang, J.; Zhang, W.; Hu, W.; Zhang, Q.; Shen, C.; Chen, X.; Zheng, L. Evaluating Drug Interactions between Ritonavir and Opioid Analgesics: Implications from Physiologically Based Pharmacokinetic Simulation. Pharmaceuticals 2024, 17, 640. https://doi.org/10.3390/ph17050640
Ni L, Cao Z, Jiang J, Zhang W, Hu W, Zhang Q, Shen C, Chen X, Zheng L. Evaluating Drug Interactions between Ritonavir and Opioid Analgesics: Implications from Physiologically Based Pharmacokinetic Simulation. Pharmaceuticals. 2024; 17(5):640. https://doi.org/10.3390/ph17050640
Chicago/Turabian StyleNi, Liang, Zhihai Cao, Jiakang Jiang, Wei Zhang, Wei Hu, Qian Zhang, Chaozhuang Shen, Xijing Chen, and Liang Zheng. 2024. "Evaluating Drug Interactions between Ritonavir and Opioid Analgesics: Implications from Physiologically Based Pharmacokinetic Simulation" Pharmaceuticals 17, no. 5: 640. https://doi.org/10.3390/ph17050640
APA StyleNi, L., Cao, Z., Jiang, J., Zhang, W., Hu, W., Zhang, Q., Shen, C., Chen, X., & Zheng, L. (2024). Evaluating Drug Interactions between Ritonavir and Opioid Analgesics: Implications from Physiologically Based Pharmacokinetic Simulation. Pharmaceuticals, 17(5), 640. https://doi.org/10.3390/ph17050640