

Allosteric Modulators of Serotonin Receptors: A Medicinal Chemistry Survey




Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | G Protein/Type of Ion Channel | Distribution | Physio-Pathological Implications | Refs. |
|---|---|---|---|---|
| 5-HT1AR | Gi/o | Raphe nuclei (presynaptic receptor), hippocampus, prefrontal cortex, thalamus, lateral septum, amygdala, hypothalamic nuclei (postsynaptic receptor) | Modulation of mood, anxiety, cognition, sleep, and pain perception | [25,26] |
| 5-HT1BR | Gi/o | Substantia nigra, globus pallidus, dorsal subiculum, superior colliculi, peripheral vasculature | Migraine | [15,35] |
| 5-HT1DR | Gi/o | Substantia nigra, basal ganglia, nigrostriatal pathway, hippocampus, raphe nuclei, cortex | Migraine | [15,35] |
| 5-HT1ER | Gi/o | Hippocampus, hypothalamus | Migraine | [15,16] |
| 5-HT1FR | Gi/o | Cortex, hippocampus | Migraine | [15,16] |
| 5-HT2AR | Gq/11 | Cortex, vascular smooth muscle | Regulation of vascular tone, mood, cognition, and hallucinogenic effects of various alkaloids | [27,35] |
| 5-HT2BR | Gq/11 | Smooth muscle, cardiac tissue, endothelial cells | Cardiovascular function | [28] |
| 5-HT2CR | Gq/11 | Gastrointestinal tract, choroid plexus, basal ganglia, substantia nigra, olfactory nucleus, cingulate cortex, lateral habenula, subthalamic nucleus | Gut motility, appetite, cognition, mood, addiction, anxiety, depression, epilepsy, schizophrenia, and obesity | [17,28,35] |
| 5-HT3R | Cation channel | Cortex, hippocampus, nucleus accumbens, substantia nigra, ventral tegmental area, area postrema, nucleus tractus solitarius, PNS | Emesis, cognition, anxiety, peristaltic activity, and urinary function | [30] |
| 5-HT4R | Gs or Gi | Limbic areas, nucleus accumbens, corpus striatum, globus pallidus, substantia nigra, hippocampus colliculus, cardiac tissue, gastrointestinal tract | Motility, secretion, and visceral sensitivity | [31,35] |
| 5-HT5R | Gi/o | Raphe nuclei, cerebral cortex, hippocampus, amygdala, hypothalamus | Memory, learning, neurotransmitter release, and synaptic plasticity | [33] |
| 5-HT6R | Gs | Cortex, hippocampus, striatum | Learning, memory, addiction, and seizure control | [18,36] |
| 5-HT7R | Gs | Spinal cord, hippocampus, thalamus | Cognitive functions, circadian rhythm, and mood | [24,32] |
2. 5-HTR Allosteric Modulators
2.1. Early Discoveries
2.2. Fatty Acid Amides and Cannabinoids
2.2.1. Endocannabinoid Activity at 5-HTRs
2.2.2. Phytocannabinoid Activity at 5-HTRs
2.3. Other Natural Substances
2.4. Synthetic Allosteric Modulators
2.4.1. Allosteric Modulators of 5-HT2CR
2.4.2. Allosteric Modulators of 5-HT3R
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Monod, J.; Wyman, J.; Changeux, J.-P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Christopoulos, A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. [Google Scholar] [CrossRef] [PubMed]
- ASD Home. Available online: https://mdl.shsmu.edu.cn/ASD/module/mainpage/mainpage.jsp (accessed on 19 March 2024).
- Jakubík, J.; Randáková, A.; Chetverikov, N.; El-Fakahany, E.E.; Doležal, V. The Operational Model of Allosteric Modulation of Pharmacological Agonism. Sci. Rep. 2020, 10, 14421. [Google Scholar] [CrossRef] [PubMed]
- Fasciani, I.; Petragnano, F.; Aloisi, G.; Marampon, F.; Carli, M.; Scarselli, M.; Maggio, R.; Rossi, M. Allosteric Modulators of G Protein-Coupled Dopamine and Serotonin Receptors: A New Class of Atypical Antipsychotics. Pharmaceuticals 2020, 13, 388. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.W.; Holst, B. Ago-Allosteric Modulation and Other Types of Allostery in Dimeric 7TM Receptors. J. Recept. Signal Transduct. Res. 2006, 26, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Cong, Z.; Chen, L.-N.; Ma, H.; Zhou, Q.; Zou, X.; Ye, C.; Dai, A.; Liu, Q.; Huang, W.; Sun, X.; et al. Molecular Insights into Ago-Allosteric Modulation of the Human Glucagon-like Peptide-1 Receptor. Nat. Commun. 2021, 12, 3763. [Google Scholar] [CrossRef]
- Evans, A.K.; Lowry, C.A. Pharmacology of the Beta-Carboline FG-7,142, a Partial Inverse Agonist at the Benzodiazepine Allosteric Site of the GABA A Receptor: Neurochemical, Neurophysiological, and Behavioral Effects. CNS Drug Rev. 2007, 13, 475–501. [Google Scholar] [CrossRef]
- Lees, J.A.; Dias, J.M.; Rajamohan, F.; Fortin, J.-P.; O’Connor, R.; Kong, J.X.; Hughes, E.A.G.; Fisher, E.L.; Tuttle, J.B.; Lovett, G.; et al. An Inverse Agonist of Orphan Receptor GPR61 Acts by a G Protein-Competitive Allosteric Mechanism. Nat. Commun. 2023, 14, 5938. [Google Scholar] [CrossRef]
- Kenakin, T.; Christopoulos, A. Signalling Bias in New Drug Discovery: Detection, Quantification and Therapeutic Impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef]
- Votey, S.R.; Bosse, G.M.; Bayer, M.J.; Hoffman, J.R. Flumazenil: A New Benzodiazepine Antagonist. Ann. Emerg. Med. 1991, 20, 181–188. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Dowden, H.; Munro, J. Trends in Clinical Success Rates and Therapeutic Focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef] [PubMed]
- López-Rodríguez, M.L.; Benhamú, B.; Vázquez-Villa, H. Chapter 11—Allosteric Modulators Targeting GPCRs. In GPCRs; Jastrzebska, B., Park, P.S.-H., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 195–241. ISBN 978-0-12-816228-6. [Google Scholar]
- McCorvy, J.D.; Roth, B.L. Structure and Function of Serotonin G Protein-Coupled Receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Parajulee, A.; Kim, K. Structural Studies of Serotonin Receptor Family. BMB Rep. 2023, 56, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.M.; Ahern, G.P.; Becamel, C.; Bockaert, J.; Camilleri, M.; Chaumont-Dubel, S.; Claeysen, S.; Cunningham, K.A.; Fone, K.C.; Gershon, M.; et al. International Union of Basic and Clinical Pharmacology. CX. Classification of Receptors for 5-Hydroxytryptamine; Pharmacology and Function. Pharmacol. Rev. 2021, 73, 310–520. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.; Barnes, N.M. Central 5-HT Receptors and Their Function; Present and Future. Neuropharmacology 2020, 177, 108155. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Bécamel, C.; Chaumont-Dubel, S.; Claeysen, S.; Vandermoere, F.; Marin, P. Novel and Atypical Pathways for Serotonin Signaling. Fac. Rev. 2021, 10, 52. [Google Scholar] [CrossRef]
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wüthrich, K. The GPCR Network: A Large-Scale Collaboration to Determine Human GPCR Structure and Function. Nat. Rev. Drug Discov. 2013, 12, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.J.; Lefkowitz, R.J. Desensitization of G Protein-Coupled Receptors. Recent Prog. Horm. Res. 1996, 51, 319–351; discussion 352–353. [Google Scholar]
- Noda, M.; Higashida, H.; Aoki, S.; Wada, K. Multiple Signal Transduction Pathways Mediated by 5-HT Receptors. Mol. Neurobiol. 2004, 29, 31–39. [Google Scholar] [CrossRef]
- Huang, S.; Xu, P.; Shen, D.-D.; Simon, I.A.; Mao, C.; Tan, Y.; Zhang, H.; Harpsøe, K.; Li, H.; Zhang, Y.; et al. GPCRs Steer Gi and Gs Selectivity via TM5-TM6 Switches as Revealed by Structures of Serotonin Receptors. Mol. Cell 2022, 82, 2681–2695.e6. [Google Scholar] [CrossRef]
- Guseva, D.; Wirth, A.; Ponimaskin, E. Cellular Mechanisms of the 5-HT7 Receptor-Mediated Signaling. Front. Behav. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef]
- Altieri, S.C.; Garcia-Garcia, A.L.; Leonardo, E.D.; Andrews, A.M. Rethinking 5-HT1A Receptors: Emerging Modes of Inhibitory Feedback of Relevance to Emotion-Related Behavior. ACS Chem. Neurosci. 2013, 4, 72–83. [Google Scholar] [CrossRef]
- Aznavour, N.; Zimmer, L. [18F]MPPF as a Tool for the in Vivo Imaging of 5-HT1A Receptors in Animal and Human Brain. Neuropharmacology 2007, 52, 695–707. [Google Scholar] [CrossRef]
- Aznar, S.; Hervig, M.E.-S. The 5-HT2A Serotonin Receptor in Executive Function: Implications for Neuropsychiatric and Neurodegenerative Diseases. Neurosci. Biobehav. Rev. 2016, 64, 63–82. [Google Scholar] [CrossRef]
- De Deurwaerdère, P.; Bharatiya, R.; Chagraoui, A.; Di Giovanni, G. Constitutive Activity of 5-HT Receptors: Factual Analysis. Neuropharmacology 2020, 168, 107967. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lummis, S.C.R. 5-HT3 Receptors. Curr. Pharm. Des. 2006, 12, 3615–3630. [Google Scholar] [CrossRef]
- Chang-Halabi, Y.; Cordero, J.; Sarabia, X.; Villalobos, D.; Barrera, N.P. Crosstalking Interactions between P2X4 and 5-HT3A Receptors. Neuropharmacology 2023, 236, 109574. [Google Scholar] [CrossRef]
- Eglen, R.M.; Wong, E.H.; Dumuis, A.; Bockaert, J. Central 5-HT4 Receptors. Trends Pharmacol. Sci. 1995, 16, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.S.; Neumaier, J.F. 5-HT6 Receptors: A Novel Target for Cognitive Enhancement. Pharmacol. Ther. 2005, 108, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Grailhe, R.; Grabtree, G.W.; Hen, R. Human 5-HT5 Receptors: The 5-HT5A Receptor Is Functional but the 5-HT5B Receptor Was Lost during Mammalian Evolution. Eur. J. Pharmacol. 2001, 418, 157–167. [Google Scholar] [CrossRef]
- Thomas, D.R. 5-ht5A Receptors as a Therapeutic Target. Pharmacol. Ther. 2006, 111, 707–714. [Google Scholar] [CrossRef]
- Hoyer, D.; Clarke, D.E.; Fozard, J.R.; Hartig, P.R.; Martin, G.R.; Mylecharane, E.J.; Saxena, P.R.; Humphrey, P.P. International Union of Pharmacology Classification of Receptors for 5-Hydroxytryptamine (Serotonin). Pharmacol. Rev. 1994, 46, 157–203. [Google Scholar]
- Lesiak, A.J.; Brodsky, M.; Cohenca, N.; Croicu, A.G.; Neumaier, J.F. Restoration of Physiological Expression of 5-HT 6 Receptor into the Primary Cilia of Null Mutant Neurons Lengthens Both Primary Cilia and Dendrites. Mol. Pharmacol. 2018, 94, 731–742. [Google Scholar] [CrossRef]
- Frenken, M.; Kaumann, A.J. Allosteric Properties of the 5-HT2 Receptor System of the Rat Tail Artery: Ritanserin and Methysergide Are Not Competitive 5-HT2 Receptor Antagonists but Allosteric Modulators. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1987, 335, 359–366. [Google Scholar] [CrossRef]
- Koehler, P.J.; Tfelt-Hansen, P.C. History of Methysergide in Migraine. Cephalalgia Int. J. Headache 2008, 28, 1126–1135. [Google Scholar] [CrossRef]
- Silberstein, S.D. Methysergide. Cephalalgia Int. J. Headache 1998, 18, 421–435. [Google Scholar] [CrossRef]
- Dahlöf, C.; Maassen Van Den Brink, A. Dihydroergotamine, Ergotamine, Methysergide and Sumatriptan—Basic Science in Relation to Migraine Treatment. Headache 2012, 52, 707–714. [Google Scholar] [CrossRef]
- Rizzoli, P. Preventive Pharmacotherapy in Migraine. Headache 2014, 54, 364–369. [Google Scholar] [CrossRef]
- Janssen, P.A.J. Pharmacology of Potent and Selective S2-Serotonergic Antagonists. J. Cardiovasc. Pharmacol. 1985, 7, S2–S11. [Google Scholar] [CrossRef]
- Leysen, J.E.; Gommeren, W.; Van Gompel, P.; Wynants, J.; Janssen, P.F.; Laduron, P.M. Receptor-Binding Properties in Vitro and in Vivo of Ritanserin: A Very Potent and Long Acting Serotonin-S2 Antagonist. Mol. Pharmacol. 1985, 27, 600–611. [Google Scholar]
- Vauquelin, G.; Van Liefde, I.; Vanderheyden, P. Models and Methods for Studying Insurmountable Antagonism. Trends Pharmacol. Sci. 2002, 23, 514–518. [Google Scholar] [CrossRef]
- Mendelson, W.B.; Basile, A.S. The Hypnotic Actions of the Fatty Acid Amide, Oleamide. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2001, 25, S36–S39. [Google Scholar] [CrossRef]
- Huidobro-Toro, J.P.; Harris, R.A. Brain Lipids That Induce Sleep Are Novel Modulators of 5-Hydroxytrypamine Receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8078–8082. [Google Scholar] [CrossRef]
- Thomas, E.A.; Carson, M.J.; Neal, M.J.; Sutcliffe, J.G. Unique Allosteric Regulation of 5-Hydroxytryptamine Receptor-Mediated Signal Transduction by Oleamide. Proc. Natl. Acad. Sci. USA 1997, 94, 14115–14119. [Google Scholar] [CrossRef]
- Hedlund, P.B.; Carson, M.J.; Sutcliffe, J.G.; Thomas, E.A. Allosteric Regulation by Oleamide of the Binding Properties of 5-Hydroxytryptamine7 Receptors. Biochem. Pharmacol. 1999, 58, 1807–1813. [Google Scholar] [CrossRef]
- Alberts, G.L.; Chio, C.L.; Im, W.B. Allosteric Modulation of the Human 5-HT 7A Receptor by Lipidic Amphipathic Compounds. Mol. Pharmacol. 2001, 60, 1349–1355. [Google Scholar] [CrossRef]
- Ibarra-Lecue, I.; Diez-Alarcia, R.; Urigüen, L. Serotonin 2A Receptors and Cannabinoids. Prog. Brain Res. 2021, 259, 135–175. [Google Scholar] [CrossRef]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An Overview of the Cannabinoid Type 2 (CB2) Receptor System and Its Therapeutic Potential. Curr. Opin. Anaesthesiol. 2018, 31, 407. [Google Scholar] [CrossRef] [PubMed]
- Best, A.R.; Regehr, W.G. Serotonin Evokes Endocannabinoid Release and Retrogradely Suppresses Excitatory Synapses. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 6508–6515. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Fan, M.; An, C.; Ni, F.; Huang, W.; Luo, J. A Narrative Review of Molecular Mechanism and Therapeutic Effect of Cannabidiol (CBD). Basic Clin. Pharmacol. Toxicol. 2022, 130, 439–456. [Google Scholar] [CrossRef]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic Properties of Cannabidiol at 5-HT1a Receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- Rock, E.M.; Bolognini, D.; Limebeer, C.L.; Cascio, M.G.; Anavi-Goffer, S.; Fletcher, P.J.; Mechoulam, R.; Pertwee, R.G.; Parker, L.A. Cannabidiol, a Non-Psychotropic Component of Cannabis, Attenuates Vomiting and Nausea-like Behaviour via Indirect Agonism of 5-HT(1A) Somatodendritic Autoreceptors in the Dorsal Raphe Nucleus. Br. J. Pharmacol. 2012, 165, 2620–2634. [Google Scholar] [CrossRef]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martínez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of Cannabidiol Neuroprotection in Hypoxic-Ischemic Newborn Pigs: Role of 5HT(1A) and CB2 Receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef]
- Barata, L.; de Hoz-Rivera, M.; Romero, A.; Martínez, M.; Silva, L.; Villa, M.; Campa, L.; Jiménez-Sánchez, L.; Martínez-Orgado, J. Role of 5HT1A Receptors in the Neuroprotective and Behavioral Effects of Cannabidiol in Hypoxic-Ischemic Newborn Piglets. Front. Pharmacol. 2022, 13, 925740. [Google Scholar] [CrossRef]
- Al Kury, L.T.; Mahgoub, M.; Howarth, F.C.; Oz, M. Natural Negative Allosteric Modulators of 5-HT3 Receptors. Molecules 2018, 23, 3186. [Google Scholar] [CrossRef]
- Yang, K.-H.; Galadari, S.; Isaev, D.; Petroianu, G.; Shippenberg, T.S.; Oz, M. The Nonpsychoactive Cannabinoid Cannabidiol Inhibits 5-Hydroxytryptamine3A Receptor-Mediated Currents in Xenopus Laevis Oocytes. J. Pharmacol. Exp. Ther. 2010, 333, 547–554. [Google Scholar] [CrossRef]
- Kossakowski, R.; Schlicker, E.; Toczek, M.; Weresa, J.; Malinowska, B. Cannabidiol Affects the Bezold-Jarisch Reflex via TRPV1 and 5-HT3 Receptors and Has Peripheral Sympathomimetic Effects in Spontaneously Hypertensive and Normotensive Rats. Front. Pharmacol. 2019, 10, 500. [Google Scholar] [CrossRef]
- Yano, H.; Adhikari, P.; Naing, S.; Hoffman, A.F.; Baumann, M.H.; Lupica, C.R.; Shi, L. Positive Allosteric Modulation of the 5-HT1A Receptor by Indole-Based Synthetic Cannabinoids Abused by Humans. ACS Chem. Neurosci. 2020, 11, 1400–1405. [Google Scholar] [CrossRef]
- Vilardaga, J.-P.; Agnati, L.F.; Fuxe, K.; Ciruela, F. G-Protein-Coupled Receptor Heteromer Dynamics. J. Cell Sci. 2010, 123, 4215–4220. [Google Scholar] [CrossRef]
- Lillo, J.; Raïch, I.; Silva, L.; Zafra, D.A.; Lillo, A.; Ferreiro-Vera, C.; Sánchez de Medina, V.; Martínez-Orgado, J.; Franco, R.; Navarro, G. Regulation of Expression of Cannabinoid CB2 and Serotonin 5HT1A Receptor Complexes by Cannabinoids in Animal Models of Hypoxia and in Oxygen/Glucose-Deprived Neurons. Int. J. Mol. Sci. 2022, 23, 9695. [Google Scholar] [CrossRef]
- Viñals, X.; Moreno, E.; Lanfumey, L.; Cordomí, A.; Pastor, A.; de La Torre, R.; Gasperini, P.; Navarro, G.; Howell, L.A.; Pardo, L.; et al. Cognitive Impairment Induced by Delta9-Tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLOS Biol. 2015, 13, e1002194. [Google Scholar] [CrossRef]
- Galindo, L.; Moreno, E.; López-Armenta, F.; Guinart, D.; Cuenca-Royo, A.; Izquierdo-Serra, M.; Xicota, L.; Fernandez, C.; Menoyo, E.; Fernández-Fernández, J.M.; et al. Cannabis Users Show Enhanced Expression of CB1-5HT2A Receptor Heteromers in Olfactory Neuroepithelium Cells. Mol. Neurobiol. 2018, 55, 6347–6361. [Google Scholar] [CrossRef]
- Gallo, M.; Moreno, E.; Defaus, S.; Ortega-Alvaro, A.; Gonzalez, A.; Robledo, P.; Cavaco, M.; Neves, V.; Castanho, M.A.R.B.; Casadó, V.; et al. Orally Active Peptide Vector Allows Using Cannabis to Fight Pain While Avoiding Side Effects. J. Med. Chem. 2021, 64, 6937–6948. [Google Scholar] [CrossRef]
- DelaCuesta-Barrutia, J.; Peñagarikano, O.; Erdozain, A.M. G Protein-Coupled Receptor Heteromers as Putative Pharmacotherapeutic Targets in Autism. Front. Cell. Neurosci. 2020, 14, 588662. [Google Scholar] [CrossRef]
- Heng, H.L.; Chee, C.F.; Thy, C.K.; Tee, J.T.; Chin, S.P.; Herr, D.R.; Buckle, M.J.; Paterson, I.C.; Doughty, S.W.; Abd Rahman, N.; et al. In Vitro Functional Evaluation of Isolaureline, Dicentrine and Glaucine Enantiomers at 5-HT2 and A1 Receptors. Chem. Biol. Drug Des. 2019, 93, 132–138. [Google Scholar] [CrossRef]
- Ziemba, P.M.; Schreiner, B.S.P.; Flegel, C.; Herbrechter, R.; Stark, T.D.; Hofmann, T.; Hatt, H.; Werner, M.; Gisselmann, G. Activation and Modulation of Recombinantly Expressed Serotonin Receptor Type 3A by Terpenes and Pungent Substances. Biochem. Biophys. Res. Commun. 2015, 467, 1090–1096. [Google Scholar] [CrossRef]
- Nebrisi, E.E.; Prytkova, T.; Lorke, D.E.; Howarth, L.; Alzaabi, A.H.; Yang, K.-H.S.; Howarth, F.C.; Oz, M. Capsaicin Is a Negative Allosteric Modulator of the 5-HT3 Receptor. Front. Pharmacol. 2020, 11, 1274. [Google Scholar] [CrossRef]
- Im, W.B.; Chio, C.L.; Alberts, G.L.; Dinh, D.M. Positive Allosteric Modulator of the Human 5-HT2C Receptor. Mol. Pharmacol. 2003, 64, 78–84. [Google Scholar] [CrossRef]
- Ding, C.; Bremer, N.M.; Smith, T.D.; Seitz, P.K.; Anastasio, N.C.; Cunningham, K.A.; Zhou, J. Exploration of Synthetic Approaches and Pharmacological Evaluation of PNU-69176E and Its Stereoisomer as 5-HT2C Receptor Allosteric Modulators. ACS Chem. Neurosci. 2012, 3, 538–545. [Google Scholar] [CrossRef]
- Wild, C.T.; Miszkiel, J.M.; Wold, E.A.; Soto, C.A.; Ding, C.; Hartley, R.M.; White, M.A.; Anastasio, N.C.; Cunningham, K.A.; Zhou, J. Design, Synthesis, and Characterization of 4-Undecylpiperidine-2-Carboxamides as Positive Allosteric Modulators of the Serotonin (5-HT) 5-HT 2C Receptor. J. Med. Chem. 2019, 62, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Wold, E.A.; Garcia, E.J.; Wild, C.T.; Miszkiel, J.M.; Soto, C.A.; Chen, J.; Pazdrak, K.; Fox, R.G.; Anastasio, N.C.; Cunningham, K.A.; et al. Discovery of 4-Phenylpiperidine-2-Carboxamide Analogues as Serotonin 5-HT2C Receptor-Positive Allosteric Modulators with Enhanced Drug-like Properties. J. Med. Chem. 2020, 63, 7529–7544. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Garcia, E.J.; Merritt, C.R.; Zamora, J.C.; Bolinger, A.A.; Pazdrak, K.; Stafford, S.J.; Mifflin, R.C.; Wold, E.A.; Wild, C.T.; et al. Discovery of Novel Oleamide Analogues as Brain-Penetrant Positive Allosteric Serotonin 5-HT 2C Receptor and Dual 5-HT 2C /5-HT 2A Receptor Modulators. J. Med. Chem. 2023, 66, 9992–10009. [Google Scholar] [CrossRef] [PubMed]
- García-Cárceles, J.; Decara, J.M.; Vázquez-Villa, H.; Rodríguez, R.; Codesido, E.; Cruces, J.; Brea, J.; Loza, M.I.; Alén, F.; Botta, J.; et al. A Positive Allosteric Modulator of the Serotonin 5-HT 2C Receptor for Obesity. J. Med. Chem. 2017, 60, 9575–9584. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Sona, C.; Ojha, V.; Singh, M.; Mishra, A.; Kumar, A.; Siddiqi, M.I.; Tripathi, R.P.; Yadav, P.N. Identification of Dual Role of Piperazine-Linked Phenyl Cyclopropyl Methanone as Positive Allosteric Modulator of 5-HT2C and Negative Allosteric Modulator of 5-HT2B Receptors. Eur. J. Med. Chem. 2019, 164, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Sandeep Kumar, J.; Naimisha, R.; Thirupataiah, B.; Sujeevan Reddy, G.; Bung, N.; Roy, A.; Bulusu, G.; Mishra, A.; Yadav, P.N.; Misra, P.; et al. Sonochemical Synthesis and Biological Evaluation of Isoquinolin-1(2H)-One/Isoindolin-1-One Derivatives: Discovery of a Positive Ago-Allosteric Modulator (PAAM) of 5HT2CR. Bioorg. Chem. 2022, 129, 106202. [Google Scholar] [CrossRef] [PubMed]
- Kooyman, A.R.; van Hooft, J.A.; Vanderheijden, P.M.; Vijverberg, H.P. Competitive and Non-Competitive Effects of 5-Hydroxyindole on 5-HT3 Receptors in N1E-115 Neuroblastoma Cells. Br. J. Pharmacol. 1994, 112, 541–546. [Google Scholar] [CrossRef] [PubMed]
- van Hooft, J.A.; van der Haar, E.; Vijverberg, H.P. Allosteric Potentiation of the 5-HT3 Receptor-Mediated Ion Current in N1E-115 Neuroblastoma Cells by 5-Hydroxyindole and Analogues. Neuropharmacology 1997, 36, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Grønlien, J.H.; Ween, H.; Thorin-Hagene, K.; Cassar, S.; Li, J.; Briggs, C.A.; Gopalakrishnan, M.; Malysz, J. Importance of M2-M3 Loop in Governing Properties of Genistein at the A7 Nicotinic Acetylcholine Receptor Inferred from A7/5-HT3A Chimera. Eur. J. Pharmacol. 2010, 647, 37–47. [Google Scholar] [CrossRef]
- Newman, A.S.; Batis, N.; Grafton, G.; Caputo, F.; Brady, C.A.; Lambert, J.J.; Peters, J.A.; Gordon, J.; Brain, K.L.; Powell, A.D.; et al. 5-Chloroindole: A Potent Allosteric Modulator of the 5-HT3 Receptor. Br. J. Pharmacol. 2013, 169, 1228–1238. [Google Scholar] [CrossRef]
- De Oliveira-Pierce, A.N.; Zhang, R.; Machu, T.K. Colchicine: A Novel Positive Allosteric Modulator of the Human 5-Hydroxytryptamine 3A Receptor. J. Pharmacol. Exp. Ther. 2009, 329, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.F.; Lester, H.A.; Dougherty, D.A. Allosteric Activation of the 5-HT3AB Receptor by mCPBG. Neuropharmacology 2015, 91, 103–108. [Google Scholar] [CrossRef]
- Gasiorek, A.; Trattnig, S.M.; Ahring, P.K.; Kristiansen, U.; Frølund, B.; Frederiksen, K.; Jensen, A.A. Delineation of the Functional Properties and the Mechanism of Action of TMPPAA, an Allosteric Agonist and Positive Allosteric Modulator of 5-HT3 Receptors. Biochem. Pharmacol. 2016, 110–111, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Pandhare, A.; Pappu, A.S.; Wilms, H.; Blanton, M.P.; Jansen, M. The Antidepressant Bupropion Is a Negative Allosteric Modulator of Serotonin Type 3A Receptors. Neuropharmacology 2017, 113, 89–99. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural Insights into G-Protein-Coupled Receptor Allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Isberg, V.; Vroling, B.; Van Der Kant, R.; Li, K.; Vriend, G.; Gloriam, D. GPCRDB: An Information System for G Protein-Coupled Receptors. Nucleic Acids Res. 2014, 42, D422–D425. [Google Scholar] [CrossRef]
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsøe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An Information System for G Protein-Coupled Receptors. Nucleic Acids Res. 2016, 44, D356–D364. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The Pocketome of G-Protein-Coupled Receptors Reveals Previously Untargeted Allosteric Sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef]
- Olson, K.M.; Traynor, J.R.; Alt, A. Allosteric Modulator Leads Hiding in Plain Site: Developing Peptide and Peptidomimetics as GPCR Allosteric Modulators. Front. Chem. 2021, 9, 671483. [Google Scholar] [CrossRef]
- Rousselle, J.-C.; Plantefol, M.; Fillion, M.-P.; Massot, O.; Pauwels, P.J.; Fillion, G. Specific Interaction of 5-HT-Moduline with Human 5-HT1b as Well as 5-HT1d Receptors Expressed in Transfected Cultured Cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 358, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Plantefol, M.; Rousselle, J.C.; Massot, O.; Bernardi, E.; Schoofs, A.R.; Pourrias, B.; Ollivier, R.; Fillion, G. Structural Requirements of 5-Hydroxytryptamine-Moduline Analogues to Interact with the 5-hydroxytryptamine1B Receptor. J. Neurochem. 1999, 73, 2617–2620. [Google Scholar] [CrossRef] [PubMed]
- Anastasio, N.C.; Gilbertson, S.R.; Bubar, M.J.; Agarkov, A.; Stutz, S.J.; Jeng, Y.; Bremer, N.M.; Smith, T.D.; Fox, R.G.; Swinford, S.E.; et al. Peptide Inhibitors Disrupt the Serotonin 5-HT2C Receptor Interaction with Phosphatase and Tensin Homolog to Allosterically Modulate Cellular Signaling and Behavior. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; He, J.; Wu, C.; Zhang, M.; Liu, X.; Zhang, J. Designing Drugs and Chemical Probes with the Dualsteric Approach. Chem. Soc. Rev. 2023, 52, 8651–8677. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhou, Q.; Yan, W.; Sun, J.; Kozikowski, A.P.; Zhao, S.; Huang, X.-P.; Cheng, J. Design and Synthesis of Bitopic 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as Selective Dopamine D3 Receptor Ligands. J. Med. Chem. 2020, 63, 4579–4602. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, B.I.; Danielsen, M.; Marcher-Rørsted, E.; Røpke Jørgensen, K.; Wróbel, T.M.; Frykman, M.; Johansson, H.; Bräuner-Osborne, H.; Gloriam, D.E.; Mathiesen, J.M.; et al. Probing the Existence of a Metastable Binding Site at the Β2-Adrenergic Receptor with Homobivalent Bitopic Ligands. J. Med. Chem. 2019, 62, 7806–7839. [Google Scholar] [CrossRef] [PubMed]
- Egyed, A.; Kelemen, Á.A.; Vass, M.; Visegrády, A.; Thee, S.A.; Wang, Z.; de Graaf, C.; Brea, J.; Loza, M.I.; Leurs, R.; et al. Controlling the Selectivity of Aminergic GPCR Ligands from the Extracellular Vestibule. Bioorg. Chem. 2021, 111, 104832. [Google Scholar] [CrossRef]
- Leysen, J.E.; Niemegeers, C.J.; Van Nueten, J.M.; Laduron, P.M. [3H]Ketanserin (R 41 468), a Selective 3H-Ligand for Serotonin2 Receptor Binding Sites. Binding Properties, Brain Distribution, and Functional Role. Mol. Pharmacol. 1982, 21, 301–314. [Google Scholar]
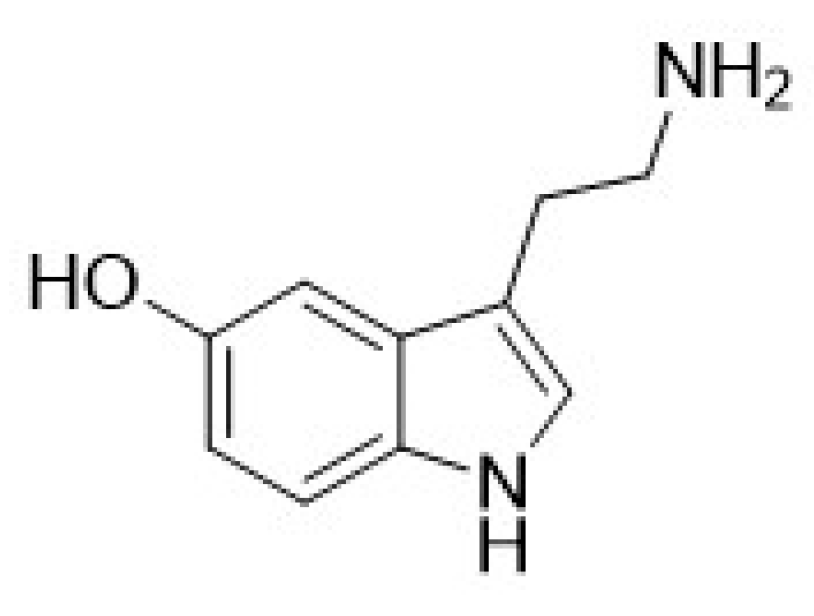
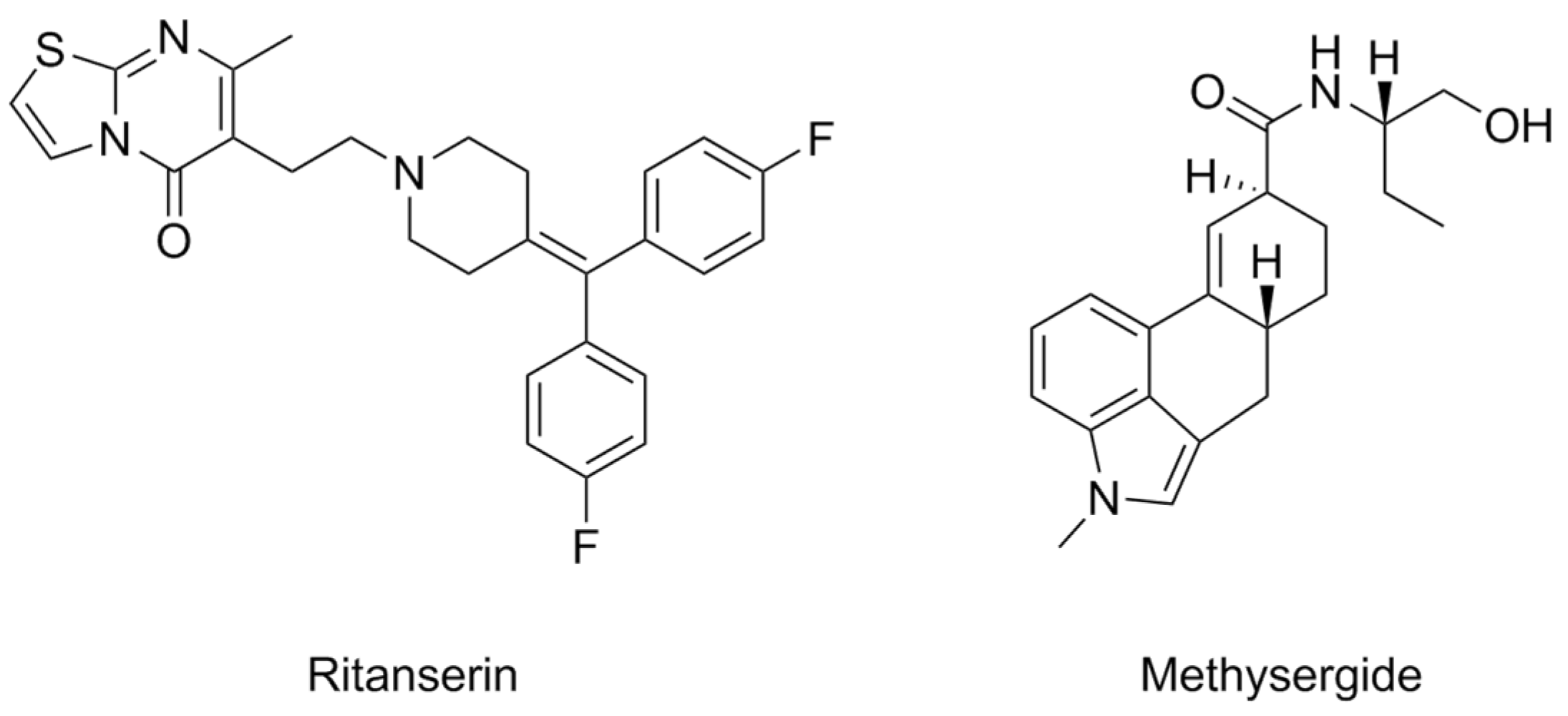
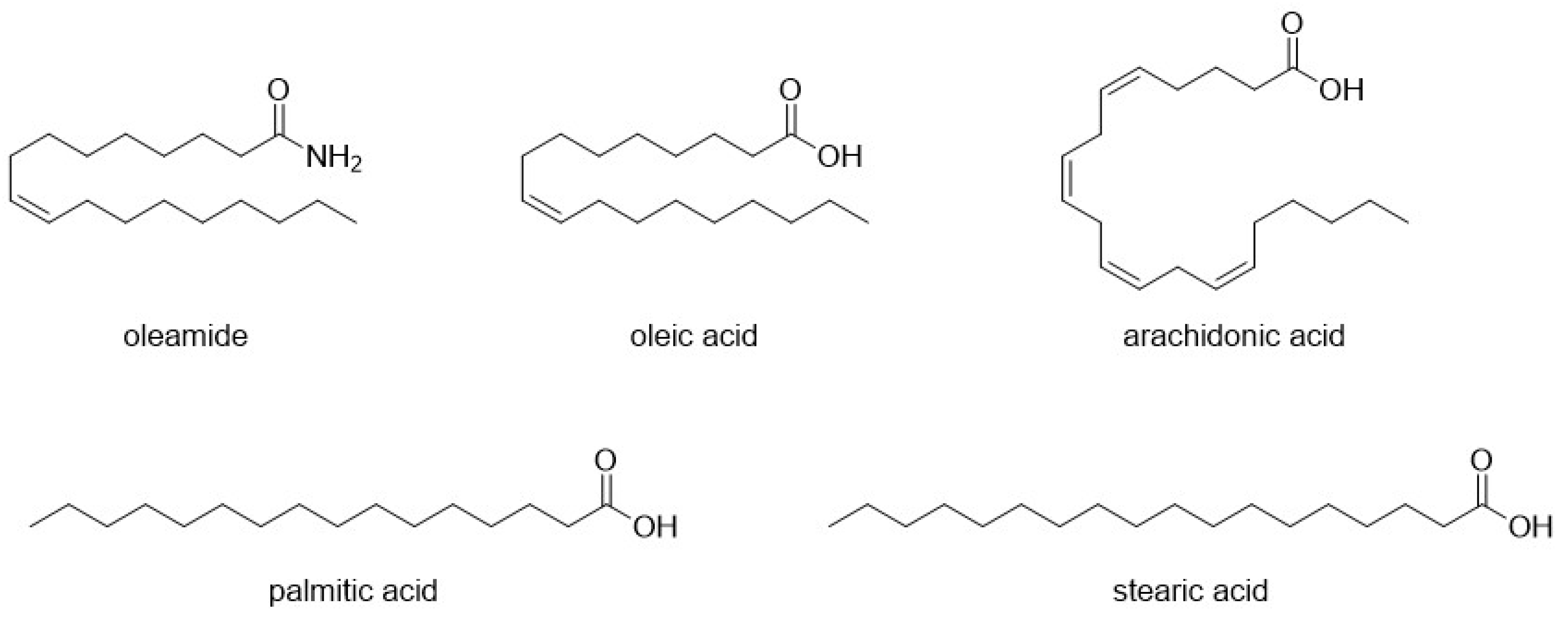


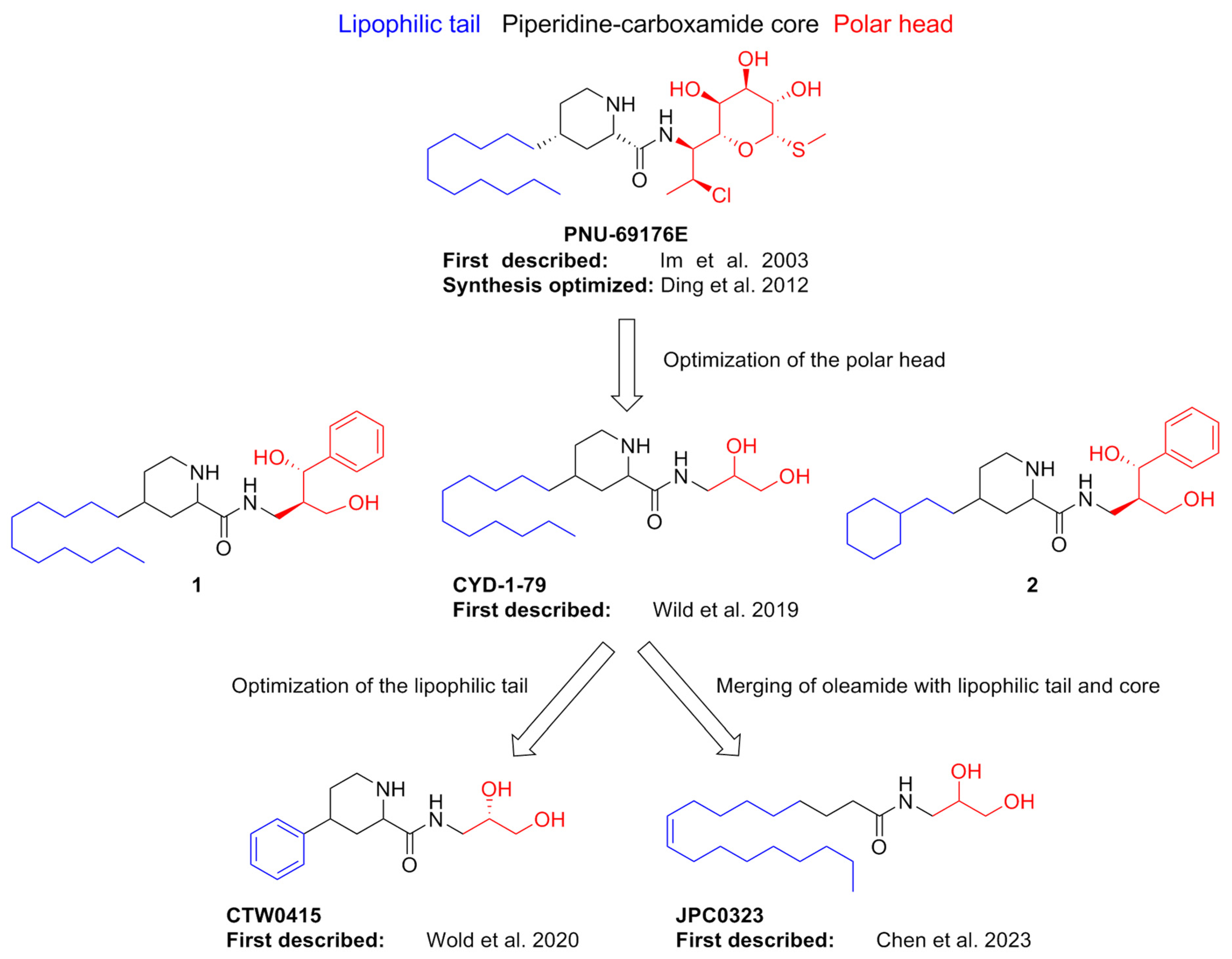

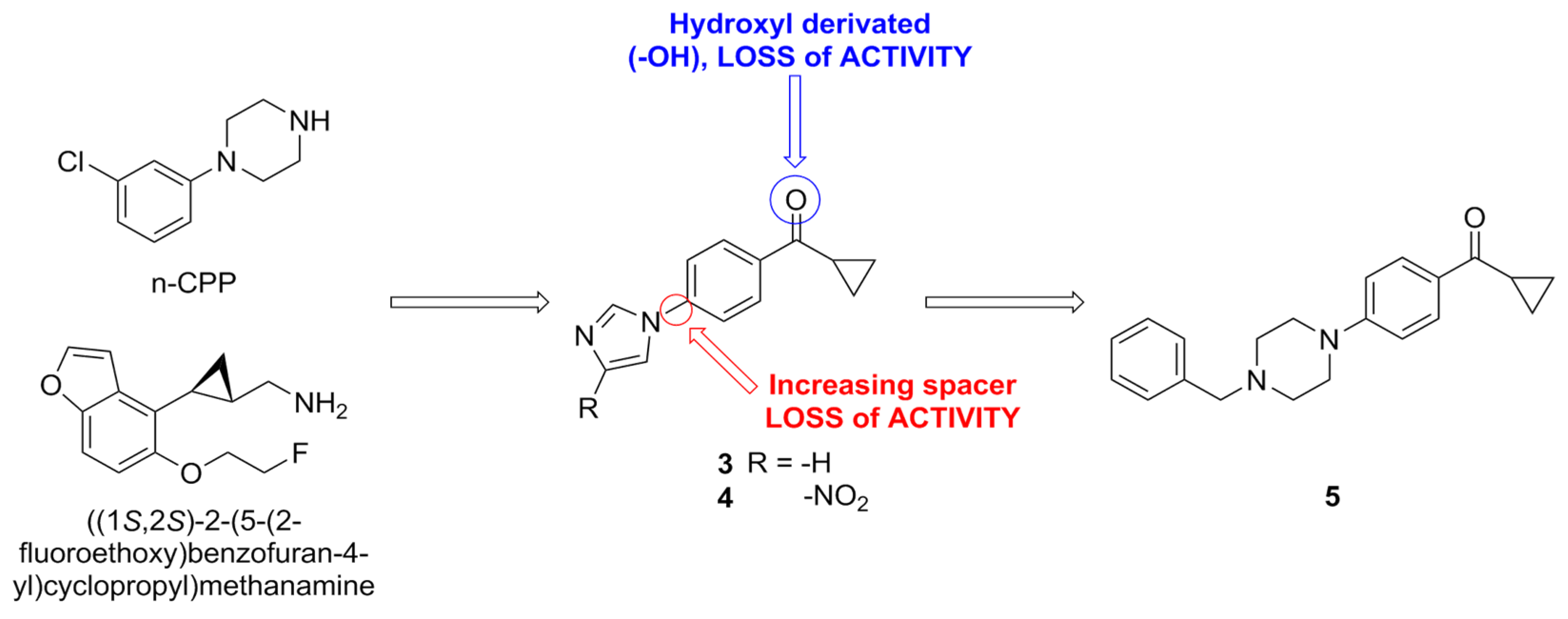


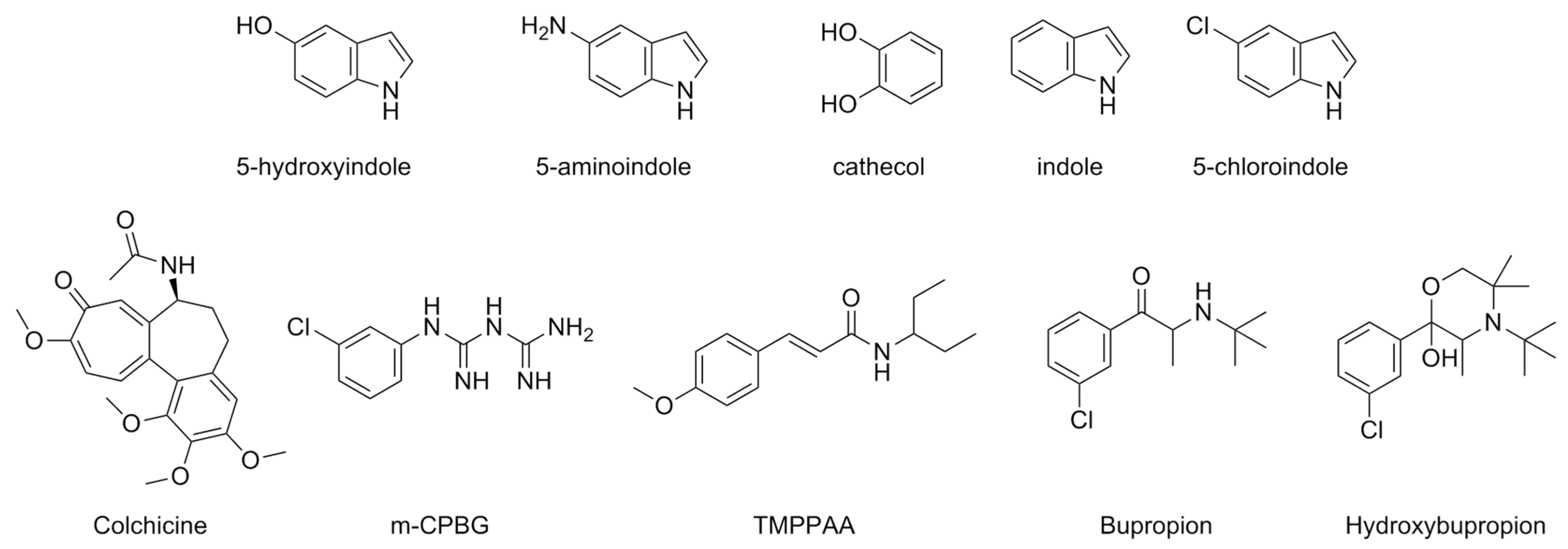
| Compound | Type of Modulator | Cell Model | Animal Model | Effects on Animal Model | Reference |
|---|---|---|---|---|---|
| PNU-69176E | PAM (PAM-agonist?). It induces slow conformational changes in the receptor population leading to constitutive activation. | CHO | N.T. a | N.T. a | [72] |
| 1 | NAM | CHO | N.T. a | N.T.a | [73] |
| CYD-1-79 | PAM | CHO | Sprague Dawley rats | Suppression of spontaneous ambulations; attenuation of cocaine cue reactivity | |
| 2 | Mild NAM | CHO | N.T. a | N.T. a | |
| CTW0415 | PAM | CHO | N.T. a | N.T. a | [74] |
| JPC0323 | Dual PAM (5-HT2CR/5-HT2AR) | CHO | Sprague Dawley rats | Suppression of spontaneous ambulations | [75] |
| VA240 | PAM | CHO | N.T. a | N.T. a | [76] |
| VA012 | PAM | CHO | Wistar rats | Reduction in food intake; reduction in weight gain; hyperlocomotion (side effect) | |
| 3 | Dual PAM (5-HT2CR/5-HT2AR) | HEK293T cells | N.T. a | N.T. a | [77] |
| 4 | Dual PAM (5-HT2CR/5-HT2AR) | HEK293T cells | N.T. a | N.T. a | |
| 5 | PAM (5-HT2CR) NAM (5-HT2BR) | HEK293T cells | Sprague Dawley rats | Reduction in food intake | |
| 6 | PAAM | HEK293T cells | Sprague Dawley rats | Reduction in food intake | [78] |
| Compound | Type of Modulator | Cell Model | Reference | |
|---|---|---|---|---|
| 5-Hydroxyindole (5-HI) | PAM | N1E-115 | [80] | |
| 5-Aminoindole | PAM | N1E-115 | ||
| Cathecol | PAM | N1E-115 | ||
| Indole | PAM | N1E-115 | ||
| 5-Chloroindole (Cl-indole) | PAM | HEK293 | [82] | |
| Colchicine | m5-HT3ARs: NAM | Xenopus laevis oocytes | [83] | |
| h5-HT3ARs: | -PAM low concentrations -NAM high concentrations | |||
| Meta-chlorophenylbiguanide (mCPBG) | PAM | Xenopus laevis oocytes | [84] | |
| Trans-3-(4-methoxyphenyl)-N-(pentan-3-yl)acrylamide (TMPPAA) | PAM | COS-7 Xenopus laevis oocytes HEK293 | [85] | |
| Bupropion | NAM | Xenopus laevis oocytes | [86] | |
| Hydroxybrupropion | NAM | Xenopus laevis oocytes | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, L.; Francavilla, F.; Leopoldo, M.; Lacivita, E. Allosteric Modulators of Serotonin Receptors: A Medicinal Chemistry Survey. Pharmaceuticals 2024, 17, 695. https://doi.org/10.3390/ph17060695
Brunetti L, Francavilla F, Leopoldo M, Lacivita E. Allosteric Modulators of Serotonin Receptors: A Medicinal Chemistry Survey. Pharmaceuticals. 2024; 17(6):695. https://doi.org/10.3390/ph17060695
Chicago/Turabian StyleBrunetti, Leonardo, Fabio Francavilla, Marcello Leopoldo, and Enza Lacivita. 2024. "Allosteric Modulators of Serotonin Receptors: A Medicinal Chemistry Survey" Pharmaceuticals 17, no. 6: 695. https://doi.org/10.3390/ph17060695
APA StyleBrunetti, L., Francavilla, F., Leopoldo, M., & Lacivita, E. (2024). Allosteric Modulators of Serotonin Receptors: A Medicinal Chemistry Survey. Pharmaceuticals, 17(6), 695. https://doi.org/10.3390/ph17060695