
Synthesis, Biological, and Computational Evaluations of Conformationally Restricted NAD-Mimics as Discriminant Inhibitors of Human NMN-Adenylyltransferase Isozymes
 , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
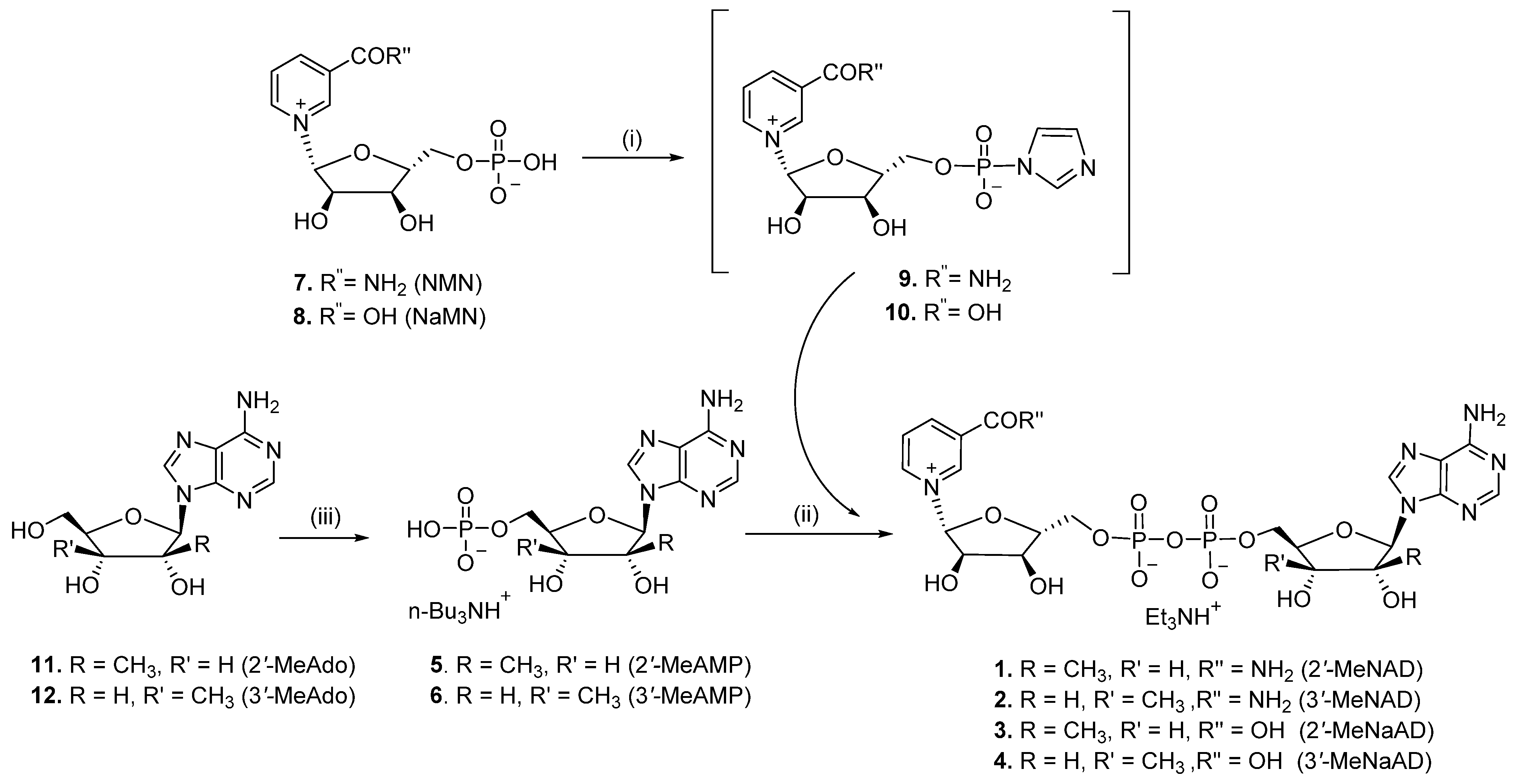
2.1. Chemistry
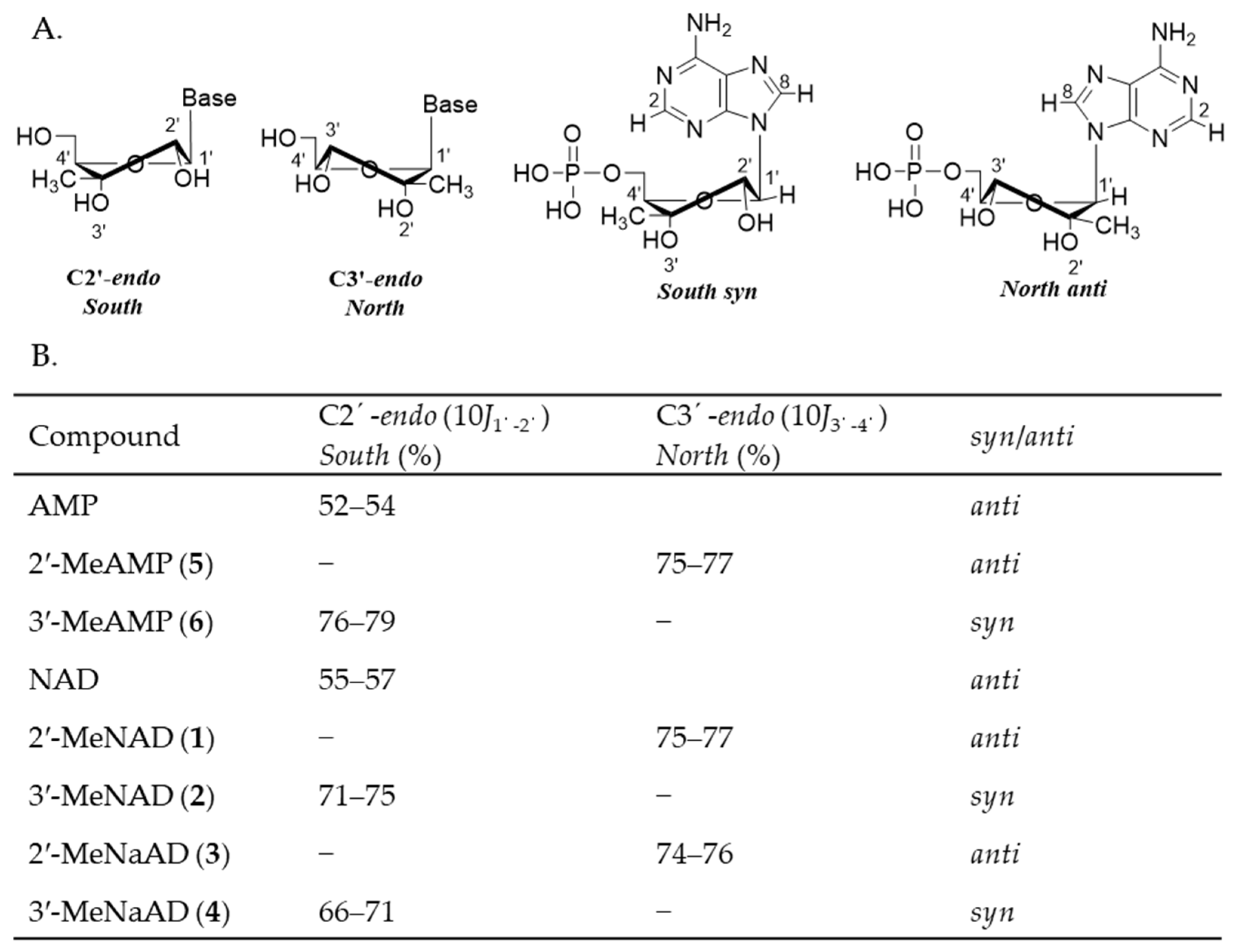
2.2. Conformational Analysis
2.3. Biological Evaluation
2.4. Mechanistic Analysis of hNMNAT Inhibition by 2′-MeNAD and 2′-MeNaAD
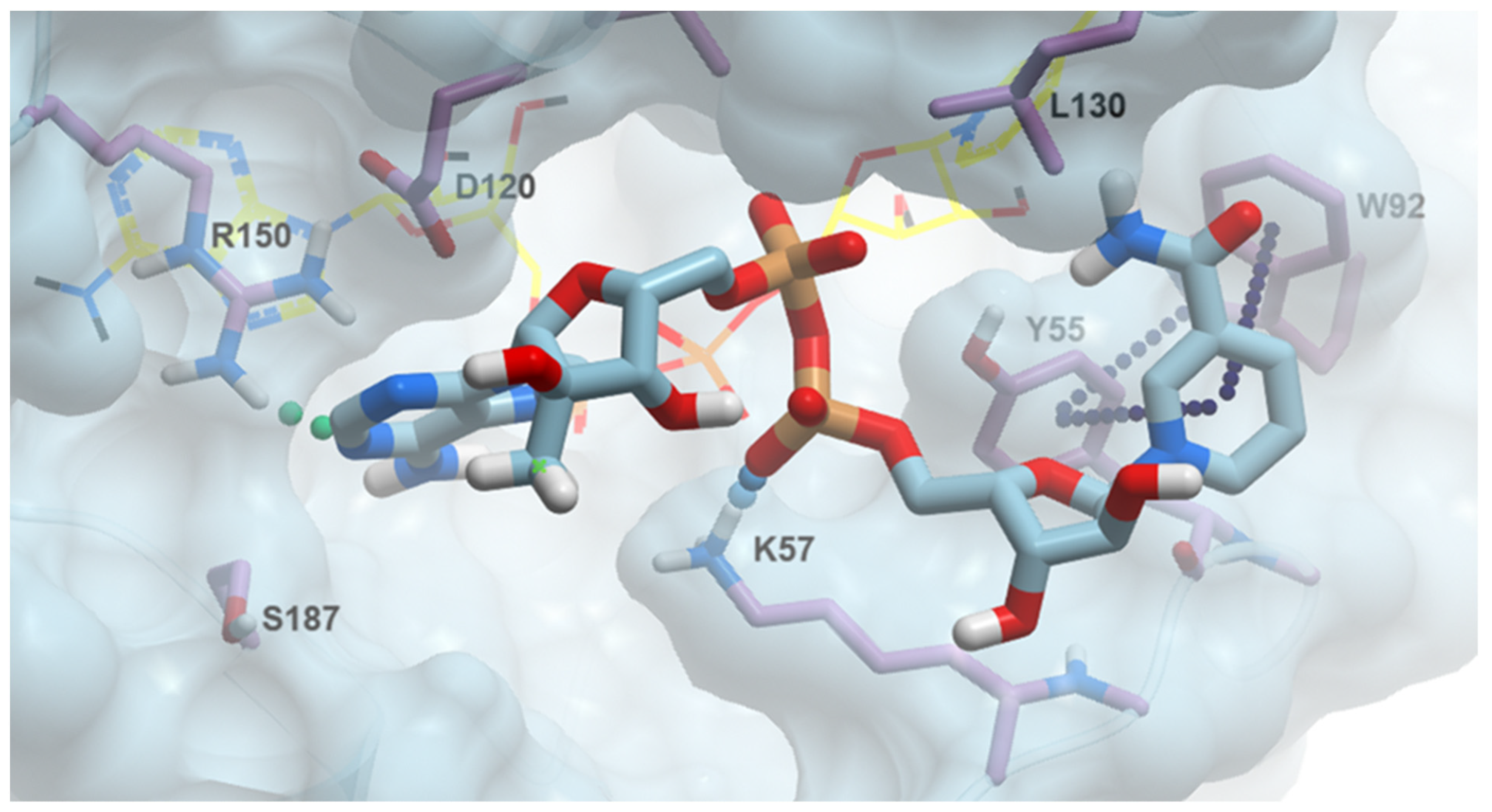
2.5. The Structural Rationale of 2′-Methylated NAD Analogues’ Bioactivity
2.6. 2′-MeNAD Binding Mode
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure
3.1.2. P1-[5′-(2′-C-Methyl-β-D-ribofuranosyl)adenine]-P2-[5′-(β-D-ribofuranosyl) nicotinamide] Pyrophosphate (Triethylammonium Salt) (2′-MeNAD, 1)
3.1.3. P1-[5′-(3′-C-Methyl-β-D-ribofuranosyl)adenine]-P2-[5′-(β-D-ribofuranosyl) nicotinamide] Pyrophospshate (Triethylammonium Salt) (3′-MeNAD, 2)
3.1.4. P1-[5′-(2′-C-Methyl-β-D-ribofuranosyl)adenine]-P2-[5′-(β-D-ribofuranosyl) nicotinic acid] Pyrophospshate (Triethylammonium Salt) (2′-MeNaAD, 3)
3.1.5. P1-[5′-(3′-C-methyl-β-D-ribofuranosyl)adenine]-P2-[5′-(β-D-ribofuranosyl) Nicotinic acid] Pyrophospshate (Triethylammonium Salt) (3′-MeNaAD, 4)
3.1.6. 9H-(2′-C-Methyl-β-D-ribofuranosyl)adenine-5′-monophosphate Tributyl Ammonium Salt (5)
3.1.7. 9H-(3′-C-Methyl-β-D-ribofuranosyl)adenine-5′-monophosphate Tributyl Ammonium Salt (6)
3.2. Biochemical and Computational Methods
3.2.1. NMN Adenylyltransferase Activity Assays
3.2.2. Protein Structural Modeling
3.2.3. Flexible Docking, Sequence, and Structural Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Wu, S. the Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolà, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Perez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD+ homeostasis in human health and disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef] [PubMed]
- Rodionova, I.A.; Schuster, B.M.; Guinn, K.M.; Sorci, L.; Scott, D.A.; Li, X.; Kheterpal, I.; Shoen, C.; Cynamon, M.; Locher, C.; et al. Metabolic and bactericidal effects of targeted suppression of NadD and NadE enzymes in mycobacteria. mBio 2014, 5, e00747-13. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, M.S.; Caffa, I.; Monacelli, F.; Nencioni, A. Inhibitors of NAD+ Production in Cancer Treatment: State of the Art and Perspectives. Int. J. Mol. Sci. 2024, 25, 2092. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.A.; Forouhar, F.; Tao, X.; Tong, L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert. Opin. Ther. Targets 2007, 11, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Fortunato, C.; Mazzola, F.; Raffaelli, N. The key role of the NAD biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase in regulating cell functions. IUBMB Life 2022, 74, 562–572. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef] [PubMed]
- Sorci, L.; Cimadamore, F.; Scotti, S.; Petrelli, R.; Cappellacci, L.; Franchetti, P.; Orsomando, G.; Magni, G. Initial-rate kinetics of human NMN-adenylyltransferases: Substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry 2007, 46, 4912–4922. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.N.; Zhen, W.; Gharehbaghi, K. Biochemical consequences of resistance to tiazofurin in human myelogenous leukemic K562 cells. Cancer Res. 1993, 53 (Suppl. 10), 2344–2348. [Google Scholar] [PubMed]
- Buonvicino, D.; Mazzola, F.; Zamporlini, F.; Resta, F.; Ranieri, G.; Camaioni, E.; Muzzi, M.; Zecchi, R.; Pieraccini, G.; Dolle, C.; et al. Identification of the Nicotinamide Salvage Pathway as a New Toxification Route for Antimetabolites. Cell Chem. Biol. 2018, 25, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Cui, C.; Deng, Q.; Wang, L.; Chen, R.; Zhai, D.; Xie, L.; Yu, J. Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence, depth and stage of colorectal cancer. Oncol. Lett. 2018, 16, 5829–5837. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Raduly, A.P.; Regdon, Z.; Polgar, Z.; Tarapcsak, S.; Sturniolo, I.; El-Hamoly, T.; Virag, L.; Hegedus, C. Targeting Nuclear NAD+ Synthesis Inhibits DNA Repair, Impairs Metabolic Adaptation and Increases Chemosensitivity of U-2OS Osteosarcoma Cells. Cancers 2020, 12, 1180. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jiang, Y.; Kitano, A.; Hu, T.; Murdaugh, R.L.; Li, Y.; Hoegenauer, K.A.; Chen, R.; Takahashi, K.; Nakada, D. Nuclear NAD+ homeostasis governed by NMNAT1 prevents apoptosis of acute myeloid leukemia stem cells. Sci. Adv. 2021, 7, eabf3895. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Yang, L.; Kabra, N.; Chen, L.; Koomen, J.; Haura, E.B.; Chen, J. The NAD+ synthesis enzyme nicotinamide mononucleotide adenylyltransferase (NMNAT1) regulates ribosomal RNA transcription. J. Biol. Chem. 2013, 288, 20908–20917. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.W.; Chen, L.; Petrelli, R.; Felczak, K.; Gao, G.; Bonnac, L.; Yu, J.S.; Bennett, E.M. Nicotinamide adenine dinucleotide based therapeutics. Curr. Med. Chem. 2008, 15, 650–670. [Google Scholar] [CrossRef]
- Petrelli, R.; Felczak, K.; Cappellacci, L. NMN/NaMN adenylyltransferase (NMNAT) and NAD kinase (NADK) inhibitors: Chemistry and potential therapeutic applications. Curr. Med. Chem. 2011, 18, 1973–1992. [Google Scholar] [CrossRef]
- Pankiewicz, K.W.; Petrelli, R.; Singh, R.; Felczak, K. Nicotinamide Adenine Dinucleotide Based Therapeutics, Update. Curr. Med. Chem. 2015, 22, 3991–4028. [Google Scholar] [CrossRef] [PubMed]
- Franchetti, P.; Cappellacci, L.; Marchetti, S.; Trincavelli, L.; Martini, C.; Mazzoni, M.R.; Lucacchini, A.; Grifantini, M. 2′-C-Methyl analogues of selective adenosine receptor agonists: Synthesis and binding studies. J. Med. Chem. 1998, 41, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Franchetti, P.; Cappellacci, L.; Pasqualini, M.; Petrelli, R.; Vita, P.; Jayaram, H.N.; Horvath, Z.; Szekeres, T.; Grifantini, M. Antitumor activity of C-methyl-beta-D-ribofuranosyladenine nucleoside ribonucleotide reductase inhibitors. J. Med. Chem. 2005, 48, 4983–4989. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Kato, T.; Takenishi, T. A novel method for phosphorylation of nucleosides to 5′-nucleotides. Tetrahedron Lett. 1967, 50, 5065–5068. [Google Scholar] [CrossRef] [PubMed]
- Altona, C.; Sundaralingam, M. Conformational analysis of the sugar ring in nucleosides and nucleotides. Improved method for the interpretation of proton magnetic resonance coupling constants. J. Am. Chem. Soc. 1973, 95, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, M.; Uesugi, S.; Yoshida, K. Studies on the conformation of purine nucleosides and their 5′-phosphates. Biochemistry 1972, 11, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, C.F.G.C.; Santos, H. Solvent effects on the conformation of nucleotides. Part 1. The conformation of 5′-adenosine monophosphate in water–dimethyl sulphoxide using nuclear Overhauser effects and lanthanide relaxation probes. J. Chem. Soc. Perkin Trans. 2 1983, 9, 1693–1697. [Google Scholar] [CrossRef]
- Rohde, K.H.; Sorci, L. The Prospective Synergy of Antitubercular Drugs with NAD Biosynthesis Inhibitors. Front. Microbiol. 2020, 11, 634640. [Google Scholar] [CrossRef] [PubMed]
- Osterman, A.L.; Rodionova, I.; Li, X.; Sergienko, E.; Ma, C.T.; Catanzaro, A.; Pettigrove, M.E.; Reed, R.W.; Gupta, R.; Rohde, K.H.; et al. Novel Antimycobacterial Compounds Suppress NAD Biogenesis by Targeting a Unique Pocket of NaMN Adenylyltransferase. ACS Chem. Biol. 2019, 14, 949–958. [Google Scholar] [CrossRef]
- Rodionova, I.A.; Zuccola, H.J.; Sorci, L.; Aleshin, A.E.; Kazanov, M.D.; Ma, C.T.; Sergienko, E.; Rubin, E.J.; Locher, C.P.; Osterman, A.L. Mycobacterial nicotinate mononucleotide adenylyltransferase: Structure, mechanism, and implications for drug discovery. J. Biol. Chem. 2015, 290, 7693–7706. [Google Scholar] [CrossRef]
- Orsomando, G.; Agostinelli, S.; Bramucci, M.; Cappellacci, L.; Damiano, S.; Lupidi, G.; Maggi, F.; Ngahang Kamte, S.L.; Biapa Nya, P.C.; Papa, F.; et al. Mexican sunflower (Tithonia diversifolia, Asteraceae) volatile oil as a selective inhibitor of Staphylococcus aureus nicotinate mononucleotide adenylyltransferase (NadD). Ind. Crops Prod. 2016, 85, 181–189. [Google Scholar] [CrossRef]
- Sorci, L.; Pan, Y.; Eyobo, Y.; Rodionova, I.; Huang, N.; Kurnasov, O.; Zhong, S.; MacKerell, A.D., Jr.; Zhang, H.; Osterman, A.L. Targeting NAD biosynthesis in bacterial pathogens: Structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem. Biol. 2009, 16, 849–861. [Google Scholar] [CrossRef]
- O’Hara, J.K.; Kerwin, L.J.; Cobbold, S.A.; Tai, J.; Bedell, T.A.; Reider, P.J.; Llinas, M. Targeting NAD+ metabolism in the human malaria parasite Plasmodium falciparum. PLoS ONE 2014, 9, e94061. [Google Scholar] [CrossRef]
- Schultz, M.D.; Dadali, T.; Jacques, S.A.; Muller-Steffner, H.; Foote, J.B.; Sorci, L.; Kellenberger, E.; Botta, D.; Lund, F.E. Inhibition of the NAD salvage pathway in schistosomes impairs metabolism, reproduction, and parasite survival. PLoS Pathog. 2020, 16, e1008539. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Kolhatkar, R.; Eyobo, Y.; Sorci, L.; Rodionova, I.; Osterman, A.L.; Mackerell, A.D.; Zhang, H. Complexes of bacterial nicotinate mononucleotide adenylyltransferase with inhibitors: Implication for structure-based drug design and improvement. J. Med. Chem. 2010, 53, 5229–5239. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 1–538. [Google Scholar]
- Brunetti, L.; Di Stefano, M.; Ruggieri, S.; Cimadamore, F.; Magni, G. Homology modeling and deletion mutants of human nicotinamide mononucleotide adenylyltransferase isozyme 2: New insights on structure and function relationship. Protein Sci. 2010, 19, 2440–2450. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Lau, C.; Dolle, C.; Gossmann, T.I.; Agledal, L.; Niere, M.; Ziegler, M. Isoform-specific targeting and interaction domains in human nicotinamide mononucleotide adenylyltransferases. J. Biol. Chem. 2010, 285, 18868–18876. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product a | hNMNAT-1 | hNMNAT-2 | hNMNAT-3 | |||
|---|---|---|---|---|---|---|
| NMN | ATP | NMN | ATP | NMN | ATP | |
| Ki (μM) a | Ki (μM) | Ki (μM) | Ki (μM) | Ki (μM) | Ki (μM) | |
| 2′-MeNAD (1) | N.D. b | N.D. | 15 ± 3 (M) | 21 ± 3 (M) | 312 ± 51 (C) | 182 ± 53 (M) |
| 2′-MeNaAD (3) | 168 ± 19 (C) | 82 ± 14 (U) | 60 ± 18 (M) | 41 ± 14 (U) | 224 ± 17 (C) | 223 ± 22 (C) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matteucci, F.; Ferrati, M.; Spinozzi, E.; Piergentili, A.; Del Bello, F.; Giorgioni, G.; Sorci, L.; Petrelli, R.; Cappellacci, L. Synthesis, Biological, and Computational Evaluations of Conformationally Restricted NAD-Mimics as Discriminant Inhibitors of Human NMN-Adenylyltransferase Isozymes. Pharmaceuticals 2024, 17, 739. https://doi.org/10.3390/ph17060739
Matteucci F, Ferrati M, Spinozzi E, Piergentili A, Del Bello F, Giorgioni G, Sorci L, Petrelli R, Cappellacci L. Synthesis, Biological, and Computational Evaluations of Conformationally Restricted NAD-Mimics as Discriminant Inhibitors of Human NMN-Adenylyltransferase Isozymes. Pharmaceuticals. 2024; 17(6):739. https://doi.org/10.3390/ph17060739
Chicago/Turabian StyleMatteucci, Federica, Marta Ferrati, Eleonora Spinozzi, Alessia Piergentili, Fabio Del Bello, Gianfabio Giorgioni, Leonardo Sorci, Riccardo Petrelli, and Loredana Cappellacci. 2024. "Synthesis, Biological, and Computational Evaluations of Conformationally Restricted NAD-Mimics as Discriminant Inhibitors of Human NMN-Adenylyltransferase Isozymes" Pharmaceuticals 17, no. 6: 739. https://doi.org/10.3390/ph17060739