
Remapping the Chemical Space and the Pharmacological Space of Drugs: What Can We Expect from the Road Ahead?

 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Data Curation
- (i)
- Approved drugs.
- (ii)
- Drugs approved after 2020.
- (iii)
- Drug candidates in clinical development.
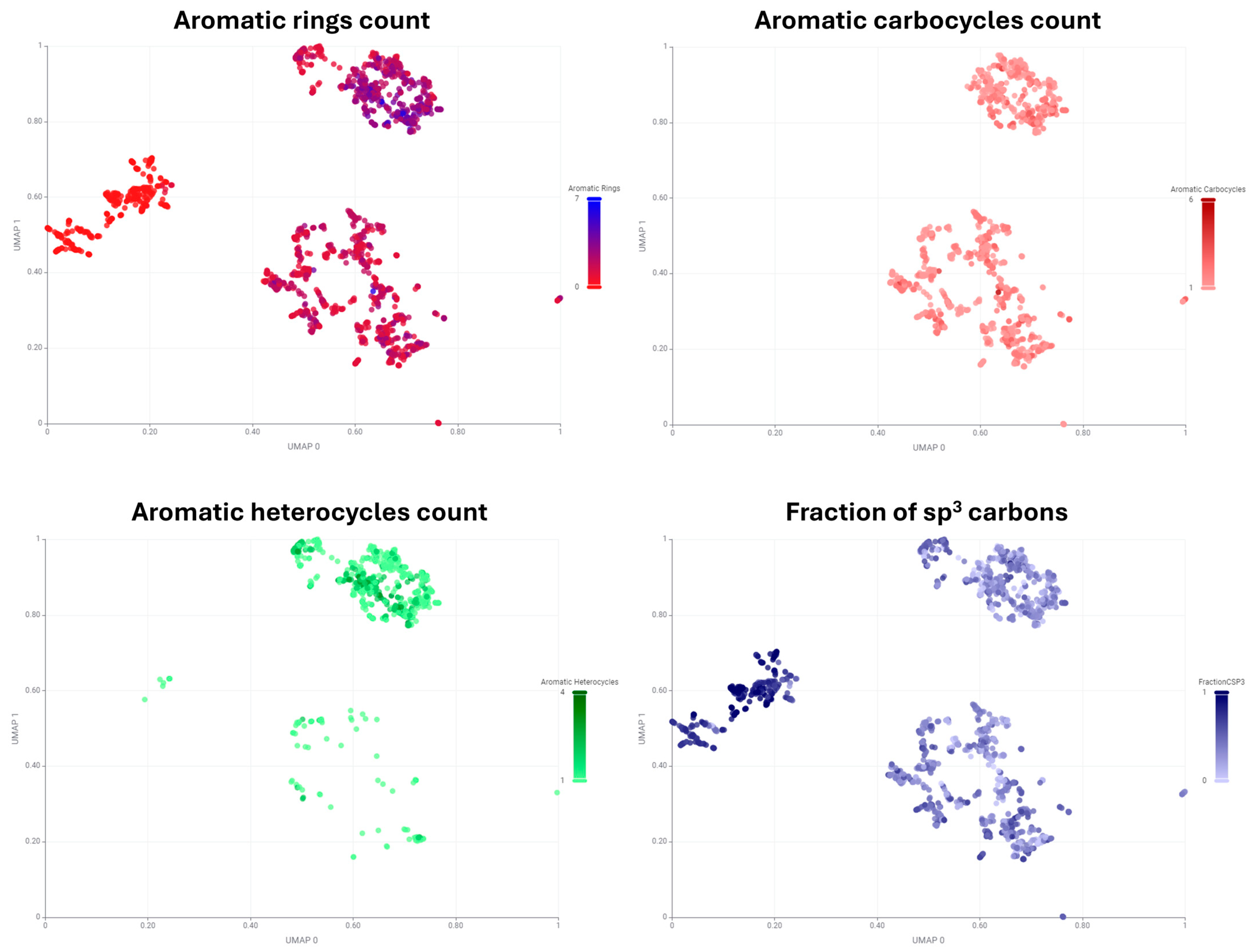
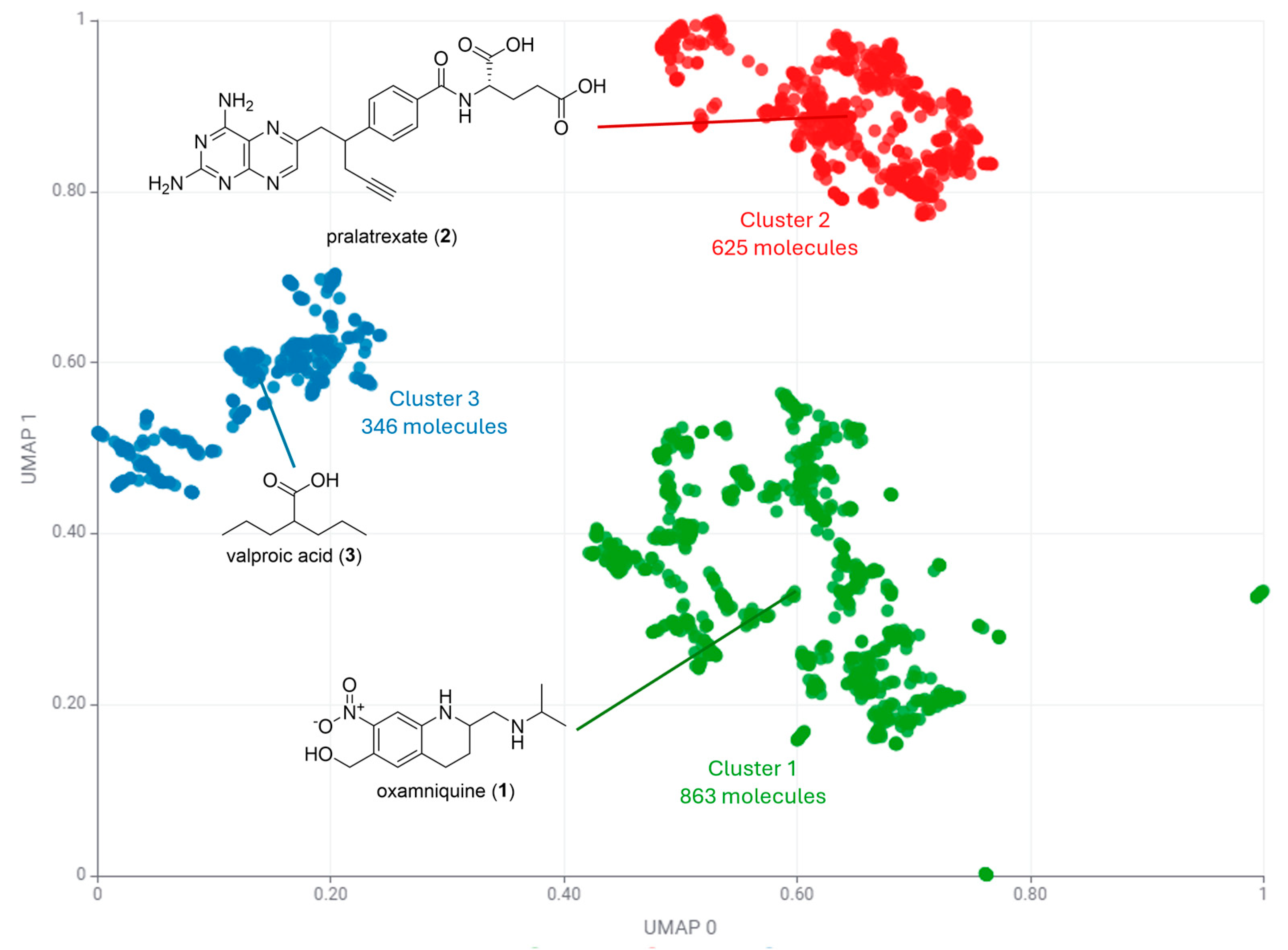
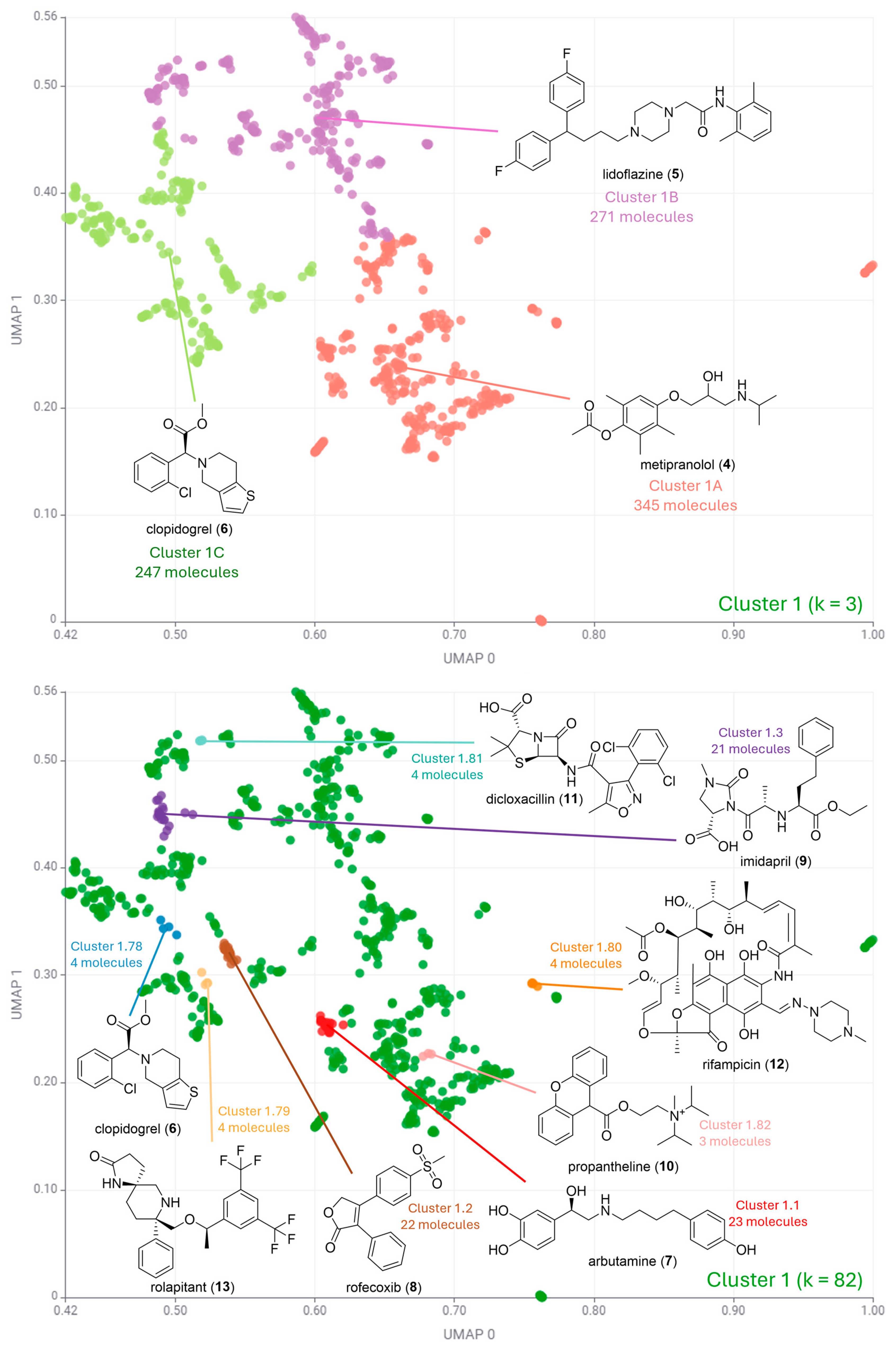
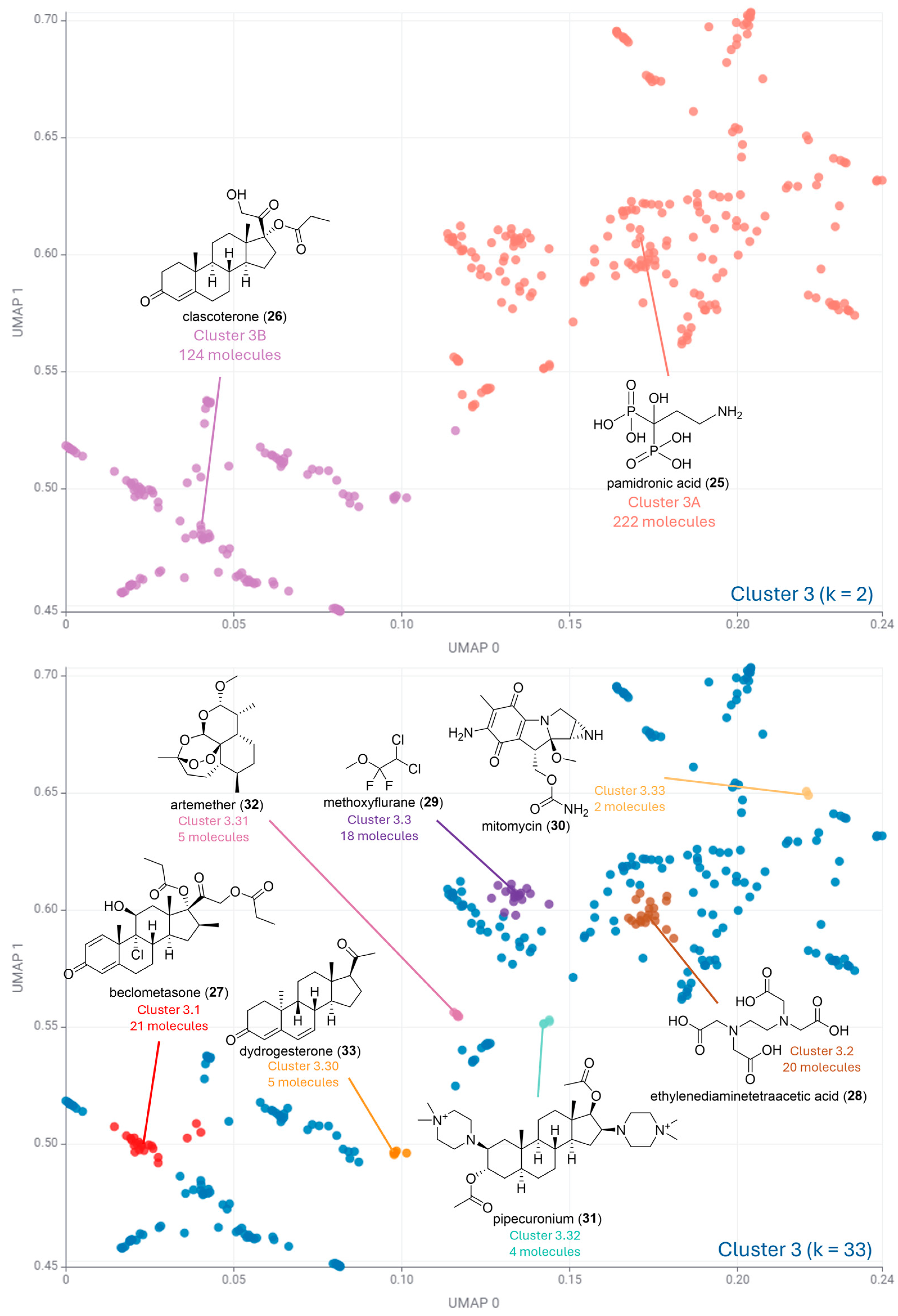
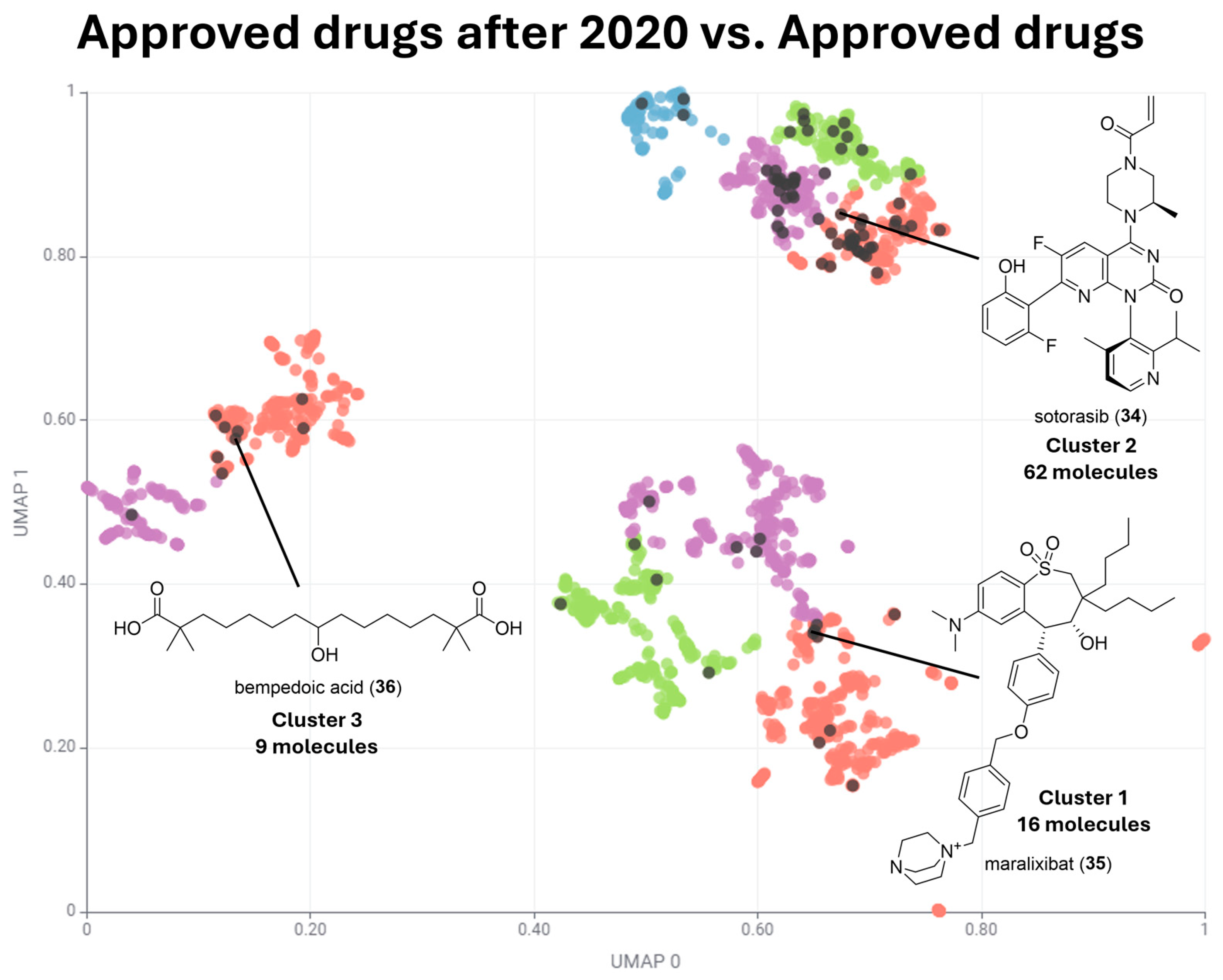
2.2. Chemical Space Analysis
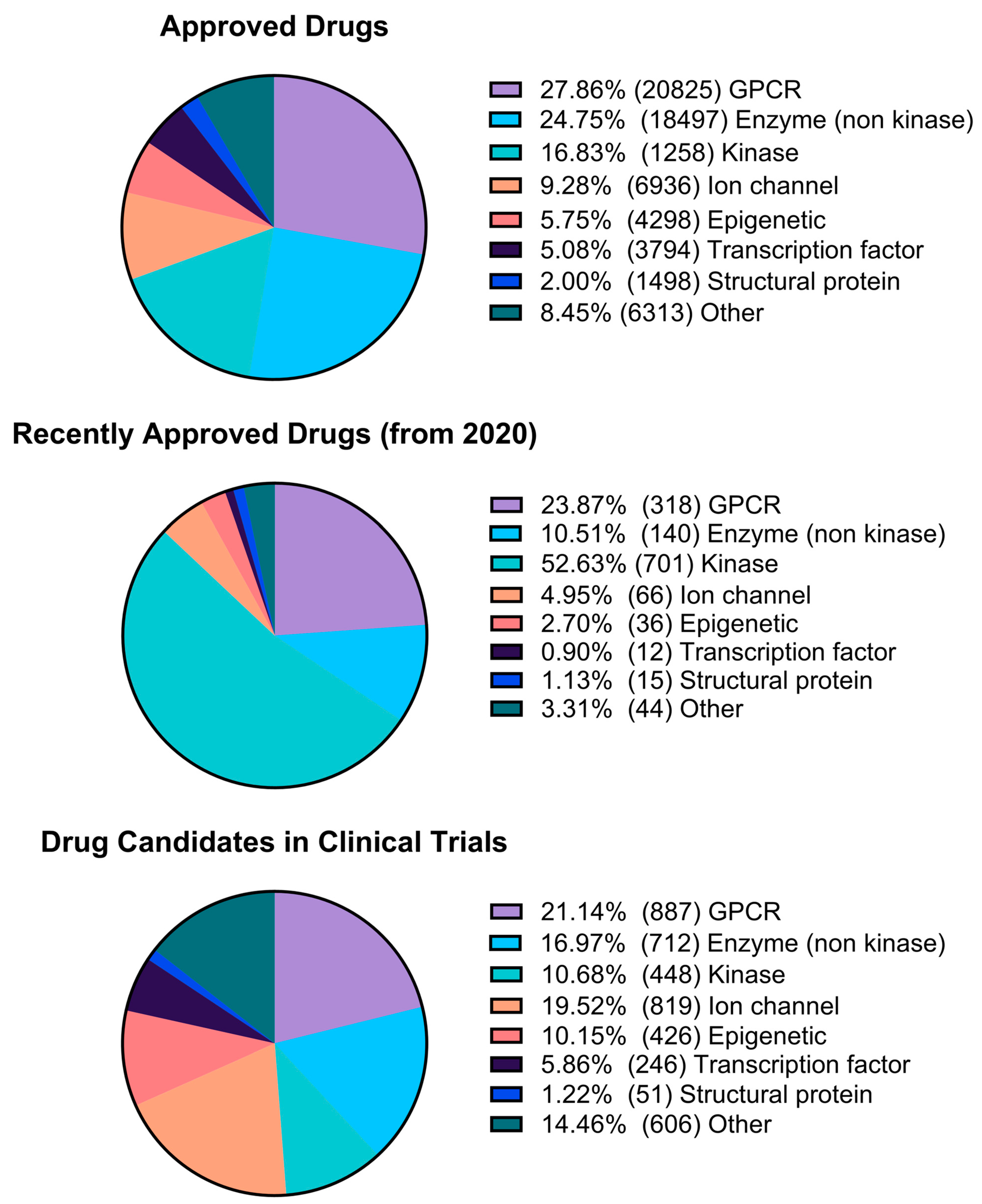
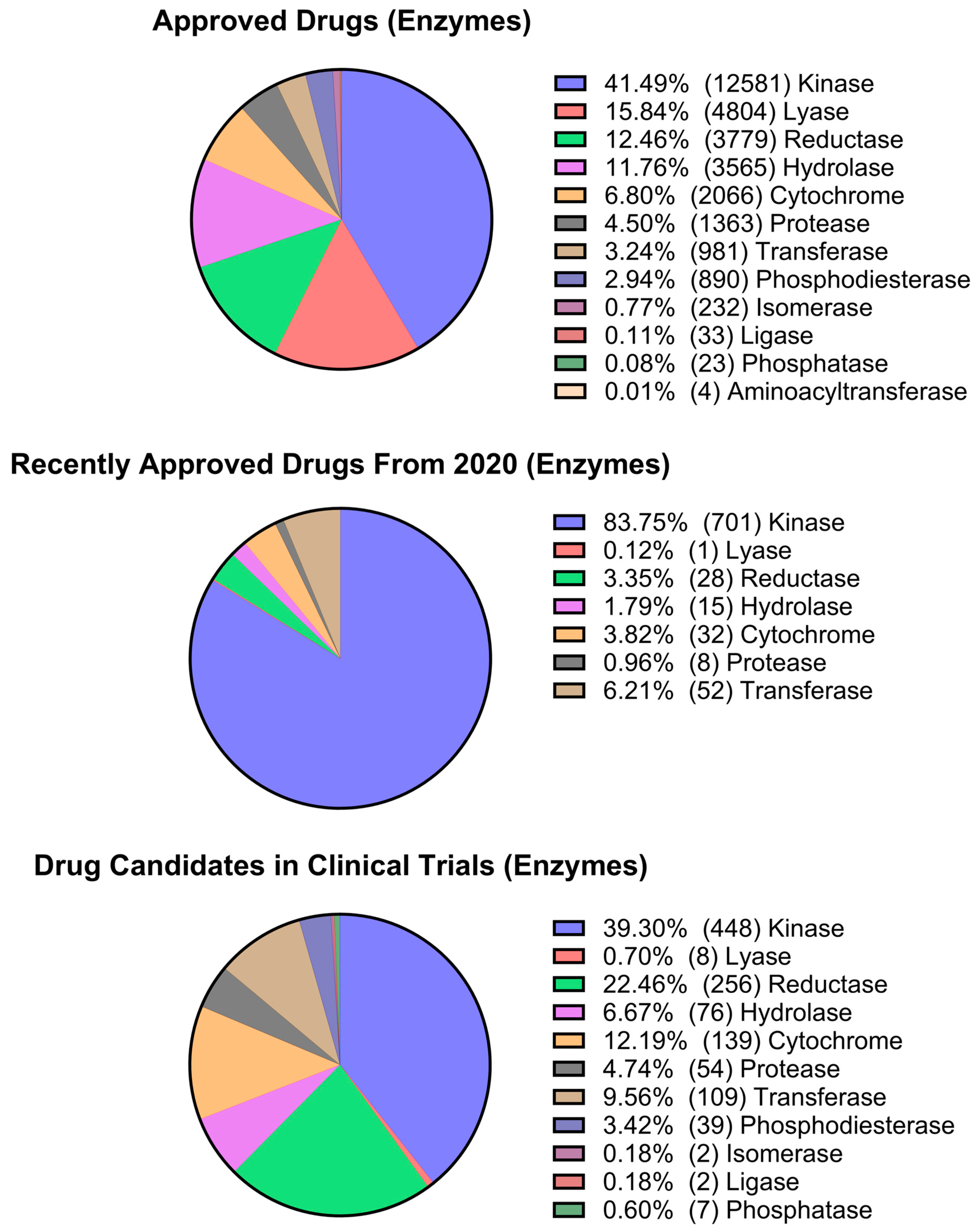
2.3. Pharmacological Space Analysis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lipinski, C.; Hopkins, A. Navigating Chemical Space for Biology and Medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P. Virtual Chemical Libraries. J. Med. Chem. 2019, 62, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M. Exploring Chemical Space—Generative Models and Their Evaluation. Artif. Intell. Life Sci. 2023, 3, 100064. [Google Scholar] [CrossRef]
- Reymond, J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; López-López, E. What Is the Plausibility That All Drugs Will Be Designed by Computers by the End of the Decade? Expert Opin. Drug Discov. 2024, 19, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Foerster, S.; Gustafsson, T.N.; Brochado, A.R.; Desilvestro, V.; Typas, A.; Unemo, M. The First Wide-Scale Drug Repurposing Screen Using the Prestwick Chemical Library (1200 Bioactive Molecules) against Neisseria gonorrhoeae Identifies High In Vitro Activity of Auranofin and Many Additional Drugs. APMIS 2020, 128, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; van Helden, J.; Decroly, E.; de Lamballerie, X.; Coutard, B. In Vitro Screening of a FDA Approved Chemical Library Reveals Potential Inhibitors of SARS-CoV-2 Replication. Sci. Rep. 2020, 10, 13093. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The Druggable Genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Radoux, C.J.; Vianello, F.; McGreig, J.; Desai, N.; Bradley, A.R. The Druggable Genome: Twenty Years Later. Front. Bioinform. 2022, 2, 958378. [Google Scholar] [CrossRef]
- Russ, A.P.; Lampel, S. The Druggable Genome: An Update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A Drug Discovery Platform Spanning Multiple Bioactivity Data Types and Time Periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Bellmann, L.; Penner, P.; Gastreich, M.; Rarey, M. Comparison of Combinatorial Fragment Spaces and Its Application to Ultralarge Make-on-Demand Compound Catalogs. J. Chem. Inf. Model. 2022, 62, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.V.; Katritch, V. Computational Approaches Streamlining Drug Discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Boldini, D.; Ballabio, D.; Consonni, V.; Todeschini, R.; Grisoni, F.; Sieber, S.A. Effectiveness of Molecular Fingerprints for Exploring the Chemical Space of Natural Products. J. Cheminform. 2024, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Macdonald, S.J.F. The Impact of Aromatic Ring Count on Compound Developability—Are Too Many Aromatic Rings a Liability in Drug Design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Flaxel, C.; John, R.S. Metipranolol. J. Toxicol. Cutaneous Ocul. Toxicol. 1991, 10, 171–174. [Google Scholar] [CrossRef]
- RIDLEY, J. Lidoflazine Is a High Affinity Blocker of the HERG K+channel. J. Mol. Cell Cardiol. 2004, 36, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Shahzad, D.; Faisal, M.; Larik, F.A.; El-Seedi, H.R.; Channar, P.A. Developments in the Synthesis of the Antiplatelet and Antithrombotic Drug (S)-clopidogrel. Chirality 2017, 29, 684–707. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, G.O.; Ogueta, S.B.; de Urtiaga, A.C.; Mollerach, M.E.; de Torres, R.A. Participation of PBP 3 in the Acquisition of Dicloxacillin Resistance in Listeria Monocytogenes. J. Antimicrob. Chemother. 1990, 25, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Floss, H.G.; Yu, T.-W. RifamycinMode of Action, Resistance, and Biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Rana, M.; Mishra, A.; Kaur, C.; Pan, C.-H.; Nepali, K. Recent Advances and Future Directions on Small Molecule VEGFR Inhibitors in Oncological Conditions. Eur. J. Med. Chem. 2024, 272, 116472. [Google Scholar] [CrossRef] [PubMed]
- Crestani, F.; Martin, J.R.; Möhler, H.; Rudolph, U. Mechanism of Action of the Hypnotic Zolpidem In Vivo. Br. J. Pharmacol. 2000, 131, 1251–1254. [Google Scholar] [CrossRef]
- Pauli, C. Bücher-Anzeigen. Arch. Psychiatr. Nervenkr. 1877, 7, 386–392. [Google Scholar] [CrossRef]
- Jallow, S.; Govender, N.P. Ibrexafungerp: A First-in-Class Oral Triterpenoid Glucan Synthase Inhibitor. J. Fungi 2021, 7, 163. [Google Scholar] [CrossRef]
- Coppée, R.; Bailly, J.; Sarrasin, V.; Vianou, B.; Zinsou, B.E.; Mazars, E.; Georges, H.; Hamane, S.; Lavergne, R.A.; Dannaoui, E.; et al. Circulation of an Artemisinin-Resistant Malaria Lineage in a Traveler Returning from East Africa to France. Clin. Infect. Dis. 2022, 75, 1242–1244. [Google Scholar] [CrossRef]
- Watts, N.B. Bisphosphonate Treatment for Osteoporosis. In The Osteoporotic Syndrome; Elsevier: Amsterdam, The Netherlands, 2000; pp. 121–132. [Google Scholar]
- Dhillon, S. Clascoterone: First Approval. Drugs 2020, 80, 1745–1750. [Google Scholar] [CrossRef]
- Barnes, P.J. Inhaled Corticosteroids. Pharmaceuticals 2010, 3, 514–540. [Google Scholar] [CrossRef]
- Huang, S.; Dai, Y.; Zhang, C.; Yang, C.; Huang, Q.; Hao, W.; Shen, H. Higher Impulsivity and Lower Grey Matter Volume in the Bilateral Prefrontal Cortex in Long-Term Abstinent Individuals with Severe Methamphetamine Use Disorder. Drug Alcohol. Depend. 2020, 212, 108040. [Google Scholar] [CrossRef]
- Tobias, J.D.; Sauder, R.A.; Hirshman, C.A. Pulmonary Reactivity to Methacholine during β-Adrenergic Blockade. Anesthesiology 1990, 73, 132–136. [Google Scholar] [CrossRef]
- Dows, D.A. On the Origin of Characteristic Group Frequencies in Infrared Spectra. J. Chem. Educ. 1958, 35, 629. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS G12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, D.-S.; Liu, H.; Zhang, S.-N.; Yang, W.-G.; Lv, M.; Zhou, Y.-X.; Zhang, S.-Y.; Song, J.; Liu, H.-M. New Drug Approvals for 2021: Synthesis and Clinical Applications. Eur. J. Med. Chem. 2023, 245, 114898. [Google Scholar] [CrossRef]
- Nguyen, H.; Akamnonu, I.; Yang, T. Bempedoic Acid: A Cholesterol Lowering Agent with a Novel Mechanism of Action. Expert Rev. Clin. Pharmacol. 2021, 14, 545–551. [Google Scholar] [CrossRef]
- Masoudi-Nejad, A.; Mousavian, Z.; Bozorgmehr, J.H. Drug-Target and Disease Networks: Polypharmacology in the Post-Genomic Era. In Silico Pharmacol. 2013, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network Pharmacology: The next Paradigm in Drug Discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- de Sena Murteira Pinheiro, P.; Franco, L.S.; Montagnoli, T.L.; Fraga, C.A.M. Molecular Hybridization: A Powerful Tool for Multitarget Drug Discovery. Expert Opin. Drug Discov. 2024, 19, 451–470. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The Human Disease Network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef]
- Paolini, G.V.; Shapland, R.H.B.; van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global Mapping of Pharmacological Space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Number | Best Silhouette Score for k ≤ 5 | Best Silhouette Score for k ≤ 100 |
|---|---|---|
| Cluster 1 | k = 3 0.47 | k = 82 0.60 |
| Cluster 2 | k = 4 0.51 | k = 63 0.52 |
| Cluster 3 | k = 2 0.65 | k = 33 0.63 |
| Cluster Number | Number of Approved Drugs | Number of Approved Drugs after 2020 |
|---|---|---|
| Cluster 1 | 863 | 16 |
| Cluster 2 | 625 | 62 |
| Cluster 3 | 346 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, L.S.; de Jesus, B.d.S.M.; Pinheiro, P.d.S.M.; Fraga, C.A.M. Remapping the Chemical Space and the Pharmacological Space of Drugs: What Can We Expect from the Road Ahead? Pharmaceuticals 2024, 17, 742. https://doi.org/10.3390/ph17060742
Franco LS, de Jesus BdSM, Pinheiro PdSM, Fraga CAM. Remapping the Chemical Space and the Pharmacological Space of Drugs: What Can We Expect from the Road Ahead? Pharmaceuticals. 2024; 17(6):742. https://doi.org/10.3390/ph17060742
Chicago/Turabian StyleFranco, Lucas Silva, Bárbara da Silva Mascarenhas de Jesus, Pedro de Sena Murteira Pinheiro, and Carlos Alberto Manssour Fraga. 2024. "Remapping the Chemical Space and the Pharmacological Space of Drugs: What Can We Expect from the Road Ahead?" Pharmaceuticals 17, no. 6: 742. https://doi.org/10.3390/ph17060742
APA StyleFranco, L. S., de Jesus, B. d. S. M., Pinheiro, P. d. S. M., & Fraga, C. A. M. (2024). Remapping the Chemical Space and the Pharmacological Space of Drugs: What Can We Expect from the Road Ahead? Pharmaceuticals, 17(6), 742. https://doi.org/10.3390/ph17060742