
Dichloroacetate for Cancer Treatment: Some Facts and Many Doubts


Abstract
:1. Introduction
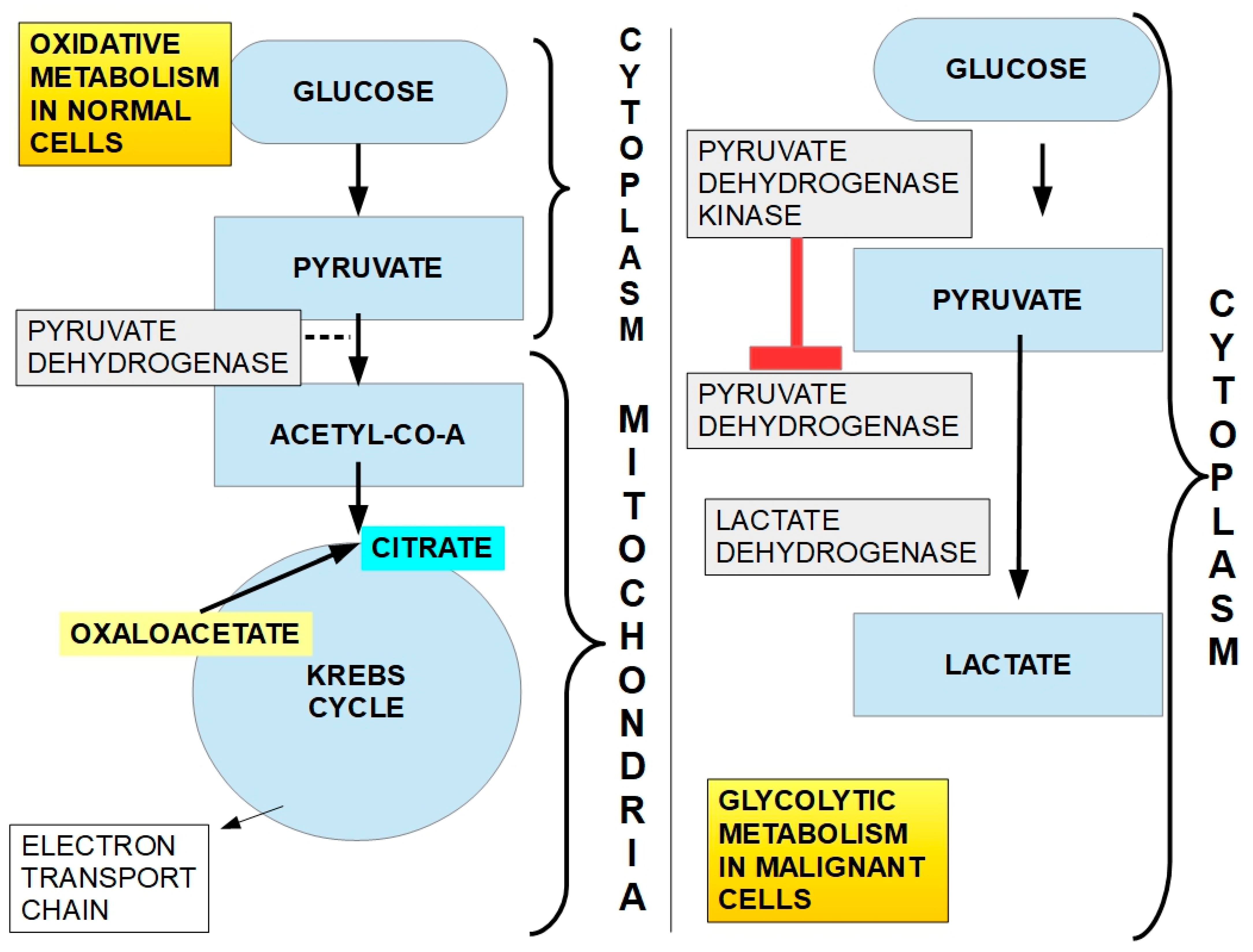
1.1. Lactate and Cancer
- (1)
- Malignant cells’ increased glucose uptake and metabolism;
- (2)
- Glycolytic metabolism, which occurs even in the presence of oxygen (Warburg effect);
- (3)
- Decreased activity of the pyruvate dehydrogenase (PDH) complex;
- (4)
- Increased activity of pyruvate dehydrogenase kinases (PDKs);
- (5)
- Increased expression and activity of the lactate extrusion proteins MCT1 and MCT4;
- (6)
- Increased expression and activity of the glycolytic enzymes.
- (1)
- (2)
- (3)
- (4)
- (5)
- (6)
- (7)
- Lactate activates HIF-1α in non-glycolytic cells (but not in glycolytic cells), which in turn further stimulates glycolysis and angiogenesis [50].
- (8)
- (9)
- Lactate has a positive correlation with radioresistance [53].
- (10)
- It increases hyaluronan production involved in migration and growth [54].
- (11)
- Lactate modulates the tumor microenvironment [55].
- (12)
- (13)
- Lactylation has also been found to be an important mechanism of post-translational modification of proteins associated with a poor prognosis of cancer progression [59].
- (14)
- Lactate is an important source of energy for oxidative lactophagic cells [60].
- (15)
- Pyruvate-to-lactate conversion by lactate dehydrogenase regenerates nicotinamide adenine dinucleotide (NAD+) in the cytoplasm [61], and NAD+ is an important metabolite for redox processes.
1.2. Dichloroacetate (DCA)
1.2.1. Therapeutic History of Dichloroacetate
1.2.2. DCA Enters the Oncology Terrain
1.2.3. Chemistry and Pharmacology of DCA
- (1)
- Doubts about the possibility of translating findings in other animals to humans;
- (2)
- The importance of the inter-species differences in the clearance mechanism.
1.2.4. Mechanism of Action
- (a)
- An inhibitory system represented by the four isoforms of PDK;
- (b)
- A “disinhibitory” system represented by the two PDH phosphatases (see below).
2. The PDH Complex
2.1. PDH Complex
2.2. PDK Family of Enzymes
2.3. Inhibition of PDKs

2.4. Pyruvate Dehydrogenase Phosphatases (PDPs)
2.5. Mechanism of Action of DCA
Lactylation and DCA
- (1)
- (2)
- Can external lactate that enters the cell through MCT1 activity be integrated into lactylation, or must it be converted into pyruvate? If external lactate (from the lactate shuttle) participates significantly in lactylation, DCA would be ineffective in preventing it. Thus far, there is only evidence of lactylation that originates from lactate metabolically produced by the cell [149]. However, for now, there is no evidence that excludes lactate imported into cells from participating in lactylation.
3. Experimental Evidence for DCA Activity in Cancer
3.1. Breast Cancer
3.2. Prostate Cancer
- (1)
- In prostate cancer cells, oxaloacetic acid is not regenerated during the Krebs cycle but is produced from imported aspartate [175];
- (2)
- Prostate cancer is initially not a glycolytic type of tumor, rather it adopts a lipogenic phenotype until some later critical point during its progression when it becomes glycolytic. This last peculiarity in metabolism contrasts with breast cancer, where glycolysis is the predominant metabolic feature from the very early stages. This also explains the reasons why fluorodeoxyglucose positron emission scans are of little help in the initial stages of prostate cancer [176] and become useful in the advanced stages [177].
3.3. Colon Cancer
3.4. Melanoma
3.5. Glioblastoma
3.6. Hematopoietic Tumors
3.6.1. Myeloma
3.6.2. Lymphoma
3.6.3. Leukemia
3.7. Ovarian, Cervical and Uterine Cancer
3.8. Lung Cancer
3.8.1. Non-Small Cell Lung Cancer (NSCLC)
3.8.2. Small Cell Lung Cancer (SCLC)
3.9. Head and Neck Squamous Cell Carcinoma (HNSCC)
3.10. Renal Tumors
3.11. Pancreatic Cancer
3.12. Hepatocarcinoma
3.13. Other Tumors
4. Resistance to DCA
5. DCA and Some Interesting Associations
5.1. DCA and Metformin
5.2. DCA and COX2 Inhibitors
5.3. DCA and Lipoic Acid
5.4. DCA and 2D-Deoxy Glucose (2DG)
5.5. DCA and Bicarbonate
5.6. DCA and Sulindac
5.7. Mitaplatin
5.8. Thiamin
5.9. DCA and Betulinic Acid
5.10. DCA and Rapamycin
5.11. DCA and Vemurafenib
5.12. DCA and Ivermectin
5.13. DCA and TRAIL Liposomes
5.14. DCA and 5-Fluorouracil (5-FU)
5.15. DCA and Chemotherapeutic Drugs in General
5.16. DCA and Salinomycin
5.17. DCA and Propranolol
5.18. DCA and All-Transretinoic Acid (ATRA)
5.19. DCA and Radiotherapy
5.20. DCA and Omeprazol
5.21. DCA and 2-Methoxiestradiol
5.22. DCA and Sirtinol
5.23. DCA and EGFR Tyrosine Kinase Inhibitors
6. DCA and T-Cells
7. Side Effects, Toxicity, and Doses
8. DCA Dosage
9. DCA Concentrations in Humans and Animals: A Key Issue
10. DCA Derivatives
11. Clinical Cases
11.1. Clinical Trials
Clinical Trials with Poor Results
12. Negative Results with DCA
13. Discussion
13.1. DCA as a Metabolic Modifier
13.2. DCA Concentrations
13.3. Effects of Glycolytic Inhibition on Tumor Growth
- (a)
- Cells that survive can switch to another type of metabolism, such as mitochondrial oxidative metabolism;
- (b)
- There may be a persistence of oxidative cells that are not affected by DCA;
- (c)
- DCA is cytostatic rather than cytotoxic, and both require very high concentrations;
- (d)
- The tumor is predominantly oxidative.
13.4. DCA Is not a Stand-Alone Drug
13.5. Other Antitumor Effects of DCA
13.6. DCA Associated with Other Pharmaceuticals
- (1)
- DCA responders—the glycolytic phenotype is due to the upregulation of PDK;
- (2)
- DCA non-responders—the glycolytic phenotype is not dependent on PDK upregulation.
14. Future Perspectives
15. General Conclusions
16. Main Conclusions
Funding
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.-Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical Role for Mitochondrial Oxidative Phosphorylation in the Activation of Tumor Suppressors Bax and Bak. JNCI J. Natl. Cancer Inst. 2006, 98, 1462–1473. [Google Scholar] [CrossRef]
- Dey, R.; Moraes, C.T. Lack of Oxidative Phosphorylation and Low Mitochondrial Membrane Potential Decrease Susceptibility to Apoptosis and Do Not Modulate the Protective Effect of Bcl-xL in Osteosarcoma Cells. J. Biol. Chem. 2000, 275, 7087–7094. [Google Scholar] [CrossRef]
- Park, S.Y.; Chang, I.; Kim, J.-Y.; Kang, S.W.; Park, S.-H.; Singh, K.; Lee, M.-S. Resistance of Mitochondrial DNA-depleted Cells against Cell Death: Role of mitochondrial superoxide dismutase. J. Biol. Chem. 2004, 279, 7512–7520. [Google Scholar] [CrossRef]
- Harris, M.H.; Heiden, M.G.V.; Kron, S.J.; Thompson, C.B. Role of Oxidative Phosphorylation in Bax Toxicity. Mol. Cell. Biol. 2000, 20, 3590–3596. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Nature 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anti-Cancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Bashir, A.H.H.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, J.D.P.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef]
- Harguindey, S.; Reshkin, S.J. “The new pH-centric anticancer paradigm in Oncology and Medicine”; SCB, 2017. Semin. Cancer Biol. 2017, 43, 1–4. [Google Scholar] [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Newell, K.; Franchi, A.; Pouysségur, J.; Tannock, I. Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc. Natl. Acad. Sci. USA 1993, 90, 1127–1131. [Google Scholar] [CrossRef]
- Yamagata, M.; Hasuda, K.; Stamato, T.; Tannock, I. The contribution of lactic acid to acidification of tumours: Studies of variant cells lacking lactate dehydrogenase. Br. J. Cancer 1998, 77, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Meredith, D.; Fox, J.E.M.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) Is the Target for Organomercurial Inhibition of Monocarboxylate Transporter Isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [CrossRef]
- Polański, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the Monocarboxylate Transporter 1 Inhibitor AZD3965 in Small Cell Lung Cancer. Clin. Cancer Res. 2013, 20, 926–937. [Google Scholar] [CrossRef]
- Bueno, V.; Binet, I.; Steger, U.; Bundick, R.; Ferguson, D.; Murray, C.; Donald, D.; Wood, K. The Specific Monocarboxylate Transporter (MCT1) Inhibitor, AR-C117977, a Novel Immunosuppressant, Prolongs Allograft Survival in the Mouse. Transplantation 2007, 84, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Ovens, M.J.; Davies, A.J.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem. J. 2010, 425, 523–530. [Google Scholar] [CrossRef]
- Murray, C.M.; Hutchinson, R.; Bantick, J.R.; Belfield, G.P.; Benjamin, A.D.; Brazma, D.; Bundick, R.V.; Cook, I.D.; Craggs, R.I.; Edwards, S.; et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 2005, 1, 371–376. [Google Scholar] [CrossRef]
- Kobayashi, M.; Otsuka, Y.; Itagaki, S.; Hirano, T.; Iseki, K. Inhibitory effects of statins on human monocarboxylate transporter 4. Int. J. Pharm. 2006, 317, 19–25. [Google Scholar] [CrossRef]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by TGF-β2–dependent regulation of matrix metalloproteinase-2. Neuro-Oncology 2009, 11, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Hussien, R.; Oommen, S.; Gohil, K.; Brooks, G.A. Lactate sensitive transcription factor network in L6 cells: Activation of MCT1 and mitochondrial biogenesis. FASEB J. 2007, 21, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor–macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.C.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Puig-Kröger, A.; Muñiz-Pello, O.; Selgas, R.; Criado, G.; Bajo, M.-A.; Sánchez-Tomero, J.A.; Alvarez, V.; del Peso, G.; Sánchez-Mateos, P.; Holmes, C.; et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: Effect of lactate and glucose-degradation products. J. Leukoc. Biol. 2003, 73, 482–492. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Constant, J.S.; Feng, J.J.; Zabel, D.D.; Yuan, H.; Suh, D.Y.; Scheuenstuhl, H.; Hunt, T.K.; Hussain, M.Z. Lactate elicits vascular endothelial growth factor from macrophages: A possible alternative to hypoxia. Wound Repair Regen. 2000, 8, 353–360. [Google Scholar] [CrossRef]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Lactate Stimulates Vasculogenic Stem Cells via the Thioredoxin System and Engages an Autocrine Activation Loop Involving Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 2008, 28, 6248–6261. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically Derived Lactate Stimulates Revascularization and Tissue Repair via Redox Mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; Scheuenstuhl, H.; Königsrainer, A.; Hussain, M.Z.; Hunt, T.K. Lactate stimulates endothelial cell migration. Wound Repair Regen. 2006, 14, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-κB/IL-8 Pathway that Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Walenta, S.; Salameh, A.; Lyng, H.; Evensen, J.F.; Mitze, M.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am. J. Pathol. 1997, 150, 409–415. [Google Scholar] [PubMed]
- Schwickert, G.; Walenta, S.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in human cervical cancer with incidence of metastasis. Cancer Res. 1995, 55, 4757–4759. [Google Scholar] [PubMed]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef]
- Shegay, P.V.; Zabolotneva, A.A.; Shatova, O.P.; Shestopalov, A.V.; Kaprin, A.D. Evolutionary View on Lactate-Dependent Mechanisms of Maintaining Cancer Cell Stemness and Reprimitivization. Cancers 2022, 14, 4552. [Google Scholar] [CrossRef]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate Activates HIF-1 in Oxidative but Not in Warburg-Phenotype Human Tumor Cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef]
- Samuvel, D.J.; Sundararaj, K.P.; Nareika, A.; Lopes-Virella, M.F.; Huang, Y. Lactate Boosts TLR4 Signaling and NF-κB Pathway-Mediated Gene Transcription in Macrophages via Monocarboxylate Transporters and MD-2 Up-Regulation. J. Immunol. 2009, 182, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-Secreted Lactic Acid Promotes IL-23/IL-17 Proinflammatory Pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Stern, R. Hyaluronidases in cancer biology. In Hyaluronan in Cancer Biology; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2009; pp. 207–220. [Google Scholar]
- DE LA Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is Lactate an Oncometabolite? Evidence Supporting a Role for Lactate in the Regulation of Transcriptional Activity of Cancer-Related Genes in MCF7 Breast Cancer Cells. Front. Oncol. 2020, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Izzo, L.T.; Wellen, K.E. Histone lactylation links metabolism and gene regulation. Nature 2019, 574, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Xin, Q.; Wang, H.; Li, Q.; Liu, S.; Qu, K.; Liu, C.; Zhang, J. Lactylation: A Passing Fad or the Future of Posttranslational Modification. Inflammation 2022, 45, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, L.; Gu, Y.; Cang, W.; Sun, P.; Xiang, Y. Lactate-Lactylation Hands between Metabolic Reprogramming and Immunosuppression. Int. J. Mol. Sci. 2022, 23, 11943. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The tortuous path of lactate shuttle discovery: From cinders and boards to the lab and ICU. J. Sport Health Sci. 2020, 9, 446–460. [Google Scholar] [CrossRef]
- Hopp, A.-K.; Grüter, P.; Hottiger, M.O. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 890. [Google Scholar] [CrossRef]
- Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wan, J.; Cai, K.; Song, H.; Wang, Y.; Sun, W.; Hu, J. Understanding the Contribution of Lactate Metabolism in Cancer Progress: A Perspective from Isomers. Cancers 2022, 15, 87. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Harman, E.M.; Curry, S.H.; Baumgartner, T.G.; Misbin, R.I. Treatment of Lactic Acidosis with Dichloroacetate. N. Engl. J. Med. 1983, 309, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. Dichloroacetate in the Treatment of Lactic Acidosis. Ann. Intern. Med. 1988, 108, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Kerr, D.S.; Barnes, C.; Bunch, S.T.; Carney, P.R.; Fennell, E.M.; Felitsyn, N.M.; Gilmore, R.L.; Greer, M.; Henderson, G.N.; et al. Controlled Clinical Trial of Dichloroacetate for Treatment of Congenital Lactic Acidosis in Children. Pediatrics 2006, 117, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Irsigler, K.; Brändle, J.; Kaspar, L.; Kritz, H.; Lageder, H.; Regal, H. Treament of biguanide-induced lactic acidosis with dichloroacetate. 3 case histories. Arzneimittel-Forschung 1979, 29, 555–559. [Google Scholar] [PubMed]
- Stacpoole, P.W.; Wright, E.C.; Baumgartner, T.G.; Bersin, R.M.; Buchalter, S.; Curry, S.H.; Duncan, C.A.; Harman, E.M.; Henderson, G.N.; Jenkinson, S.; et al. A Controlled Clinical Trial of Dichloroacetate for Treatment of Lactic Acidosis in Adults. N. Engl. J. Med. 1992, 327, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Agbenyega, T.; Angus, B.J.; Bedu-Addo, G.; Ofori-Amanfo, G.; Henderson, G.; Szwandt, I.; O’Brien, R.; Stacpoole, P. Pharmacokinetics and pharmacodynamics of dichloroacetate in children with lactic acidosis due to severe malaria. QJM Int. J. Med. 1995, 88, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Barnes, C.L.; Hurbanis, M.D.; Cannon, S.L.; Kerr, D.S. Treatment of congenital lactic acidosis with dichloroacetate. Arch. Dis. Child. 1997, 77, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Plas, D.R.; Thompson, C.B. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol. Metab. 2002, 13, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Agbenyega, T.; Planche, T.; Bedu-Addo, G.; Ansong, D.; Owusu-Ofori, A.; Bhattaram, V.A.; Nagaraja, N.V.; Shroads, A.L.; Henderson, G.N.; Hutson, A.D.; et al. Population Kinetics, Efficacy, and Safety of Dichloroacetate for Lactic Acidosis Due to Severe Malaria in Children. J. Clin. Pharmacol. 2003, 43, 386–396. [Google Scholar] [CrossRef]
- World Health Organization. Dichloroacetic Acid in Drinking-Water Background Document for Development of WHO Guidelines for Drinking-Water Quality. 2005. Available online: https://cdn.who.int/media/docs/default-source/wash-documents/wash-chemicals/dichloroaceticacid0505.pdf?sfvrsn=2246389b_4 (accessed on 26 December 2023).
- Stacpoole, P.W. Clinical physiology and pharmacology of GSTZ1/MAAI. Biochem. Pharmacol. 2023, 217, 115818. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.H.; Lorenz, A.; Pei-ichu; Limacher, M.; Stacpoole, P.W. Disposition and pharmacodynamics of dichloroacetate (DCA) and oxalate following oral DCA doses. Biopharm. Drug Dispos. 1991, 12, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.-I. Pharmacokinetics of Sodium Dichloroacetate. Ph.D. Dissertation, University of Florida, Gainesville, FL, USA, 1987. Available online: https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Chu+P-I.+Pharmacokinetics+of+Sodium+Dichloroacetate.+PhD+dissertation.+Gainesville%2C+FL%3AUniversity+of+Florida%2C+1987.&btnG= (accessed on 20 February 2023).
- Evans, O.B. Dichloroacetate tissue concentrations and its relationship to hypolactatemia and pyruvate dehydrogenase activation. Biochem. Pharmacol. 1982, 31, 3124–3126. [Google Scholar] [CrossRef] [PubMed]
- Shroads, A.L.; Guo, X.; Dixit, V.; Liu, H.-P.; James, M.O.; Stacpoole, P.W. Age-Dependent Kinetics and Metabolism of Dichloroacetate: Possible Relevance to Toxicity. J. Pharmacol. Exp. Ther. 2008, 324, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.C.; Smeltz, M.G.; Hu, Z.; Rowland-Faux, L.; Zhong, G.; Lorenzo, R.J.; Cisneros, K.V.; Stacpoole, P.W.; James, M.O. Regulation of dichloroacetate biotransformation in rat liver and extrahepatic tissues by GSTZ1 expression and chloride concentration. Biochem. Pharmacol. 2018, 152, 236–243. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- James, M.O.; Yan, Z.; Cornett, R.; Jayanti, V.M.; Henderson, G.N.; Davydova, N.; Katovich, M.J.; Pollock, B.; Stacpoole, P.W. Pharmacokinetics and metabolism of [14C] dichloroacetate in male sprague-dawley rats: Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab. Dispos. 1998, 26, 1134–1143. [Google Scholar] [PubMed]
- Wells, P.G.; Moore, G.W.; Rabin, D.; Wilkinson, G.R.; Oates, J.A.; Stacpoole, P.W. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia 1980, 19, 109–113. [Google Scholar] [CrossRef]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metab.-Clin. Exp. 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Moore, G.W.; Kornhauser, D.M. Metabolic Effects of Dichloroacetate in Patients with Diabetes Mellitus and Hyperlipoproteinemia. N. Engl. J. Med. 1978, 298, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Leon, A.; Schultz, I.R.; Xu, G.; Bull, R.J. Pharmacokinetics and Metabolism of Dichloroacetate in the F344 Rat after Prior Administration in Drinking Water. Toxicol. Appl. Pharmacol. 1997, 146, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Lukas, G.; Vyas, K.H.; Brindle, S.D.; Le Sher, A.R.; Wagner, W.E. Biological Disposition of Sodium Dichloroacetate in Animals and Humans after Intravenous Administration. J. Pharm. Sci. 1980, 69, 419–421. [Google Scholar] [CrossRef]
- Maisenbacher, H.W.; Shroads, A.L.; Zhong, G.; Daigle, A.D.; Abdelmalak, M.M.; Samper, I.S.; Mincey, B.D.; James, M.O.; Stacpoole, P.W. Pharmacokinetics of Oral Dichloroacetate in Dogs. J. Biochem. Mol. Toxicol. 2013, 27, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.J.; Lane, J.R.; Turkel, C.C.; Capparelli, E.V.; Dziewanowska, Z.; Fox, A.W.; Turkel, F.C.C. Dichloroacetate: Population Pharmacokinetics with a Pharmacodynamic Sequential Link Model. J. Clin. Pharmacol. 2001, 41, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Henderson, G.N.; Yan, Z.; James, M.O. Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 1998, 106, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef]
- Constantin-Teodosiu, D. Regulation of Muscle Pyruvate Dehydrogenase Complex in Insulin Resistance: Effects of Exercise and Dichloroacetate. Diabetes Metab. J. 2013, 37, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.; McCormack, J.; Rutter, G.; Burnett, P.; Edgell, N.; Moule, S.; Diggle, T. The hormonal regulation of pyruvate dehydrogenase complex. Adv. Enzym. Regul. 1996, 36, 183–198. [Google Scholar] [CrossRef]
- Schoenmann, N.; Tannenbaum, N.; Hodgeman, R.M.; Raju, R.P. Regulating mitochondrial metabolism by targeting pyruvate dehydrogenase with dichloroacetate, a metabolic messenger. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166769. [Google Scholar] [CrossRef] [PubMed]
- Popov, K.; Zhao, Y.; Shimomura, Y.; Kuntz, M.; Harris, R. Branched-chain alpha-ketoacid dehydrogenase kinase. Molecular cloning, expression, and sequence similarity with histidine protein kinases. J. Biol. Chem. 1992, 267, 13127–13130. [Google Scholar] [CrossRef] [PubMed]
- Gudi, R.; Melissa, M.B.-K.; Kedishvili, N.Y.; Zhao, Y.; Popov, K.M. Diversity of the Pyruvate Dehydrogenase Kinase Gene Family in Humans. J. Biol. Chem. 1995, 270, 28989–28994. [Google Scholar] [CrossRef] [PubMed]
- Rowles, J.; Scherer, S.W.; Xi, T.; Majer, M.; Nickle, D.C.; Rommens, J.M.; Popov, K.M.; Harris, R.A.; Riebow, N.L.; Xia, J.; et al. Cloning and Characterization of PDK4 on 7q21.3 Encoding a Fourth Pyruvate Dehydrogenase Kinase Isoenzyme in Human. J. Biol. Chem. 1996, 271, 22376–22382. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C. Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab. 2006, 3, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Degenhardt, T.; Saramäki, A.; Malinen, M.; Rieck, M.; Väisänen, S.; Huotari, A.; Herzig, K.-H.; Müller, R.; Carlberg, C. Three Members of the Human Pyruvate Dehydrogenase Kinase Gene Family Are Direct Targets of the Peroxisome Proliferator-activated Receptor β/δ. J. Mol. Biol. 2007, 372, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Wua, P.; Peters, J.M.; Harris, R.A. Adaptive Increase in Pyruvate Dehydrogenase Kinase 4 during Starvation Is Mediated by Peroxisome Proliferator-Activated Receptor α. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res 2011, 72, 560–567. [Google Scholar] [CrossRef]
- Yeaman, S.J.; Hutcheson, E.T.; Roche, T.E.; Pettit, F.H.; Brown, J.R.; Reed, L.J.; Watson, D.C.; Dixon, G.H. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 1978, 17, 2364–2370. [Google Scholar] [CrossRef]
- Bowker-Kinley, M.M.; Davis, I.W.; Wu, P.; Harris, A.R.; Popov, M.K. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329, 191–196. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. p53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharmacol. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef]
- Kato, M.; Li, J.; Chuang, J.L.; Chuang, D.T. Distinct Structural Mechanisms for Inhibition of Pyruvate Dehydrogenase Kinase Isoforms by AZD7545, Dichloroacetate, and Radicicol. Structure 2007, 15, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Morley, A. An evolutionary perspective on the Crabtree effect. Front. Mol. Biosci. 2014, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Holness, M.; Sugden, M. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Whitehouse, S.; Cooper, R.H.; Randle, P.J. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 1974, 141, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Whitehouse, S.; Randle, P.J. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem. J. 1973, 134, 651–653. [Google Scholar] [CrossRef]
- Lorini, M.; Ciman, M. Hypoglycaemic action of diisopropylammonium salts in experimental diabetes. Biochem. Pharmacol. 1962, 11, 823–827. [Google Scholar] [CrossRef]
- Albatany, M.; Li, A.; Meakin, S.; Bartha, R. Dichloroacetate induced intracellular acidification in glioblastoma: In vivo detection using AACID-CEST MRI at 9.4 Tesla. J. Neuro-Oncology 2018, 136, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Anemone, A.; Consolino, L.; Conti, L.; Reineri, F.; Cavallo, F.; Aime, S.; Longo, D.L. In vivo evaluation of tumour acidosis for assessing the early metabolic response and onset of resistance to dichloroacetate by using magnetic resonance pH imaging. Int. J. Oncol. 2017, 51, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Albatany, M.; Li, A.; Meakin, S.; Bartha, R. In vivo detection of acute intracellular acidification in glioblastoma multiforme following a single dose of cariporide. Int. J. Clin. Oncol. 2018, 23, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Xiong, H.; Zhou, P.; Ni, Z.; Yang, T.; Zhang, Y.; Zeng, Y.; He, J.; Yang, F.; et al. Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells. Oncotarget 2017, 8, 51748–51757. [Google Scholar] [CrossRef] [PubMed]
- Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Treading slowly through hypoxic waters: Dichloroacetate to the rescue! J. Physiol. 2018, 596, 2957–2958. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase: Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef]
- Zhang, X.; Lee, W.D.; Leitner, B.P.; Zhu, W.; Fosam, A.; Li, Z.; Gaspar, R.C.; Halberstam, A.A.; Robles, B.; Rabinowitz, J.D.; et al. Dichloroacetate as a novel pharmaceutical treatment for cancer-related fatigue in melanoma. Am. J. Physiol. Metab. 2023, 325, E363–E375. [Google Scholar] [CrossRef]
- Abdel-Razek, E.A.-N.; Mahmoud, H.M.; Azouz, A.A. Management of ulcerative colitis by dichloroacetate: Impact on NFATC1/NLRP3/IL1B signaling based on bioinformatics analysis combined with in vivo experimental verification. Inflammopharmacology 2023, 32, 667–682. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Felts, J.M. Diisopropylammonium dichloroacetate (DIPA) and sodium dichloroacetate (DCA): Effect on glucose and fat metabolism in normal and diabetic tissue. Metabolism 1970, 19, 71–78. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Harwood, H.J.; Varnado, C.E. Regulation of rat liver hydroxymethylglutaryl coenzyme A reductase by a new class of noncompetitive inhibitors. Effects of dichloroacetate and related carboxylic acids on enzyme activity. J. Clin. Investig. 1983, 72, 1575–1585. [Google Scholar] [CrossRef]
- Moore, G.W.; Swift, L.L.; Rabinowitz, D.; Crofford, O.B.; Oates, J.A.; Stacpoole, P.W. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis 1979, 33, 285–293. [Google Scholar] [CrossRef]
- Stakišaitis, D.; Kapočius, L.; Kilimaitė, E.; Gečys, D.; Šlekienė, L.; Balnytė, I.; Palubinskienė, J.; Lesauskaitė, V. Preclinical Study in Mouse Thymus and Thymocytes: Effects of Treatment with a Combination of Sodium Dichloroacetate and Sodium Valproate on Infectious Inflammation Pathways. Pharmaceutics 2023, 15, 2715. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Qu, J.; Li, P.; Sun, Z. Histone lactylation regulates cancer progression by reshaping the tumor microenvironment. Front. Immunol. 2023, 14, 1284344. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; et al. Lactate Modulates Cellular Metabolism Through Histone Lactylation-Mediated Gene Expression in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 647559. [Google Scholar] [CrossRef]
- Wang, T.; Ye, Z.; Li, Z.; Jing, D.; Fan, G.; Liu, M.; Zhuo, Q.; Ji, S.; Yu, X.; Xu, X.; et al. Lactate-induced protein lactylation: A bridge between epigenetics and metabolic reprogramming in cancer. Cell Prolif. 2023, 56, e13478. [Google Scholar] [CrossRef]
- Lv, X.; Lv, Y.; Dai, X. Lactate, histone lactylation and cancer hallmarks. Expert Rev. Mol. Med. 2023, 25, e7. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, Q.; Zheng, J.; Yang, Y.; Zhang, X.; Ma, A.; Qin, Y.; Qin, Z.; Zheng, X. The function and mechanism of lactate and lactylation in tumor metabolism and microenvironment. Genes Dis. 2023, 10, 2029–2037. [Google Scholar] [CrossRef]
- Yang, H.; Zou, X.; Yang, S.; Zhang, A.; Li, N.; Ma, Z. Identification of lactylation related model to predict prognostic, tumor infiltrating immunocytes and response of immunotherapy in gastric cancer. Front. Immunol. 2023, 14, 1149989. [Google Scholar] [CrossRef]
- Qiao, Z.; Li, Y.; Li, S.; Liu, S.; Cheng, Y. Hypoxia-induced SHMT2 protein lactylation facilitates glycolysis and stemness of esophageal cancer cells. Mol. Cell. Biochem. 2024, 1–14. [Google Scholar] [CrossRef]
- Su, J.; Zheng, Z.; Bian, C.; Chang, S.; Bao, J.; Yu, H.; Xin, Y.; Jiang, X. Functions and mechanisms of lactylation in carcinogenesis and immunosuppression. Front. Immunol. 2023, 14, 1253064. [Google Scholar] [CrossRef]
- Wang, X.; Ying, T.; Yuan, J.; Wang, Y.; Su, X.; Chen, S.; Zhao, Y.; Zhao, Y.; Sheng, J.; Teng, L.; et al. BRAFV600E restructures cellular lactylation to promote anaplastic thyroid cancer proliferation. Endocr.-Relat. Cancer 2023, 30, e220344. [Google Scholar] [CrossRef]
- Miao, Z.; Zhao, X.; Liu, X. Hypoxia induced β-catenin lactylation promotes the cell proliferation and stemness of colorectal cancer through the wnt signaling pathway. Exp. Cell Res. 2023, 422, 113439. [Google Scholar] [CrossRef]
- Rho, H.; Terry, A.R.; Chronis, C.; Hay, N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. 2023, 35, 1406–1423.e8. [Google Scholar] [CrossRef]
- Wang, X.; Fan, W.; Li, N.; Ma, Y.; Yao, M.; Wang, G.; He, S.; Li, W.; Tan, J.; Lu, Q.; et al. YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2. Genome Biol. 2023, 24, 87. [Google Scholar] [CrossRef]
- Wang, N.; Wang, W.; Wang, X.; Mang, G.; Chen, J.; Yan, X.; Tong, Z.; Yang, Q.; Wang, M.; Chen, L.; et al. Histone Lactylation Boosts Reparative Gene Activation Post–Myocardial Infarction. Circ. Res. 2022, 131, 893–908. [Google Scholar] [CrossRef]
- He, Y.; Ji, Z.; Gong, Y.; Fan, L.; Xu, P.; Chen, X.; Miao, J.; Zhang, K.; Zhang, W.; Ma, P.; et al. Numb/Parkin-directed mitochondrial fitness governs cancer cell fate via metabolic regulation of histone lactylation. Cell Rep. 2023, 42, 112033. [Google Scholar] [CrossRef]
- Cheng, Z.; Huang, H.; Li, M.; Chen, Y. Proteomic analysis identifies PFKP lactylation in SW480 colon cancer cells. iScience 2024, 27, 108645. [Google Scholar] [CrossRef]
- Wang, M.; He, T.; Meng, D.; Lv, W.; Ye, J.; Cheng, L.; Hu, J. BZW2 Modulates Lung Adenocarcinoma Progression through Glycolysis-Mediated IDH3G Lactylation Modification. J. Proteome Res. 2023, 22, 3854–3865. [Google Scholar] [CrossRef]
- Wang, J.-H.; Mao, L.; Zhang, X.; Wu, M.; Wen, Q.; Yu, S.-C. Beyond metabolic waste: Lysine lactylation and its potential roles in cancer progression and cell fate determination. Cell. Oncol. 2023, 46, 465–480. [Google Scholar] [CrossRef]
- Varner, E.L.; Trefely, S.; Bartee, D.; von Krusenstiern, E.; Izzo, L.; Bekeova, C.; O’Connor, R.S.; Seifert, E.L.; Wellen, K.E.; Meier, J.L.; et al. Quantification of lactoyl-CoA (lactyl-CoA) by liquid chromatography mass spectrometry in mammalian cells and tissues. Open Biol. 2020, 10, 200187. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Bæk, M.; Monda, F.; Olsen, C.A. Chiral Posttranslational Modification to Lysine ε-Amino Groups. Accounts Chem. Res. 2022, 55, 1456–1466. [Google Scholar] [CrossRef]
- Patel, R.; Kumar, A.; Lokhande, K.B.; Swamy, K.V.; Sharma, N.K. Molecular Docking and Simulation Studies Predict Lactyl-CoA as the Substrate for P300 Directed Lactylation. Available online: https://chemrxiv.org/engage/api-gateway/chemrxiv/assets/orp/resource/item/60c74e8d4c8919380fad3a55/original/molecular-docking-and-simulation-studies-predict-lactyl-co-a-as-the-substrate-for-p300-directed-lactylation.pdf (accessed on 21 February 2024).
- Wu, H.; Huang, H.; Zhao, Y. Interplay between metabolic reprogramming and post-translational modifications: From glycolysis to lactylation. Front. Immunol. 2023, 14, 1211221. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 728759. [Google Scholar] [CrossRef]
- Han, J.; Chen, X.; Wang, J.; Liu, B. Glycolysis-related lncRNA TMEM105 upregulates LDHA to facilitate breast cancer liver metastasis via sponging miR-1208. Cell Death Dis. 2023, 14, 80. [Google Scholar] [CrossRef]
- Schreier, A.; Zappasodi, R.; Serganova, I.; Brown, K.A.; Demaria, S.; Andreopoulou, E. Facts and Perspectives: Implications of tumor glycolysis on immunotherapy response in triple negative breast cancer. Front. Oncol. 2023, 12, 1061789. [Google Scholar] [CrossRef]
- Godoy, A.; Ulloa, V.; Rodríguez, F.; Reinicke, K.; Yañez, A.J.; García, M.d.L.A.; Medina, R.A.; Carrasco, M.; Barberis, S.; Castro, T.; et al. Differential subcellular distribution of glucose transporters GLUT1–6 and GLUT9 in human cancer: Ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J. Cell. Physiol. 2006, 207, 614–627. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef] [PubMed]
- Littleflower, A.B.; Parambil, S.T.; Antony, G.R.; Subhadradevi, L. The determinants of metabolic discrepancies in aerobic glycolysis: Providing potential targets for breast cancer treatment. Biochimie 2024, 220, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.L.; Eubank, W.B.; Mankoff, D.A. FDG PET, PET/CT, and Breast Cancer Imaging. RadioGraphics 2007, 27 (Suppl. 1), S215–S229. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2010, 120, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Gang, B.P.; Dilda, P.J.; Hogg, P.J.; Blackburn, A.C. Dichloroacetate reverses the Warburg effect, inhibiting growth and sensitizing breast cancer cells towards apoptosis. In Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research, Chicago, IL, USA, 31 March–4 April 2012. [Google Scholar]
- Harting, T.P.; Stubbendorff, M.; Hammer, S.C.; Schadzek, P.; Ngezahayo, A.; Escobar, H.M.; Nolte, I. Dichloroacetate affects proliferation but not apoptosis in canine mammary cell lines. PLoS ONE 2017, 12, e0178744. [Google Scholar] [CrossRef] [PubMed]
- De Preter, G.; Neveu, M.-A.; Danhier, P.; Brisson, L.; Payen, V.L.; Porporato, P.E.; Jordan, B.F.; Sonveaux, P.; Gallez, B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 2016, 7, 2910–2920. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, A.C.; Rooke, M.; Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Board, P.G. Reversal of the glycolytic phenotype with dichloroacetate in a mouse mammary adenocarcinoma model. Pathology 2010, 42, S61–S62. [Google Scholar] [CrossRef]
- Wang, M.; Liao, C.; Hu, Y.; Pan, Q.; Jiang, J. Sensitization of breast cancer cells to paclitaxel by dichloroacetate through inhibiting autophagy. Biochem. Biophys. Res. Commun. 2017, 489, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.H.; Seo, S.-K.; Park, Y.; Kim, E.-K.; Seong, M.-K.; Kim, H.-A.; Song, J.-Y.; Hwang, S.-G.; Lee, J.K.; Noh, W.C.; et al. Dichloroacetate potentiates tamoxifen-induced cell death in breast cancer cells via downregulation of the epidermal growth factor receptor. Oncotarget 2016, 7, 59809–59819. [Google Scholar] [CrossRef]
- Haugrud, A.B.; Zhuang, Y.; Coppock, J.D.; Miskimins, W.K. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 539–550. [Google Scholar] [CrossRef]
- Sun, R.C.; Board, P.G.; Blackburn, A.C. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol. Cancer 2011, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Lam, Y.-M.; Leung, Y.-C.; Hu, X.; Chen, X.; Cheung, E.; Tam, K.Y. Combined use of arginase and dichloroacetate exhibits anti-proliferative effects in triple negative breast cancer cells. J. Pharm. Pharmacol. 2019, 71, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F.; Martin, N.K. Bicarbonate and dichloroacetate: Evaluating pH altering therapies in a mouse model for metastatic breast cancer. BMC Cancer 2011, 11, 235. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-E.; Shin, K.-S.; Lee, Y.-H.; Seo, S.-K.; Yun, S.-M.; Choe, T.-B.; Kim, H.-A.; Kim, E.-K.; Noh, W.C.; Kim, J.-I.; et al. Inhibition of S6K1 enhances dichloroacetate-induced cell death. J. Cancer Res. Clin. Oncol. 2015, 141, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. [Google Scholar] [CrossRef] [PubMed]
- Lefort, N.; Brown, A.; Lloyd, V.; Ouellette, R.; Touaibia, M.; Culf, A.S.; Cuperlovic-Culf, M. 1H NMR metabolomics analysis of the effect of dichloroacetate and allopurinol on breast cancers. J. Pharm. Biomed. Anal. 2014, 93, 77–85. [Google Scholar] [CrossRef] [PubMed]
- de Mey, S.; Dufait, I.; Jiang, H.; Corbet, C.; Wang, H.; Van De Gucht, M.; Kerkhove, L.; Law, K.L.; Vandenplas, H.; Gevaert, T.; et al. Dichloroacetate Radiosensitizes Hypoxic Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9367. [Google Scholar] [CrossRef]
- Gholami, L.; Attari, F.; Talkhabi, M.; Saadatpour, F. MDA-MB-231. Available online: https://scholar.google.com/scholar?cluster=12666777980647358402&hl=en&as_sdt=0,5&as_ylo=2023 (accessed on 24 January 2024).
- Cutruzzolà, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: Connecting the dots. Mol. Cancer 2006, 5, 17. [Google Scholar] [CrossRef]
- Salminen, E.; Hogg, A.; Binns, D.; Frydenberg, M.; Hicks, R. Investigations with FDG-PET Scanning in Prostate Cancer Show Limited Value for Clinical Practice. Acta Oncol. 2002, 41, 425–429. [Google Scholar] [CrossRef]
- Morris, M.J.; Akhurst, T.; Osman, I.; Nunez, R.; Macapinlac, H.; Siedlecki, K.; Verbel, D.; Schwartz, L.; Larson, S.M.; Scher, H.I. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology 2002, 59, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.; Withers, H.; Garbens, A.; Love, H.; Magnoni, L.; Hayward, S.; Moyes, C. Hypoxia and the metabolic phenotype of prostate cancer cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Harting, T.; Stubbendorff, M.; Willenbrock, S.; Wagner, S.; Schadzek, P.; Ngezahayo, A.; Escobar, H.M.; Nolte, I. The effect of dichloroacetate in canine prostate adenocarcinomas and transitional cell carcinomas in vitro. Int. J. Oncol. 2016, 49, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Liang, H.; Guan, G. Dichloroacetate enhances the cytotoxic effect of Cisplatin via decreasing the level of FOXM1 in prostate cancer. Int. J. Clin. Med. 2016, 9, 11044–11050, ISSN 1940-5901/IJCEM0020574. [Google Scholar]
- Olszewski, U.; Poulsen, T.T.; Ulsperger, E.; Poulsen, H.S.; Geissler, K.; Hamilton, G. In vitro cytotoxicity of combinations of dichloroacetate with anticancer platinum compounds. Clin. Pharmacol. Adv. Appl. 2010, 2, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.K.; Bhat, T.A.; Kumar, S.; Kumar, A.; Kumar, R.; Underwood, W.; Koochekpour, S.; Shourideh, M.; Yadav, N.; Dhar, S.; et al. Mitochondrial dysfunction-mediated apoptosis resistance associates with defective heat shock protein response in African–American men with prostate cancer. Br. J. Cancer 2016, 114, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.W.; Kasai, M.; Yamamoto, S.; Fukuhara, H.; Karashima, T.; Kurabayashi, A.; Furihata, M.; Hanazaki, K.; Inoue, K.; Ogura, S.-I. Metabolic shift towards oxidative phosphorylation reduces cell-density-induced cancer-stem-cell-like characteristics in prostate cancer in vitro. Biol. Open 2023, 12, 059615. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xia, L.; Oyang, L.; Liang, J.; Tan, S.; Wu, N.; Yi, P.; Pan, Q.; Rao, S.; Han, Y.; et al. The POU2F1-ALDOA axis promotes the proliferation and chemoresistance of colon cancer cells by enhancing glycolysis and the pentose phosphate pathway activity. Oncogene 2022, 41, 1024–1039. [Google Scholar] [CrossRef]
- Yeh, C.-S.; Wang, J.-Y.; Chung, F.-Y.; Lee, S.-C.; Huang, M.-Y.; Kuo, C.-W.; Yang, M.-J.; Lin, S.-R. Significance of the glycolytic pathway and glycolysis related-genes in tumorigenesis of human colorectal cancers. Oncol. Rep. 2008, 19, 81–91. [Google Scholar] [CrossRef]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br. J. Cancer 2010, 102, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Hill, D.K.; Andrejeva, G.; Boult, J.K.R.; Troy, H.; Fong, A.-C.L.F.W.T.; Orton, M.R.; Panek, R.; Parkes, H.G.; Jafar, M.; et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br. J. Cancer 2014, 111, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Delaney, L.M.; Ho, N.; Morrison, J.; Farias, N.R.; Mosser, D.D.; Coomber, B.L. Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis 2015, 20, 63–74. [Google Scholar] [CrossRef]
- Sun, J.; Cheng, X.; Pan, S.; Wang, L.; Dou, W.; Liu, J.; Shi, X. Dichloroacetate attenuates the stemness of colorectal cancer cells via trigerring ferroptosis through sequestering iron in lysosomes. Environ. Toxicol. 2021, 36, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Xie, G.; He, J.; Li, J.; Pan, F.; Liang, H. Synergistic Antitumor Effect of Dichloroacetate in Combination with 5-Fluorouracil in Colorectal Cancer. J. Biomed. Biotechnol. 2011, 2011, 740564. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhu, D.; Zhu, L.; Hou, Y.; Hou, L.; Huang, X.; Li, L.; Wang, Y.; Li, L.; Zou, H.; et al. Dichloroacetate Overcomes Oxaliplatin Chemoresistance in Colorectal Cancer through the miR-543/PTEN/Akt/mTOR Pathway. J. Cancer 2019, 10, 6037–6047. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Andrews, D.; Blackburn, A.C. Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy. World, J. Clin. Cases 2016, 4, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the V600EBRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef]
- Falkenius, J.; Lundeberg, J.; Johansson, H.; Tuominen, R.; Frostvik-Stolt, M.; Hansson, J.; Brage, S.E. High expression of glycolytic and pigment proteins is associated with worse clinical outcome in stage III melanoma. Melanoma Res. 2013, 23, 452–460. [Google Scholar] [CrossRef]
- Koch, A.; Ebert, E.V.; Seitz, T.; Dietrich, P.; Berneburg, M.; Bosserhoff, A.; Hellerbrand, C. Characterization of glycolysis-related gene expression in malignant melanoma. Pathol.-Res. Pract. 2020, 216, 152752. [Google Scholar] [CrossRef]
- Franco-Molina, M.A.; Mendoza-Gamboa, E.; Sierra-Rivera, C.A.; Zapata-Benavides, P.; Hernandez, D.F.; Chávez-Reyes, A.; Rivera-Morales, L.G.; Tamez-Guerra, R.; Rodríguez-Padilla, C. In vitro and in vivo antitumoral activitiy of sodium dichloroacetate (DCA-Na) against murine melanoma. Afr. J. Microbiol. Res. 2012, 6, 4782–4796. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Vikram Singh, S.; Muhammad, N.; Singh Meena, A.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281–37299. [Google Scholar] [CrossRef]
- Pópulo, H.; Caldas, R.; Lopes, J.M.; Pardal, J.; Máximo, V.; Soares, P. Overexpression of pyruvate dehydrogenase kinase supports dichloroacetate as a candidate for cutaneous melanoma therapy. Expert Opin. Ther. Targets 2015, 19, 733–745. [Google Scholar] [CrossRef]
- Abildgaard, C.; Dahl, C.; Basse, A.L.; Ma, T.; Guldberg, P. Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAFV600E inhibition. J. Transl. Med. 2014, 12, 247. [Google Scholar] [CrossRef]
- Zheng, M.-F.; Shen, S.-Y.; Huang, W.-D. DCA increases the antitumor effects of capecitabine in a mouse B16 melanoma allograft and a human non-small cell lung cancer A549 xenograft. Cancer Chemother. Pharmacol. 2013, 72, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Andrews, D.; Shainhouse, J.; Blackburn, A.C. Long-term stabilization of metastatic melanoma with sodium dichloroacetate. World, J. Clin. Oncol. 2017, 8, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Reuss, A.M.; Groos, D.; Buchfelder, M.; Savaskan, N. The Acidic Brain—Glycolytic Switch in the Microenvironment of Malignant Glioma. Int. J. Mol. Sci. 2021, 22, 5518. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Sanzey, M.; Rahim, S.A.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive Analysis of Glycolytic Enzymes as Therapeutic Targets in the Treatment of Glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Stanke, K.M.; Wilson, C.; Kidambi, S. High Expression of Glycolytic Genes in Clinical Glioblastoma Patients Correlates with Lower Survival. Front. Mol. Biosci. 2021, 8, 752404. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11, 633210. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.-L.; Mackey, J.R.; Fulton, D.; et al. Metabolic Modulation of Glioblastoma with Dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Duan, Y.; Zhao, X.; Ren, W.; Wang, X.; Yu, K.-F.; Li, D.; Zhang, Q. Antitumor activity of dichloroacetate on C6 glioma cell: In vitro and in vivo evaluation. OncoTargets Ther. 2013, 6, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Wigfield, S.; Gee, H.E.; Devlin, C.M.; Singleton, D.; Li, J.-L.; Buffa, F.; Huffman, M.; Sinn, A.L.; Silver, J.; et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. 2013, 91, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Morfouace, M.; Lalier, L.; Oliver, L.; Cheray, M.; Pecqueur, C.; Cartron, P.-F.; Vallette, F.M. Control of glioma cell death and differentiation by PKM2–Oct4 interaction. Cell Death Dis. 2014, 5, e1036. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Boichuk, I.V.; Solyanik, G.I. Hypoxia enhances antitumor activity of dichloroacetate. Exp. Oncol. 2014, 36, 231–235. [Google Scholar] [PubMed]
- Vella, S.; Conti, M.; Tasso, R.; Cancedda, R.; Pagano, A. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int. J. Cancer 2012, 130, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Sradhanjali, S.; Tripathy, D.; Rath, S.; Mittal, R.; Reddy, M.M. Overexpression of pyruvate dehydrogenase kinase 1 in retinoblastoma: A potential therapeutic opportunity for targeting vitreous seeds and hypoxic regions. PLoS ONE 2017, 12, e0177744. [Google Scholar] [CrossRef]
- Park, J.M.; Recht, L.D.; Josan, S.; Merchant, M.; Jang, T.; Yen, Y.-F.; Hurd, R.E.; Spielman, D.M.; Mayer, D. Metabolic response of glioma to dichloroacetate measured in vivo by hyperpolarized 13C magnetic resonance spectroscopic imaging. Neuro-Oncology 2013, 15, 433–441. [Google Scholar] [CrossRef]
- Fedorchuk, A.G.; Pyaskovskaya, O.N.; Gorbik, G.V.; Prokhorova, I.V.; Kolesnik, D.L.; Solyanik, G.I. Effectiveness of sodium dichloroacetate against glioma C6 depends on administration schedule and dosage. Exp. Oncol. 2016, 38, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Wicks, R.T.; Azadi, J.; Mangraviti, A.; Zhang, I.; Hwang, L.; Joshi, A.; Bow, H.; Hutt-Cabezas, M.; Martin, K.L.; Rudek, M.A.; et al. Local delivery of cancer-cell glycolytic inhibitors in high-grade glioma. Neuro-Oncology 2014, 17, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Korsakova, L.; Krasko, J.A.; Stankevicius, E. Metabolic-targeted Combination Therapy with Dichloroacetate and Metformin Suppresses Glioblastoma Cell Line Growth In Vitro and In Vivo. In Vivo 2021, 35, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, I.V.; Pyaskovskaya, O.N.; Kolesnik, D.L.; Solyanik, G.I. Influence of metformin, sodium dichloroacetate and their combination on the hematological and biochemical blood parameters of rats with gliomas C6. Exp. Oncol. 2018, 40, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Yurchenko, O.V.; Solyanik, G.I. Metformin enhances antitumor action of sodium dichloroacetate against glioma C6. Exp. Oncol. 2019, 41, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, M.; Tsoli, M.; Chang, C.; Joshi, S.; Liu, J.; Ryall, S.; Chornenkyy, Y.; Siddaway, R.; Hawkins, C.; et al. Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas. Neuro-Oncology 2020, 22, 139–151. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Zhang, L.; Zhao, C.; Wang, D. Phosphorylated form of pyruvate dehydrogenase α1 mediates tumor necrosis factor α-induced glioma cell migration. Oncol. Lett. 2021, 21, 12437. [Google Scholar] [CrossRef]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; de Mendivil, A.O.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose Metabolism Heterogeneity in Human and Mouse Malignant Glioma Cell Lines. J. Neuro-Oncol. 2005, 74, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Shibao, S.; Minami, N.; Koike, N.; Fukui, N.; Yoshida, K.; Saya, H.; Sampetrean, O. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro-Oncology 2018, 20, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Hoffmann, K.; Cummings, A.J.; de Klerk, N.H.; Kees, U.R. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia 2006, 20, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Hulleman, E.; Kazemier, K.M.; Holleman, A.; VanderWeele, D.J.; Rudin, C.M.; Broekhuis, M.J.C.; Evans, W.E.; Pieters, R.; Boer, M.L.D. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 2009, 113, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Buentke, E.; Nordström, A.; Lin, H.; Björklund, A.-C.; Laane, E.; Harada, M.; Lu, L.; Tegnebratt, T.; Stone-Elander, S.; Heyman, M.; et al. Glucocorticoid-induced cell death is mediated through reduced glucose metabolism in lymphoid leukemia cells. Blood Cancer J. 2011, 1, e31. [Google Scholar] [CrossRef] [PubMed]
- Samuels, A.L.; Heng, J.Y.; Beesley, A.H.; Kees, U.R. Bioenergetic modulation overcomes glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 165, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Mala Shanmugam, M.; McBrayer, S.K.; Rosen, S.T. Targeting the Warburg effect in hematological malignancies: From PET to therapy. Curr. Opin. Oncol. 2009, 21, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef]
- Gu, Z.; Xia, J.; Xu, H.; Frech, I.; Tricot, G.; Zhan, F. NEK2 Promotes Aerobic Glycolysis in Multiple Myeloma through Regulating Splicing of Pyruvate Kinase. J. Hematol. Oncol. 2017, 10, 17. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Paschou, S.A.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Metabolic disorders in multiple myeloma. Int. J. Mol. Sci. 2021, 22, 11430. [Google Scholar] [CrossRef]
- Sanchez, W.Y.; McGee, S.L.; Connor, T.; Mottram, B.; Wilkinson, A.; Whitehead, J.P.; Vuckovic, S.; Catley, L. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br. J. Cancer 2013, 108, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Sasano, T.; Arima, Y.; Kushima, S.; Tsujita, K.; Matsuoka, M.; Hata, H. A novel PDK1 inhibitor, JX06, inhibits glycolysis and induces apoptosis in multiple myeloma cells. Biochem. Biophys. Res. Commun. 2022, 587, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170–178. [Google Scholar] [CrossRef]
- Tian, D.D.; Bennett, S.K.; Coupland, L.A.; Forwood, K.; Lwin, Y.; Pooryousef, N.; Tea, I.; Truong, T.T.; Neeman, T.; Crispin, P.; et al. GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial. Pharmacol. Res. Perspect. 2019, 7, e00526. [Google Scholar] [CrossRef]
- Voltan, R.; Rimondi, E.; Melloni, E.; Gilli, P.; Bertolasi, V.; Casciano, F.; Rigolin, G.M.; Zauli, G.; Secchiero, P. Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1. Oncotarget 2016, 7, 18965–18977. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kant, S.; Singh, S.M. Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: A role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation. Toxicol. Appl. Pharmacol. 2013, 273, 196–208. [Google Scholar] [CrossRef]
- Flavin, D.F. Non-Hodgkin’s Lymphoma Reversal with Dichloroacetate. J. Oncol. 2010, 2010, 414726. [Google Scholar] [CrossRef]
- Strum, S.B.; Adalsteinsson, Ö.; Black, R.R.; Segal, D.; Peress, N.L.; Waldenfels, J. Case Report: Sodium dichloroacetate (DCA) inhibition of the “Warburg Effect” in a human cancer patient: Complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP. J. Bioenerg. Biomembr. 2013, 45, 307–315. [Google Scholar] [CrossRef]
- Abramek, J.; Bogucki, J.; Ziaja-Sołtys, M.; Stępniewski, A.; Bogucka-Kocka, A. Effect of sodium dichloroacetate on apoptotic gene expression in human leukemia cell lines. Pharmacol. Rep. 2019, 71, 248–256. [Google Scholar] [CrossRef]
- Agnoletto, C.; Melloni, E.; Casciano, F.; Rigolin, G.M.; Rimondi, E.; Celeghini, C.; Brunelli, L.; Cuneo, A.; Secchiero, P.; Zauli, G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget 2014, 5, 4347–4360. [Google Scholar] [CrossRef] [PubMed]
- Agnoletto, C.; Brunelli, L.; Melloni, E.; Pastorelli, R.; Casciano, F.; Rimondi, E.; Rigolin, G.M.; Cuneo, A.; Secchiero, P.; Zauli, G. The anti-leukemic activity of sodium dichloroacetate in p53mutated/null cells is mediated by a p53-independent ILF3/p21 pathway. Oncotarget 2015, 6, 2385–2396. [Google Scholar] [CrossRef]
- Emadi, A.; Sadowska, M.; Carter-Cooper, B.; Bhatnagar, V.; van der Merwe, I.; Levis, M.J.; Sausville, E.A.; Lapidus, R.G. Perturbation of cellular oxidative state induced by dichloroacetate and arsenic trioxide for treatment of acute myeloid leukemia. Leuk. Res. 2015, 39, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Queckborner, S.; Turnbull, A.; Michie, C.O.; Williams, A.R.W.; Rye, T.; Gourley, C.; Langdon, S.P. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer 2018, 18, 636. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, L.; Chai, W.; Zhao, T.; Jin, X.; Guo, X.; Han, L.; Yuan, C. Dichloroacetic acid upregulates apoptosis of ovarian cancer cells by regulating mitochondrial function. OncoTargets Ther. 2019, 12, 1729–1739. [Google Scholar] [CrossRef]
- Saed, G.M.; Fletcher, N.M.; Jiang, Z.L.; Abu-Soud, H.M.; Diamond, M.P. Dichloroacetate Induces Apoptosis of Epithelial Ovarian Cancer Cells Through a Mechanism Involving Modulation of Oxidative Stress. Reprod. Sci. 2011, 18, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Štarha, P.; Trávníček, Z.; Vančo, J.; Dvořák, Z. Half-Sandwich Ru(II) and Os(II) Bathophenanthroline Complexes Containing a Releasable Dichloroacetato Ligand. Molecules 2018, 23, 420. [Google Scholar] [CrossRef]
- Priego-Hernández, V.D.; Arizmendi-Izazaga, A.; Soto-Flores, D.G.; Santiago-Ramón, N.; Feria-Valadez, M.D.; Navarro-Tito, N.; Jiménez-Wences, H.; Martínez-Carrillo, D.N.; Salmerón-Bárcenas, E.G.; Leyva-Vázquez, M.A.; et al. Expression of HIF-1α and Genes Involved in Glucose Metabolism Is Increased in Cervical Cancer and HPV-16-Positive Cell Lines. Pathogens 2022, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, B.-S.; Yu, D.-H.; Lu, Q.; Ma, J.; Qi, H.; Fang, C.; Chen, H.-Z. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int. J. Oncol. 2011, 38, 409–417. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Zhang, Y.-Z.; Wang, Y.-S.; Ma, X.-X. Identification of novel cell glycolysis related gene signature predicting survival in patients with endometrial cancer. Cancer Cell Int. 2019, 19, 296. [Google Scholar] [CrossRef]
- Wong, J.Y.; Huggins, G.S.; Debidda, M.; Munshi, N.C.; De Vivo, I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol. Oncol. 2008, 109, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.H.; Zhou, C.; Bae-Jump, V. Abstract 2993: Dichloroacetate inhibits cell proliferation and induces apoptosis in human endometrial cancer cell lines. Cancer Res. 2016, 76 (Suppl. 14), 2993. [Google Scholar] [CrossRef]
- Li, X.; Gu, J.; Zhou, Q. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Smolle, E.; Leko, P.; Stacher-Priehse, E.; Brcic, L.; El-Heliebi, A.; Hofmann, L.; Quehenberger, F.; Hrzenjak, A.; Popper, H.H.; Olschewski, H.; et al. Distribution and prognostic significance of gluconeogenesis and glycolysis in lung cancer. Mol. Oncol. 2020, 14, 2853–2867. [Google Scholar] [CrossRef]
- Xu, J.-Q.; Fu, Y.-L.; Zhang, J.; Zhang, K.-Y.; Ma, J.; Tang, J.-Y.; Zhang, Z.-W.; Zhou, Z.-Y. Targeting glycolysis in non-small cell lung cancer: Promises and challenges. Front. Pharmacol. 2022, 13, 1037341. [Google Scholar] [CrossRef] [PubMed]
- Oylumlu, E.; Yin, N.Y. Evaluation of Antiproliferative Effects of Sodium Dichloroacetate on Lung Adenocarcinoma. Available online: https://openurl.ebsco.com/EPDB%3Agcd%3A11%3A2866491/detailv2?sid=ebsco%3Aplink%3Ascholar&id=ebsco%3Agcd%3A140410362&crl=c (accessed on 20 December 2023).
- Allen, K.T.; Chin-Sinex, H.; DeLuca, T.; Pomerening, J.R.; Sherer, J.; Watkins, J.B.; Foley, J.; Jesseph, J.M.; Mendonca, M.S. Dichloroacetate alters Warburg metabolism, inhibits cell growth, and increases the X-ray sensitivity of human A549 and H1299 NSC lung cancer cells. Free Radic. Biol. Med. 2015, 89, 263–273. [Google Scholar] [CrossRef]
- Lu, X.; Zhou, D.; Hou, B.; Liu, Q.-X.; Chen, Q.; Deng, X.-F.; Yu, Z.-B.; Dai, J.-G.; Zheng, H. Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer. Cancer Manag. Res. 2018, 10, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, A.; Zhou, X.; Wang, F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget 2017, 8, 52642–52650. [Google Scholar] [CrossRef]
- Yang, Z.; Tam, K.Y. Anti-cancer synergy of dichloroacetate and EGFR tyrosine kinase inhibitors in NSCLC cell lines. Eur. J. Pharmacol. 2016, 789, 458–467. [Google Scholar] [CrossRef]
- Al-Azawi, A.; Sulaiman, S.; Arafat, K.; Yasin, J.; Nemmar, A.; Attoub, S. Impact of Sodium Dichloroacetate Alone and in Combination Therapies on Lung Tumor Growth and Metastasis. Int. J. Mol. Sci. 2021, 22, 12553. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, Y.; Wu, P.; Ran, M.; Xu, N.; Shan, W.; Sha, O.; Tam, K.Y. Dichloroacetophenone biphenylsulfone ethers as anticancer pyruvate dehydrogenase kinase inhibitors in non-small cell lung cancer models. Chem. Interact. 2023, 378, 110467. [Google Scholar] [CrossRef] [PubMed]
- Fiebiger, W.; Olszewski, U.; Ulsperger, E.; Geissler, K.; Hamilton, G. In vitro cytotoxicity of novel platinum-based drugs and dichloroacetate against lung carcinoid cell lines. Clin. Transl. Oncol. 2011, 13, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Boychuk, I.V.; Dasyukevich, O.I.; Melnikov, O.R.; Tarasov, A.S.; Solyanik, G.I. Effect of dichloroacetate on Lewis lung carcinoma growth and metastasis. Exp. Oncol. 2015, 37, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Wang, J.; Zhou, J. Unraveling the therapeutic mechanisms of dichloroacetic acid in lung cancer through integrated multi-omics approaches: Metabolomics and transcriptomics. Front. Genet. 2023, 14, 1199566. [Google Scholar] [CrossRef] [PubMed]
- Penticuff, J.C.; Woolbright, B.L.; Sielecki, T.M.; Weir, S.J.; Taylor, J.A. MIF family proteins in genitourinary cancer: Tumorigenic roles and therapeutic potential. Nat. Rev. Urol. 2019, 16, 318–328. [Google Scholar] [CrossRef]
- Nobre, C.C.G.; de Araújo, J.M.G.; Fernandes, T.A.A.d.M.; Cobucci, R.N.O.; Lanza, D.C.F.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244. [Google Scholar] [CrossRef]
- Yasasever, V.; Camlica, H.; Duranyildiz, D.; Oguz, H.; Tas, F.; Dalay, N. Macrophage Migration Inhibitory Factor in Cancer. Cancer Investig. 2007, 25, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wei, Y.; Huang, T.; Wei, X.; Sun, S.; Wang, T.; Zhao, Z. Role of macrophage migration inhibitory factor for stem cells. Chin. J. Tissue Eng. Res. 2023, 27, 2395. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef]
- Nomura, M.; Morita, M.; Tanuma, N. A metabolic vulnerability of small-cell lung cancer. Oncotarget 2018, 9, 32278–32279. [Google Scholar] [CrossRef]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576. [Google Scholar] [CrossRef] [PubMed]
- Grass, G.D.; Scott, K.E.N.; Fernandez, M.R.; Stewart, P.A.; Kang, Y.P.; Yang, C.; DeNicola, G.M.; Bannister, T.D.; Cubitt, C.L.; Haura, E.B.; et al. Targeting Lactate Transport in Small-Cell Lung Cancer. Int. J. Radiat. Oncol. 2018, 102, E182. [Google Scholar] [CrossRef]
- Choi, J.-E.; Sebastian, C.; Ferrer, C.M.; Lewis, C.A.; Sade-Feldman, M.; LaSalle, T.; Gonye, A.; Lopez, B.G.C.; Abdelmoula, W.M.; Regan, M.S.; et al. A unique subset of glycolytic tumour-propagating cells drives squamous cell carcinoma. Nat. Metab. 2021, 3, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Somers, K.D.; Merrick, M.A.; Lopez, M.E.; Incognito, L.S.; Schechter, G.L.; Casey, G. Frequent p53 mutations in head and neck cancer. Cancer Res. 1992, 52, 5997–6000. [Google Scholar] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent Mutation of the PI3K Pathway in Head and Neck Cancer Defines Predictive Biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- McBride, S.M.; Rothenberg, S.M.; Faquin, W.C.; Chan, A.W.; Clark, J.R.; Ellisen, L.W.; Wirth, L.J. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma. Head Neck 2014, 36, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant Epidermal Growth Factor Receptor (EGFRvIII) Contributes to Head and Neck Cancer Growth and Resistance to EGFR Targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.A.; Boyle, J.O.; Koch, W.M.; Goodman, S.N.; Hruban, R.H.; Eby, Y.J.; Couch, M.J.; Forastiere, A.A.; Sidransky, D. Association between Cigarette Smoking and Mutation of the p53 Gene in Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 1995, 332, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Landor, S.K.-J.; Mutvei, A.P.; Mamaeva, V.; Jin, S.; Busk, M.; Borra, R.; Grönroos, T.J.; Kronqvist, P.; Lendahl, U.; Sahlgren, C.M. Hypo- and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 18814–18819. [Google Scholar] [CrossRef]
- Slaninova, V.; Krafcikova, M.; Perez-Gomez, R.; Steffal, P.; Trantirek, L.; Bray, S.J.; Krejci, A. Notch stimulates growth by direct regulation of genes involved in the control of glycolysis and the tricarboxylic acid cycle. Open Biol. 2016, 6, 150155. [Google Scholar] [CrossRef]
- Bi, P.; Kuang, S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol. Metab. 2015, 26, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Hüttemann, M.; Lee, I.; Ali-Fehmi, R.; et al. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530–51541. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Agriesti, F.; Scrima, R.; Laurenzana, I.; Perrone, D.; Tataranni, T.; Mazzoccoli, C.; Muzio, L.L.; Capitanio, N.; Piccoli, C. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: A metabolic perspective of treatment. Oncotarget 2015, 6, 1217–1230. [Google Scholar] [CrossRef]
- Xie, Q.; Zhang, H.-F.; Guo, Y.-Z.; Wang, P.-Y.; Liu, Z.-S.; Gao, H.-D.; Xie, W.-L. Combination of Taxol® and dichloroacetate results in synergistically inhibitory effects on Taxol-resistant oral cancer cells under hypoxia. Mol. Med. Rep. 2015, 11, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.-L.; Park, J.Y.; Kim, E.H.; Jang, H.J.; Kwon, M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016, 371, 20–29. [Google Scholar] [CrossRef]
- Powell, S.F.; Mazurczak, M.; Dib, E.G.; Bleeker, J.S.; Geeraerts, L.H.; Tinguely, M.; Lohr, M.M.; McGraw, S.C.; Jensen, A.W.; Ellison, C.A.; et al. Phase II study of dichloroacetate, an inhibitor of pyruvate dehydrogenase, in combination with chemoradiotherapy for unresected, locally advanced head and neck squamous cell carcinoma. Investig. New Drugs 2022, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Inanc, S.; Keles, D.; Eskiizmir, G.; Basbinar, Y.; Oktay, G. Metformin and Dichloroacetate Combination Exert a Synergistic Effect on Cell Viability of Oral Squamous Cell Carcinoma. ENT Updat. 2019, 9, 68–73. [Google Scholar] [CrossRef]
- Morais, M.; Dias, F.; Teixeira, A.L.; Medeiros, R. MicroRNAs and altered metabolism of clear cell renal cell carcinoma: Potential role as aerobic glycolysis biomarkers. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 2175–2185. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, M.; Liu, M.; Xu, Y.; Wu, G. Glycolysis-Related Genes Serve as Potential Prognostic Biomarkers in Clear Cell Renal Cell Carcinoma. Oxidative Med. Cell. Longev. 2021, 2021, 6699808. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Linehan, W.M.; Srinivasan, R. Aerobic glycolysis: A novel target in kidney cancer. Expert Rev. Anticancer. Ther. 2013, 13, 711–719. [Google Scholar] [CrossRef]
- Chakraborty, S.; Balan, M.; Sabarwal, A.; Choueiri, T.K.; Pal, S. Metabolic reprogramming in renal cancer: Events of a metabolic disease. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188559. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Poma, J.; Trilla-Fuertes, L.; López-Vacas, R.; López-Camacho, E.; García-Fernández, E.; Pertejo, A.; Lumbreras-Herrera, M.I.; Zapater-Moros, A.; Díaz-Almirón, M.; Dittmann, A.; et al. Proteomics Characterization of Clear Cell Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.E.; Emaldi, M.; Mingo, J.; Øyjord, T.; Mælandsmo, G.M.; Fodstad, Ø.; Errarte, P.; Larrinaga, G.; Llarena, R.; López, J.I.; et al. The expression pattern of pyruvate dehydrogenase kinases predicts prognosis and correlates with immune exhaustion in clear cell renal cell carcinoma. Sci. Rep. 2023, 13, 7339. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Dromparis, P.; Saleme, B.; Gurtu, V.; Watson, K.; Paulin, R.; Zervopoulos, S.; Stenson, T.; Sutendra, G.; Pink, D.B.; et al. Metabolic Modulation of Clear-cell Renal Cell Carcinoma with Dichloroacetate, an Inhibitor of Pyruvate Dehydrogenase Kinase. Eur. Urol. 2016, 69, 734–744. [Google Scholar] [CrossRef]
- Guo, J.Q.; Tang, H.Y.; Wang, C.D.; Sang, B.T.; Liu, X.; Yi, F.P.; Liu, G.L.; Wu, X.M. Influence of Dichloroacetate on Wilms’ Tumor in vitro. Ann. Clin. Lab. Sci. 2022, 52, 101–108. [Google Scholar]
- Kalay, S.; Dogan, A.; Turkan, A.; Demiroglu-Zergeroglu, A. Dicholoroacetate exerts anti-cancer activity on human renal cell carcinoma cells. Turk. J. Biochem. 2017, 42, 577–585. [Google Scholar] [CrossRef]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef]
- Rajeshkumar, N.; Yabuuchi, S.; Pai, S.G.; De Oliveira, E.; Kamphorst, J.J.; Rabinowitz, J.D.; Tejero, H.; Al-Shahrour, F.; Hidalgo, M.; Maitra, A.; et al. Treatment of Pancreatic Cancer Patient–Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin. Cancer Res. 2017, 23, 5639–5647. [Google Scholar] [CrossRef]
- Tavares-Valente, D.; Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Baltazar, F.; Queirós, O.; Agrimi, G.; Reshkin, S.J.; Cardone, R.A. Extracellular Matrix Collagen I Differentially Regulates the Metabolic Plasticity of Pancreatic Ductal Adenocarcinoma Parenchymal Cell and Cancer Stem Cell. Cancers 2023, 15, 3868. [Google Scholar] [CrossRef]
- Shimoni-Sebag, A.; Abramovich, I.; Agranovich, B.; Sirovsky, Y.; Stossel, C.; Atias, D.; Raitses-Gurevich, M.; Glick-Gorman, Y.; Margalit, O.; Regev, D.; et al. Abstract 2411: The pentose-phosphate pathway induces pancreatic cancer radioresistance, a preclinical study with clinical validation. Cancer Res. 2023, 83 (Suppl. 7), 2411. [Google Scholar] [CrossRef]
- Feuerecker, B.; Biechl, P.; Veltkamp, C.; Saur, D.; Eisenreich, W. Metabolic Response of Pancreatic Carcinoma Cells under Treatment with Dichloroacetate. Metabolites 2021, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Agriesti, F.; Pacelli, C.; Ruggieri, V.; Laurenzana, I.; Mazzoccoli, C.; Della Sala, G.; Panebianco, C.; Pazienza, V.; Capitanio, N.; et al. Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells 2019, 8, 478. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Xiong, X.; Huang, G.; Liu, J.; Sheng, S.; Wang, H.; Qin, W. Dichloroacetate Enhances Adriamycin-Induced Hepatoma Cell Toxicity In Vitro and In Vivo by Increasing Reactive Oxygen Species Levels. PLoS ONE 2014, 9, e92962. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-J.; Xie, P.; Dai, X.-F.; Chen, L.-X.; Sun, L.-B.; Li, T.; He, W.-H.; Xu, Z.-Z.; Huang, G.; He, F.-T.; et al. Dichloroacetate enhances the antitumor effect of pirarubicin via regulating the ROS-JNK signaling pathway in liver cancer cells. Cancer Drug Resist. 2020, 3, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Lee, M.; Park, M.; Kim, S.Y.; Shim, M.S.; Lee, C.Y.; Choi, D.H.; Cho, Y. Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1. Int. J. Mol. Sci. 2021, 22, 10027. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int. J. Cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Rooke, M.; Coupland, L.A.; Truong, T.; Blackburn, A.C. Dichloroacetate is an effective treatment for sarcoma models in vitro and in vivo. Cancer Metab. 2014, 2, P9. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ishiguro, M.; Ishiguro, R.; Iwai, S. Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors. Oncol. Lett. 2012, 3, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.R.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638–1650. [Google Scholar] [CrossRef]
- El Sayed, S.M.; Baghdadi, H.; Ahmed, N.S.; Almaramhy, H.H.; Mahmoud, A.A.-A.; El-Sawy, S.A.; Ayat, M.; Elshazley, M.; Abdel-Aziz, W.; Abdel-Latif, H.M.; et al. Dichloroacetate is an antimetabolite that antagonizes acetate and deprives cancer cells from its benefits: A novel evidence-based medical hypothesis. Med. Hypotheses 2019, 122, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Khyzhnyak, S.V.; Sorokina, L.V.; Stepanova, L.I.; Kaplia, A.A. Functional and dynamic state of inner mitochondrial membrane of sarcoma 37 in mice under administration of sodium dichloroacetate. Ukr. Biochem. J. 2014, 86, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Lim, I.K. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014, 346, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Hur, H.; Ham, I.-H.; Yun, J.; Lee, J.-Y.; Shim, W.; Kim, Y.B.; Lee, G.; Han, S.-U.; Cho, Y.K. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp. Cell Res. 2014, 321, 219–230. [Google Scholar] [CrossRef]
- Matalka, K.Z.; Badr, M.M.; Qinna, N.; Qadan, F. Dichloroacetate modulates cytokines toward T helper 1 function via induction of the interleukin-12–interferon-γ pathway. OncoTargets Ther. 2014, 7, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yuan, P.; Yu, W.; Lin, J.; Xu, A.; Xu, X.; Lou, J.; Yu, T.; Qian, C.; Liu, B.; et al. Mitochondria-Targeting Polymer Micelle of Dichloroacetate Induced Pyroptosis to Enhance Osteosarcoma Immunotherapy. ACS Nano 2022, 16, 10327–10340. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.-K.; Yan, S.; Lam, J.S.-M.; Feng, Y.; Khan, M.; Chen, C.; Ko, F.C.-F.; Ho, J.C.-M. Disturbance of the Warburg effect by dichloroacetate and niclosamide suppresses the growth of different sub-types of malignant pleural mesothelioma in vitro and in vivo. Front. Pharmacol. 2022, 13, 1020343. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zheng, G.; Li, Q.; Shen, L. Metabolic reprogramming induced by DCA enhances cisplatin sensitivity through increasing mitochondrial oxidative stress in cholangiocarcinoma. Front. Pharmacol. 2023, 14, 1128312. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Maimouni, S.; Zong, W.-X. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef]
- Klose, K.; Packeiser, E.-M.; Müller, P.; Granados-Soler, J.L.; Schille, J.T.; Goericke-Pesch, S.; Kietzmann, M.; Escobar, H.M.; Nolte, I. Metformin and sodium dichloroacetate effects on proliferation, apoptosis, and metabolic activity tested alone and in combination in a canine prostate and a bladder cancer cell line. PLoS ONE 2021, 16, e0257403. [Google Scholar] [CrossRef]
- Gurav, A.; Sivaprakasam, S.; Bhutia, Y.D.; Boettger, T.; Singh, N.; Ganapathy, V. Slc5a8, a Na+-coupled high-affinity transporter for short-chain fatty acids, is a conditional tumour suppressor in colon that protects against colitis and colon cancer under low-fibre dietary conditions. Biochem. J. 2015, 469, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Gopal, E.; Miyauchi, S.; Prasad, P. Biological functions of SLC5A8, a candidate tumour suppressor. Biochem. Soc. Trans. 2005, 33 Pt 1, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Di Bernardo, J.; Rhoden, K.J. Atlas of Genetcs and Cytogenetcs in Oncology and Haematology. 2009. Available online: https://atlasgeneticsoncology.org/gene/44089/slc5a8 (accessed on 24 January 2024).
- Whitman, S.P.; Hackanson, B.; Liyanarachchi, S.; Liu, S.; Rush, L.J.; Maharry, K.; Margeson, D.; Davuluri, R.; Wen, J.; Witte, T.; et al. DNA hypermethylation and epigenetic silencing of the tumor suppressor gene, SLC5A8, in acute myeloid leukemia with the MLL partial tandem duplication. Blood 2008, 112, 2013–2016. [Google Scholar] [CrossRef]
- Li, H.; Myeroff, L.; Smiraglia, D.; Romero, M.F.; Pretlow, T.P.; Kasturi, L.; Lutterbaugh, J.; Rerko, R.M.; Casey, G.; Issa, J.-P.; et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc. Natl. Acad. Sci. USA 2003, 100, 8412–8417. [Google Scholar] [CrossRef] [PubMed]
- Porra, V.; Ferraro-Peyret, C.; Durand, C.; Selmi-Ruby, S.; Giroud, H.; Berger-Dutrieux, N.; Decaussin, M.; Peix, J.-L.; Bournaud, C.; Orgiazzi, J.; et al. Silencing of the Tumor Suppressor Gene SLC5A8 Is Associated with BRAF Mutations in Classical Papillary Thyroid Carcinomas. J. Clin. Endocrinol. Metab. 2005, 90, 3028–3035. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Maunakea, A.; Jun, P.; Bollen, A.W.; Hodgson, J.G.; Goldenberg, D.D.; Weiss, W.A.; Costello, J.F. Shared Epigenetic Mechanisms in Human and Mouse Gliomas Inactivate Expression of the Growth SuppressorSLC5A8. Cancer Res 2005, 65, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Helm, J.F.; Zheng, W.; Ly, Q.P.; Hodul, P.J.; Centeno, B.A.; Malafa, M.P. Silencing of the Candidate Tumor Suppressor Gene Solute Carrier Family 5 Member 8 (SLC5A8) in Human Pancreatic Cancer. Pancreas 2008, 36, e32–e39. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Toyota, M.; Akino, K.; Suzuki, H.; Kusano, M.; Satoh, A.; Mita, H.; Sasaki, Y.; Nojima, M.; Yanagihara, K.; et al. Aberrant Methylation and Histone Deacetylation Associated with Silencing of SLC5A8 in Gastric Cancer. Tumor Biol. 2004, 25, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Yan, X.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; et al. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget 2016, 7, 59458–59470. [Google Scholar] [CrossRef]
- Ward, N.P.; Poff, A.M.; Koutnik, A.P.; D’agostino, D.P. Complex I inhibition augments dichloroacetate cytotoxicity through enhancing oxidative stress in VM-M3 glioblastoma cells. PLoS ONE 2017, 12, e0180061. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin Reduces Endogenous Reactive Oxygen Species and Associated DNA Damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A.J. Metformin Inhibits the Production of Reactive Oxygen Species from NADH: Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; DeBusk, B.G.; Gunsalus, I.C.; Hornberger, C.S. Crystalline α-Lipoic Acid: A Catalytic Agent Associated with Pyruvate Dehydrogenase. Science 1951, 114, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Sidhu, S.; Patel, M.S. R-Lipoic Acid Inhibits Mammalian Pyruvate Dehydrogenase Kinase. Free Radic. Res. 2004, 38, 1083–1092. [Google Scholar] [CrossRef]
- Konrad, T.; Vicini, P.; Kusterer, K.; Höflich, A.; Assadkhani, A.; Böhles, H.J.; Sewell, A.; Tritschler, H.J.; Cobelli, C.; Usadel, K.H. alpha-Lipoic acid treatment decreases serum lactate and pyruvate concentrations and improves glucose effectiveness in lean and obese patients with type 2 diabetes. Diabetes Care 1999, 22, 280–287. [Google Scholar] [CrossRef]
- Rahimifard, M.; Navaei-Nigjeh, M.; Baeeri, M.; Maqbool, F.; Abdollahi, M. Multiple protective mechanisms of alpha-lipoic acid in oxidation, apoptosis and inflammation against hydrogen peroxide induced toxicity in human lymphocytes. Mol. Cell. Biochem. 2015, 403, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Abolhassani, M.; Guais, A.; Sanders, E.; Steyaert, J.-M.; Campion, F.; Israël, M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: Preliminary results. Oncol. Rep. 2010, 23, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Nickel, A.; Daniel, H. α-lipoic acid induces apoptosis in human colon cancer cells by increasing mitochondrial respiration with a concomitant O2-generation. Apoptosis 2005, 10, 359–368. [Google Scholar] [CrossRef]
- Moungjaroen, J.; Nimmannit, U.; Callery, P.S.; Wang, L.; Azad, N.; Lipipun, V.; Chanvorachote, P.; Rojanasakul, Y. Reactive Oxygen Species Mediate Caspase Activation and Apoptosis Induced by Lipoic Acid in Human Lung Epithelial Cancer Cells through Bcl-2 Down-Regulation. J. Pharmacol. Exp. Ther. 2006, 319, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Simbula, G.; Columbano, A.; Ledda-Columbano, G.M.; Sanna, L.; Deidda, M.; Diana, A.; Pibiri, M. Increased ROS generation and p53 activation in α-lipoic acid-induced apoptosis of hepatoma cells. Apoptosis 2007, 12, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, B.; Pirsig, S.; Seidl, C.; Aichler, M.; Feuchtinger, A.; Bruchelt, G.; Senekowitsch-Schmidtke, R. Lipoic acid inhibits cell proliferation of tumor cells in vitro and in vivo. Cancer Biol. Ther. 2012, 13, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Pyaskovskaya, O.N.; Kolesnik, D.L.; Fedorchuk, A.G.; Prokhorova, I.V.; Solyanik, G.I. 2-Deoxy-D-glucose enhances dichloroacetate antitumor action against Lewis lung carcinoma. Exp. Oncol. 2016, 38, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhong, X.; Yuan, Y. Does Baking Soda Function as a Magic Bullet for Patients with Cancer? A Mini Review. Integr. Cancer Ther. 2020, 19, 1534735420922579. [Google Scholar] [CrossRef] [PubMed]
- Ayyanathan, K.; Kesaraju, S.; Dawson-Scully, K.; Weissbach, H. Combination of Sulindac and Dichloroacetate Kills Cancer Cells via Oxidative Damage. PLoS ONE 2012, 7, e39949. [Google Scholar] [CrossRef]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. USA 2009, 106, 22199–22204. [Google Scholar] [CrossRef]
- Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: A mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem. Biol. 2014, 9, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Hanberry, B.S.; Berger, R.; Zastre, J.A. High-dose vitamin B1 reduces proliferation in cancer cell lines analogous to dichloroacetate. Cancer Chemother. Pharmacol. 2014, 73, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Ghosh, M.; Dutta, S.K. A potent tumoricidal co-drug ‘Bet-CA’—An ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Sci. Rep. 2015, 5, 7762. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Wang, S.; Zheng, Y.; Wang, N.; Yang, B.; Wang, D.; Yang, D.; Mei, W.; Zhao, Z.; Wang, Z. Betulinic acid suppresses breast cancer aerobic glycolysis via caveolin-1/NF-κB/c-Myc pathway. Biochem. Pharmacol. 2019, 161, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Scaffidi, C.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998, 273, 33942–33948. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Grimmel, C.; Wagenknecht, B.; Dichgans, J.; Weller, M. Betulinic acid-induced apoptosis in glioma cells: A sequential requirement for new protein synthesis, formation of reactive oxygen species, and caspase processing. J. Pharmacol. Exp. Ther. 1999, 289, 1306–1312. [Google Scholar] [PubMed]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nunez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53- independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Fulda, S.; Debatin, K.M. Betulinic acid induces apoptosis through a direct effect on mitochondria in neuroectodermal tumors. Med. Pediatr. Oncol. 2000, 35, 616–618. [Google Scholar] [CrossRef]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Gao, Y.; Jia, Z.; Kong, X.; Li, Q.; Chang, D.Z.; Wei, D.; Le, X.; Suyun, H.; Huang, S.; Wang, L.; et al. Combining betulinic acid and mithramycin a effectively suppresses pancreatic cancer by inhibiting proliferation, invasion, and angiogenesis. Cancer Res. 2011, 71, 5182–5193. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liang, K.; Hou, C.; Piao, H.-L. Dichloroacetic acid and rapamycin synergistically inhibit tumor progression. J. Zhejiang Univ. B 2023, 24, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Bonner, M.Y.; Karlsson, I.; Rodolfo, M.; Arnold, R.S.; Vergani, E.; Arbiser, J.L. Honokiol bis-dichloroacetate (Honokiol DCA) demonstrates activity in vemurafenib-resistant melanoma in vivo. Oncotarget 2016, 7, 12857–12868. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ishiguro, R.H.; Ishiguro, M.; Toki, A.; Terunuma, H. Synergistic Anti-tumor Effect of Dichloroacetate and Ivermectin. Cureus 2022, 14, e21884. [Google Scholar] [CrossRef] [PubMed]
- Juarez, M.; Schcolnik-Cabrera, A.; Dueñas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar] [PubMed] [PubMed Central]
- Marco-Brualla, J.; de Miguel, D.; Martínez-Lostao, L.; Anel, A. DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL. J. Clin. Med. 2023, 12, 608. [Google Scholar] [CrossRef] [PubMed]
- Mironiuk-Puchalska, E.; Karatsai, O.; Żuchowska, A.; Wróblewski, W.; Borys, F.; Lehka, L.; Rędowicz, M.J.; Koszytkowska-Stawińska, M. Development of 5-fluorouracil-dichloroacetate mutual prodrugs as anticancer agents. Bioorganic Chem. 2023, 140, 106784. [Google Scholar] [CrossRef] [PubMed]
- Korga, A.; Ostrowska, M.; Iwan, M.; Herbet, M.; Dudka, J. Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes HepG2 cells to doxorubicin treatment. FEBS Open Bio 2019, 9, 959–972. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer Ther. 2018, 17, 2004–2012. [Google Scholar] [CrossRef]
- Fekir, K.; Dubois-Pot-Schneider, H.; Désert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; et al. Retrodifferentiation of Human Tumor Hepatocytes to Stem Cells Leads to Metabolic Reprogramming and Chemoresistance. Cancer Res. 2019, 79, 1869–1883. [Google Scholar] [CrossRef]
- Skeberdytė, A.; Sarapinienė, I.; Aleksander-Krasko, J.; Stankevičius, V.; Sužiedėlis, K.; Jarmalaitė, S. Dichloroacetate and Salinomycin Exert a Synergistic Cytotoxic Effect in Colorectal Cancer Cell Lines. Sci. Rep. 2018, 8, 17744. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-Y.; Huang, W.-Y.; Lin, C.-L.; Huang, T.-C.; Wu, Y.-Y.; Chen, J.-H.; Kao, C.-H. Propranolol Reduces Cancer Risk: A population-based cohort study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol. Med. Rep. 2018, 17, 5213–5221. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, H.A.; Al-Wadei, M.H.; Schuller, H.M. Prevention of pancreatic cancer by the beta-blocker propranolol. Anti-Cancer Drugs 2009, 20, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Ma, Q.; Wang, L.; Hu, H.; Li, J.; Zhang, D.; Zhang, M. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol. Rep. 2009, 22, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Propranolol as an anti-cancer agent. Ecancermedicalscience 2016, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Lucido, C.T.; Miskimins, W.K.; Vermeer, P.D. Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC. Cancers 2018, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Guha, I.; Chatterjee, A.; Banerji, A. Anti-cancer Potential of All-trans Retinoic Acid (ATRA): A Review. Proc. Zool. Soc. 2013, 66, 1–7. [Google Scholar] [CrossRef]
- Hoffman, E.; Mielicki, W. All-Trans Retinoic Acid (ATRA) in Prevention and Cancer Therapy. Advances in Hygiene and Experimental Medicine. 9 June 2010. Available online: http://www.phmd.pl/fulltxt.php?ICID=911863 (accessed on 12 June 2023).
- Abildgaard, C.; Dahl, C.; Abdul-Al, A.; Christensen, A.; Guldberg, P. Inhibition of retinoic acid receptor β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells. Oncotarget 2017, 8, 84210–84223. [Google Scholar] [CrossRef]
- Dong, G.; Chen, Q.; Jiang, F.; Yu, D.; Mao, Q.; Xia, W.; Shi, R.; Wang, J.; Xu, L. Diisopropylamine dichloroacetate enhances radiosensitization in esophageal squamous cell carcinoma by increasing mitochondria-derived reactive oxygen species levels. Oncotarget 2016, 7, 68170–68178. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, J.; Ge, S.; Su, Y.; Fan, X. Novel insight into metabolic reprogrammming in cancer radioresistance: A promising therapeutic target in radiotherapy. Int. J. Biol. Sci. 2023, 19, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Toledo, G.F.; Nagamine, M.K.; Nowosh, V.; Machado, F.T.; Massoco, C.O.; Souza-Pinto, N.C.; Dagli, M.L.Z. Antineoplastic effects of sodium dichloroacetate and omeprazole, alone or in combination, on canine oral mucosal melanoma cells. Front. Vet. Sci. 2023, 10, 1186650. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Stanciu, D.; Devesa, J.; Alfarouk, K.; Cardone, R.A.; Polo Orozco, J.D.; Devesa, P.; Rauch, C.; Orive, G.; Anitua, E.; et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 2017, 43, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Castillejos-López, M.; Romero-García, S.; Aguayo, A.S.; Herrera, I.; Garcia-Martin, M.O.; Torres-Espíndola, L.M.; Negrete-García, M.C.; Olvera, A.C.; Huerta-Cruz, J.C.; et al. Antitumor Therapy under Hypoxic Microenvironment by the Combination of 2-Methoxyestradiol and Sodium Dichloroacetate on Human Non-Small-Cell Lung Cancer. Oxidative Med. Cell. Longev. 2020, 2020, 3176375. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Gálvez, A.; González-Ávila, G.; Delgado-Tello, J.; Castillejos-López, M.; Mendoza-Milla, C.; Zúñiga, J.; Checa, M.; Maldonado-Martínez, H.A.; Trinidad-López, A.; Cisneros, J.; et al. Effects of 2-methoxyestradiol on apoptosis and HIF-1α and HIF-2α expression in lung cancer cells under normoxia and hypoxia. Oncol. Rep. 2016, 35, 577–583. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hagen, T.; D’amico, G.; Quintero, M.; Palacios-Callender, M.; Hollis, V.; Lam, F.; Moncada, S. Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochem. Biophys. Res. Commun. 2004, 322, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhao, X.; Wang, K.; Liu, J.; Huang, G. Dichloroacetic acid (DCA) synergizes with the SIRT2 inhibitor Sirtinol and AGK2 to enhance anti-tumor efficacy in non-small cell lung cancer. Cancer Biol. Ther. 2018, 19, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Dyrstad, S.E.; Lotsberg, M.L.; Tan, T.Z.; Pettersen, I.K.N.; Hjellbrekke, S.; Tusubira, D.; Engelsen, A.S.T.; Daubon, T.; Mourier, A.; Thiery, J.P.; et al. Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells. Cancers 2021, 13, 941. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Inhibition of pyruvate dehydrogenase kinase 1 enhances the anti-cancer effect of EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Eur. J. Pharmacol. 2018, 838, 41–52. [Google Scholar] [CrossRef]
- Frisch, A.; Wang, Y.; Wang, Y.; Lontos, K.; Rivadeneira, D.; Delgoffe, G. 100 Redirecting glucose flux during in vitro expansion improves the in vivo performance of adoptive T cell therapies for cancer. J. Immunother. Cancer 2021, 9 (Suppl. 2), A109. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Karioti, A.; Antoniadi, G.; Antoniadis, N.; Liakopoulos, V.; Stefanidis, I. Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. J. Basic Clin. Physiol. Pharmacol. 2013, 24, 271–276. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Calcutt, N.A.; Lopez, V.L.; Bautista, A.D.; Mizisin, L.M.; Torres, B.R.; Shroads, A.L.; Mizisin, A.P.; Stacpoole, P.W. Peripheral Neuropathy in Rats Exposed to Dichloroacetate. J. Neuropathol. Exp. Neurol. 2009, 68, 985–993. [Google Scholar] [CrossRef]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Jhung, S.; Sano, M.C.; Shungu, D.C.; Millar, W.S.; Hong, X.; Gooch, C.L.; Mao, X.; et al. Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurology 2006, 66, 324–330. [Google Scholar] [CrossRef]
- Brandsma, D.; Dorlo, T.P.C.; Haanen, J.H.; Beijnen, J.H.; Boogerd, W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J. Neurol. 2010, 257, 2099–2100. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M. Trial of dichloroacetate in MELAS: Toxicity overshadows the assessment of potential benefit. Neurology 2006, 66, 302–303. [Google Scholar] [CrossRef]
- Felitsyn, N.; Stacpoole, P.W.; Notterpek, L. Dichloroacetate causes reversible demyelination in vitro: Potential mechanism for its neuropathic effect. J. Neurochem. 2007, 100, 429–436. [Google Scholar] [CrossRef]
- Deck, M.; Van Hameren, G.; Campbell, G.; Bernard-Marissal, N.; Devaux, J.; Berthelot, J.; Lattard, A.; Médard, J.-J.; Gautier, B.; Guelfi, S.; et al. Physiology of PNS axons relies on glycolytic metabolism in myelinating Schwann cells. PLoS ONE 2022, 17, e0272097. [Google Scholar] [CrossRef]
- Babetto, E.; Wong, K.M.; Beirowski, B. A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 2020, 23, 1215–1228. [Google Scholar] [CrossRef]
- Bouçanova, F.; Chrast, R. Metabolic Interaction Between Schwann Cells and Axons Under Physiological and Disease Conditions. Front. Cell. Neurosci. 2020, 14, 148. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Martyniuk, C.J.; James, M.O.; Calcutt, N.A. Dichloroacetate-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 211–238. [Google Scholar] [CrossRef] [PubMed]
- Shroads, A.L.; Langaee, T.; Coats, B.S.; Kurtz, T.L.; Bullock, J.R.; Weithorn, D.; Gong, Y.; Wagner, D.A.; Ostrov, D.A.; Johnson, J.A.; et al. Human Polymorphisms in the Glutathione Transferase Zeta 1/Maleylacetoacetate Isomerase Gene Influence the Toxicokinetics of Dichloroacetate. J. Clin. Pharmacol. 2012, 52, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Washington, J.T.; Quintyne, N.J. Dichloroacetate Induces Different Rates of Cell Death in Cancer and Noncancer Cell Lines in Vitro. Tumori J. 2012, 98, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.J.; Sanchez, I.M.; Nelson, M.A.; Larson, J.L.; Lansing, A.J. Liver tumor induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology 1990, 63, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Bull, R. Metabolism and lipoperoxidative activity of trichloroacetate and dichloroacetate in rats and mice. Toxicol. Appl. Pharmacol. 1992, 115, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Schwab, S.; Efferth, T. Fatal Liver and Bone Marrow Toxicity by Combination Treatment of Dichloroacetate and Artesunate in a Glioblastoma Multiforme Patient: Case Report and Review of the Literature. Front. Oncol. 2016, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.M.; Bull, R.J. Early induction of reparative hyperplasia in the liver of B6C3F1 mice treated with dichloroacetate and trichloroacetate. Toxicology 1990, 64, 33–46. [Google Scholar] [CrossRef]
- Guo, X.; Dixit, V.; Liu, H.; Shroads, A.L.; Henderson, G.N.; James, M.O.; Stacpoole, P.W. Inhibition and recovery of rat hepatic glutathiones-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab. Dispos. 2006, 34, 36–42. [Google Scholar] [CrossRef]
- Hassoun, E.A.; Dey, S. Dichloroacetate- and trichloroacetate-induced phagocytic activation and production of oxidative stress in the hepatic tissues of mice after acute exposure. J. Biochem. Mol. Toxicol. 2008, 22, 27–34. [Google Scholar] [CrossRef]
- Hassoun, E.; Cearfoss, J.; Mamada, S.; Al-Hassan, N.; Brown, M.; Heimberger, K.; Liu, M.-C. The Effects of Mixtures of Dichloroacetate and Trichloroacetate on Induction of Oxidative Stress in Livers of Mice After Subchronic Exposure. J. Toxicol. Environ. Health Part A 2014, 77, 313–323. [Google Scholar] [CrossRef] [PubMed]
- James, M.O.; Stacpoole, P.W. Pharmacogenetic Considerations with Dichloroacetate Dosing. Pharmacogenomics 2016, 17, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial involvement in cisplatin resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, F.; Rocco, M.; Castelli, S.; Di Gennaro, E.; Desideri, A.; Budillon, A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol. Cancer Ther. 2009, 8, 3075–3087. [Google Scholar] [CrossRef]
- Su, L.; Zhang, H.; Yan, C.; Chen, A.; Meng, G.; Wei, J.; Yu, D.; Ding, Y. Superior anti-tumor efficacy of diisopropylamine dichloroacetate compared with dichloroacetate in a subcutaneous transplantation breast tumor model. Oncotarget 2016, 7, 65721–65731. [Google Scholar] [CrossRef] [PubMed]
- She, W.; Liu, T.; Li, H.; Wang, Z.; Guo, Z.; Liu, Y.; Liu, Y. Reprogramming Energy Metabolism with Synthesized PDK Inhibitors Based on Dichloroacetate Derivatives and Targeted Delivery Systems for Enhanced Cancer Therapy. J. Med. Chem. 2023, 66, 14683–14699. [Google Scholar] [CrossRef] [PubMed]
- Trapella, C.; Voltan, R.; Melloni, E.; Tisato, V.; Celeghini, C.; Bianco, S.; Fantinati, A.; Salvadori, S.; Guerrini, R.; Secchiero, P.; et al. Design, Synthesis, and Biological Characterization of Novel Mitochondria Targeted Dichloroacetate-Loaded Compounds with Antileukemic Activity. J. Med. Chem. 2016, 59, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Fereidoonnezhad, M.; Tabaei, S.M.H.; Sakhteman, A.; Seradj, H.; Faghih, Z.; Faghih, Z.; Mojaddami, A.; Sadeghian, B.; Rezaei, Z. Design, synthesis, molecular docking, biological evaluations and QSAR studies of novel dichloroacetate analogues as anticancer agent. J. Mol. Struct. 2020, 1221, 128689. [Google Scholar] [CrossRef]
- Yang, Y.; Shang, P.; Cheng, C.; Wang, D.; Yang, P.; Zhang, F.; Li, T.; Lu, A.; Zhao, Y. Novel N-phenyl dichloroacetamide derivatives as anticancer reagents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2010, 45, 4300–4306. [Google Scholar] [CrossRef]
- Zajac, J.; Kostrhunova, H.; Novohradsky, V.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Potentiation of mitochondrial dysfunction in tumor cells by conjugates of metabolic modulator dichloroacetate with a Pt(IV) derivative of oxaliplatin. J. Inorg. Biochem. 2016, 156, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wu, X.Y.; Luo, O.Y.; Su, L.; Ding, Y.T.; Jiang, Y.; Yu, D.C. Diisopropylamine dichloroacetate alleviates liver fibrosis through inhibiting activation and proliferation of hepatic stellate cells. Int. J. Clin. Exp. Med. 2019, 12, 3440–3448. Available online: https://e-century.us/files/ijcem/12/4/ijcem0079902.pdf (accessed on 12 June 2023).
- Yamane, K.; Indalao, I.L.; Chida, J.; Yamamoto, Y.; Hanawa, M.; Kido, H. Diisopropylamine Dichloroacetate, a Novel Pyruvate Dehydrogenase Kinase 4 Inhibitor, as a Potential Therapeutic Agent for Metabolic Disorders and Multiorgan Failure in Severe Influenza. PLoS ONE 2014, 9, e98032. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Marier, D.; Marsden, E.; Andrews, D.; Eliaz, I. A novel form of dichloroacetate therapy for patients with advanced cancer: A report of 3 cases. Altern Ther. Health Med. 2014, 20 (Suppl. 2), 21–28. [Google Scholar] [PubMed]
- Khan, A.; Ghen, M. A 15 Year Evolution of Dichloroacetate-Based Metabolic Cancer Therapy: A Review with Case Reports. Med. Res. Arch. 2023, 11, 4118. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ishiguro, R.; Ishiguro, M.; Iwai, S. Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: In vivo experiments and a case report. Hepato-Gastroenterology 2012, 59, 994–996. [Google Scholar] [PubMed]
- Lemmo, W.; Tan, G. Prolonged Survival After Dichloroacetate Treatment of Non-Small-Cell Lung Carcinoma-Related Leptomeningeal Carcinomatosis. J. Med. Cases 2016, 7, 136–142. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Christofk, H.R.; Hosmer, W.; Britten, C.D.; Bahng, A.; Crabtree, M.J.; Hong, C.S.; Kamranpour, N.; Pitts, S.; Kabbinavar, F.; et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 443–452. [Google Scholar] [CrossRef]
- Heshe, D.; Hoogestraat, S.; Brauckmann, C.; Karst, U.; Boos, J.; Lanvers-Kaminsky, C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother. Pharmacol. 2011, 67, 647–655. [Google Scholar] [CrossRef]
- Shahrzad, S.; Lacombe, K.; Adamcic, U.; Minhas, K.; Coomber, B.L. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett. 2010, 297, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Jajeh, J.; Guinan, P.; Rubenstein, M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer. Res. 2009, 29, 4579–4588. [Google Scholar] [PubMed]
- Zwicker, F.; Kirsner, A.; Peschke, P.; Roeder, F.; Debus, J.; Huber, P.; Weber, K. Dichloroacetate induces tumor-specific radiosensitivity in vitro but attenuates radiation-induced tumor growth delay in vivo. Strahlenther. Onkol. 2013, 189, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, B.; Seidl, C.; Pirsig, S.; Bruchelt, G.; Senekowitsch-Schmidtke, R. DCA promotes progression of neuroblastoma tumors in nude mice. Am. J. Cancer Res. 2015, 5, 812. [Google Scholar] [PubMed]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Sounidaki, M.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. In human alloreactive CD4+ T-cells, dichloroacetate inhibits aerobic glycolysis, induces apoptosis and favors differentiation towards the regulatory T-cell subset instead of effector T-cell subsets. Mol. Med. Rep. 2016, 13, 3370–3376. [Google Scholar] [CrossRef] [PubMed]
- Rostamian, H.; Khakpoor-Kooshe, M.; Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Tavassolifar, M.J.; Pawelek, J.M.; Mirzaei, H.R.; Hadjati, J. Restricting Tumor Lactate Metabolism using Dichloroacetate Improves T Cell Functions. BMC Cancer 2022, 22, 39. [Google Scholar] [CrossRef]
- Schoonjans, C.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-induced metabolic reprogramming in tumor cells enhances the anti-proliferative activity of the PDK inhibitor dichloroacetate. Cancer Lett. 2020, 470, 18–28. [Google Scholar] [CrossRef]
- Ferriero, R.; Iannuzzi, C.; Manco, G.; Brunetti-Pierri, N. Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate. J. Inherit. Metab. Dis. 2015, 38, 895–904. [Google Scholar] [CrossRef]
- Milić, N.; Dolinar, K.; Miš, K.; Matkovič, U.; Bizjak, M.; Pavlin, M.; Podbregar, M.; Pirkmajer, S. Suppression of Pyruvate Dehydrogenase Kinase by Dichloroacetate in Cancer and Skeletal Muscle Cells Is Isoform Specific and Partially Independent of HIF-1α. Int. J. Mol. Sci. 2021, 22, 8610. [Google Scholar] [CrossRef]
- Tiersma, J.F.; Evers, B.; Bakker, B.M.; Jalving, M.; de Jong, S. Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities. Int. J. Mol. Sci. 2022, 23, 3745. [Google Scholar] [CrossRef] [PubMed]
- Gang, B.P.; Rooke, M.; Sun, R.C.; Banskota, S.; Mishra, S.; Dahlstrom, J.E.; Blackburn, A.C. Pyruvate dehydrogenase kinase expression profile is a biomarker for cancer sensitivity to dichloroacetate-mediated growth inhibition. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Hardy, M.; Topchyan, P.; Zander, R.; Volberding, P.; Cui, W.; Kalyanaraman, B. Potent inhibition of tumour cell proliferation and immunoregulatory function by mitochondria-targeted atovaquone. Sci. Rep. 2020, 10, 17872. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Segaran, A.; Lord, S. Targeting OXPHOS and the electron transport chain in cancer; Molecular and therapeutic implications. Semin. Cancer Biol. 2022, 86, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Martinelli, R.P.; Rozados, V.R.; Scharovsky, O.G. Use of sodium dichloroacetate for cancer treatment: A scoping review. Medicina 2024, 84, 313. [Google Scholar] [PubMed]
- Gurtu, V.; Webster, L.; Kinnaird, A.; Boukouris, A.; White, C.; Nagendran, J.; Freed, D.H.; Wilkins, M.R.; Michelakis, E. Targeting Pyruvate Dehydrogenase Kinase with Dichloroacetate in Pulmonary Arterial Hypertension: A Variable Clinical Response Driven by Genetic Polymorphisms in Sirtuin-3 and Uncoupling Protein-2. Circulation 2017, 136 (Suppl. 1), A17443. [Google Scholar]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Parczyk, J.; Ruhnau, J.; Pelz, C.; Schilling, M.; Wu, H.; Piaskowski, N.N.; Eickholt, B.; Kühn, H.; Danker, K.; Klein, A. Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer 2021, 21, 481. [Google Scholar] [CrossRef]
- Li, S.; Wu, L.; Feng, J.; Li, J.; Liu, T.; Zhang, R.; Xu, S.; Cheng, K.; Zhou, Y.; Zhou, S.; et al. In vitro and in vivo study of epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic hepatocellular carcinoma cells involving inhibition of phosphofructokinase activity. Sci. Rep. 2016, 6, 28479. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Jiang, Z.; Luo, Y.; Tian, D.; Song, T.; Li, Q. PPP3CB Inhibits Cell Proliferation and the Warburg Effect in Bladder Cancer by Blocking PDHK1. Front. Biosci. 2024, 29, 48. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jiang, Y.; Yan, X.; Dai, X.; Huang, C.; Chen, L.; Li, T.; Zhang, Y.; Xiao, H.; Yang, M.; et al. Dichloroacetate enhances the anti-tumor effect of sorafenib via modulating the ROS-JNK-Mcl-1 pathway in liver cancer cells. Exp. Cell Res. 2021, 406, 112755. [Google Scholar] [CrossRef] [PubMed]
- Babu, E.; Ramachandran, S.; CoothanKandaswamy, V.; Elangovan, S.; Prasad, P.D.; Ganapathy, V.; Thangaraju, M. Role of SLC5A8, a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate. Oncogene 2011, 30, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Kailavasan, M.; Rehman, I.; Reynolds, S.; Bucur, A.; Tozer, G.; Paley, M. NMR-based evaluation of the metabolic profile and response to dichloroacetate of human prostate cancer cells. NMR Biomed. 2014, 27, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W.; Harris, R.A. Mechanism responsible for the hypoglycemic actions of dichloroacetate and 2-chloropropionate. Arch. Biochem. Biophys. 1979, 198, 145–152. [Google Scholar] [CrossRef]
- Katayama, Y.; Kawata, Y.; Moritoh, Y.; Watanabe, M. Dichloroacetate, a pyruvate dehydrogenase kinase inhibitor, ameliorates type 2 diabetes via reduced gluconeogenesis. Heliyon 2022, 8, e08889. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate Prevents and Reverses Pulmonary Hypertension by Inducing Pulmonary Artery Smooth Muscle Cell Apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.W.; Nance, S. Selective reduction of fatty acid oxidation in colonocytes: Correlation with ulcerative colitis. Lipids 1990, 25, 646–652. [Google Scholar] [CrossRef]
- Hayek, A.; Woodside, W.F. Short-term influences of dichloroacetate on genetically hyperlipemic rats. Metabolism 1980, 29, 120–124. [Google Scholar] [CrossRef]
- Nayak, M.K.; Dhanesha, N.; Doddapattar, P.; Rodriguez, O.; Sonkar, V.K.; Dayal, S.; Chauhan, A.K. Dichloroacetate, an inhibitor of pyruvate dehydrogenase kinases, inhibits platelet aggregation and arterial thrombosis. Blood Adv. 2018, 2, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Min, B.-K.; Oh, C.J.; Park, S.; Lee, J.-M.; Go, Y.; Park, B.-Y.; Kang, H.-J.; Kim, D.W.; Kim, J.-E.; Yoo, E.K.; et al. Therapeutic effect of dichloroacetate against atherosclerosis via hepatic FGF21 induction mediated by acute AMPK activation. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Horne, A.W.; Ahmad, S.F.; Carter, R.; Simitsidellis, I.; Greaves, E.; Hogg, C.; Morton, N.M.; Saunders, P.T.K. Repurposing dichloroacetate for the treatment of women with endometriosis. Proc. Natl. Acad. Sci. USA 2019, 116, 25389–25391. [Google Scholar] [CrossRef] [PubMed]
- Miquel, E.; Cassina, A.; Martinez-Palma, L.; Bolatto, C.; Trias, E.; Gandelman, M.; Radi, R.; Barbeito, L.; Cassina, P. Modulation of Astrocytic Mitochondrial Function by Dichloroacetate Improves Survival and Motor Performance in Inherited Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e34776. [Google Scholar] [CrossRef] [PubMed]
- Shangraw, R.E.; Fisher, D.M. Pharmacokinetics and pharmacodynamics of dichloroacetate in patients with cirrhosis*1. Clin. Pharmacol. Ther. 1999, 66, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Shangraw, R.E.; Jahoor, F. Mechanism of dichloroacetate-induced hypolactatemia in humans with or without cirrhosis. Metabolism 2004, 53, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.C.; Solayman, M.H.M.; Lorenzo, R.J.; Langaee, T.; Stacpoole, P.W.; James, M.O. GSTZ1 expression and chloride concentrations modulate sensitivity of cancer cells to dichloroacetate. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 1202–1210. [Google Scholar] [CrossRef]
- Kim, C.J.; Terado, T.; Tambe, Y.; Mukaisho, K.-I.; Kageyama, S.; Kawauchi, A.; Inoue, H. Cryptotanshinone, a novel PDK 4 inhibitor, suppresses bladder cancer cell invasiveness via the mTOR/β-catenin/N-cadherin axis. Int. J. Oncol. 2021, 59, 5220. [Google Scholar] [CrossRef]
- Tambe, Y.; Terado, T.; Kim, C.J.; Mukaisho, K.; Yoshida, S.; Sugihara, H.; Tanaka, H.; Chida, J.; Kido, H.; Yamaji, K.; et al. Antitumor activity of potent pyruvate dehydrogenase kinase 4 inhibitors from plants in pancreatic cancer. Mol. Carcinog. 2019, 58, 1726–1737. [Google Scholar] [CrossRef]
- Sestito, S.; Bacci, A.; Chiarugi, S.; Runfola, M.; Gado, F.; Margheritis, E.; Gul, S.; Riveiro, M.E.; Vazquez, R.; Huguet, S.; et al. Development of potent dual PDK1/AurA kinase inhibitors for cancer therapy: Lead-optimization, structural insights, and ADME-Tox profile. Eur. J. Med. Chem. 2021, 226, 113895. [Google Scholar] [CrossRef]
- Pai, S.; Yadav, V.K.; Kuo, K.-T.; Pikatan, N.W.; Lin, C.-S.; Chien, M.-H.; Lee, W.-H.; Hsiao, M.; Chiu, S.-C.; Yeh, C.-T.; et al. PDK1 Inhibitor BX795 Improves Cisplatin and Radio-Efficacy in Oral Squamous Cell Carcinoma by Downregulating the PDK1/CD47/Akt-Mediated Glycolysis Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 11492. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, C.; De Franco, M.; Carbone, D.; Bassani, D.; Pavan, M.; Cascioferro, S.; Parrino, B.; Cirrincione, G.; Dall’acqua, S.; Moro, S.; et al. 1,2,4-Amino-triazine derivatives as pyruvate dehydrogenase kinase inhibitors: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2023, 249, 115134. [Google Scholar] [CrossRef] [PubMed]
- Mitchel, J.; Bajaj, P.; Patil, K.; Gunnarson, A.; Pourchet, E.; Na Kim, Y.; Skolnick, J.; Pai, S.B. Computational Identification of Stearic Acid as a Potential PDK1 Inhibitor and In Vitro Validation of Stearic Acid as Colon Cancer Therapeutic in Combination with 5-Fluorouracil. Cancer Informatics 2021, 20, 11769351211065979. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Han, H.; Lin, F.; Yang, L.; Feng, L.; Lai, X.; Wen, Z.; Yang, M.; Wang, C.; Ma, Y.; et al. Novel shikonin derivatives suppress cell proliferation, migration and induce apoptosis in human triple-negative breast cancer cells via regulating PDK1/PDHC axis. Life Sci. 2022, 310, 121077. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody–drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 2022, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Sun, J.; Luan, W.; Hou, Z.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Natural product albiziabioside A conjugated with pyruvate dehydrogenase kinase inhibitor dichloroacetate to induce apoptosis-ferroptosis-M2-TAMs polarization for combined cancer therapy. J. Med. Chem. 2019, 62, 8760–8772. [Google Scholar] [CrossRef]
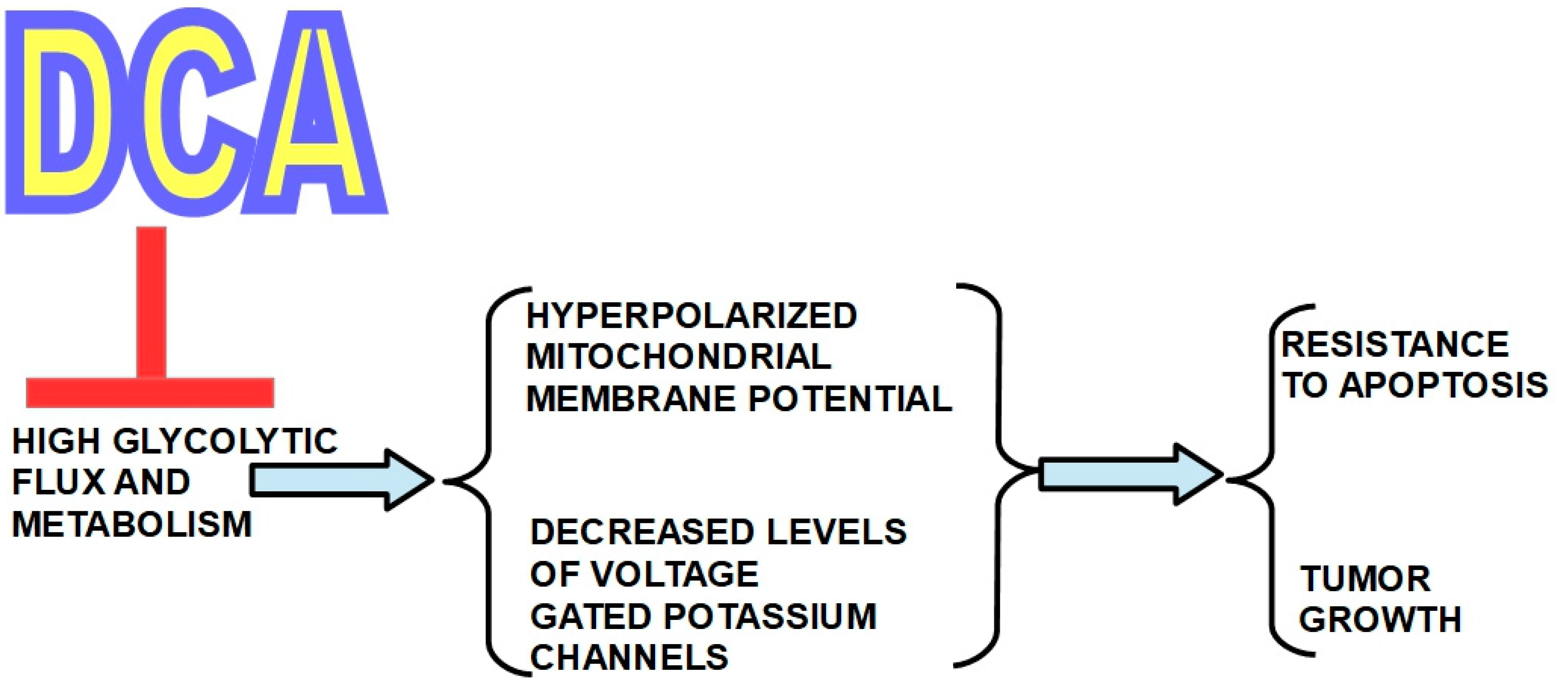

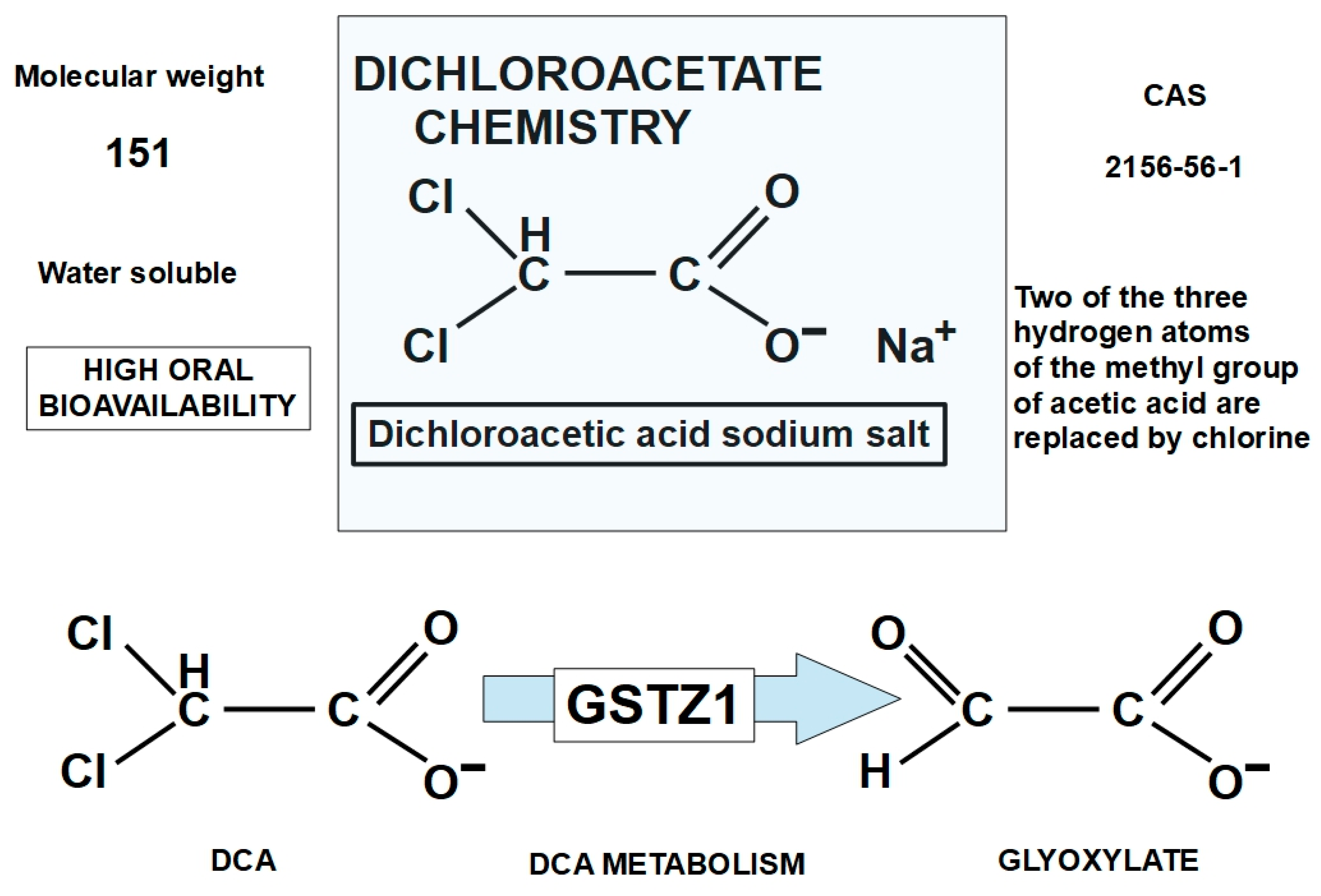
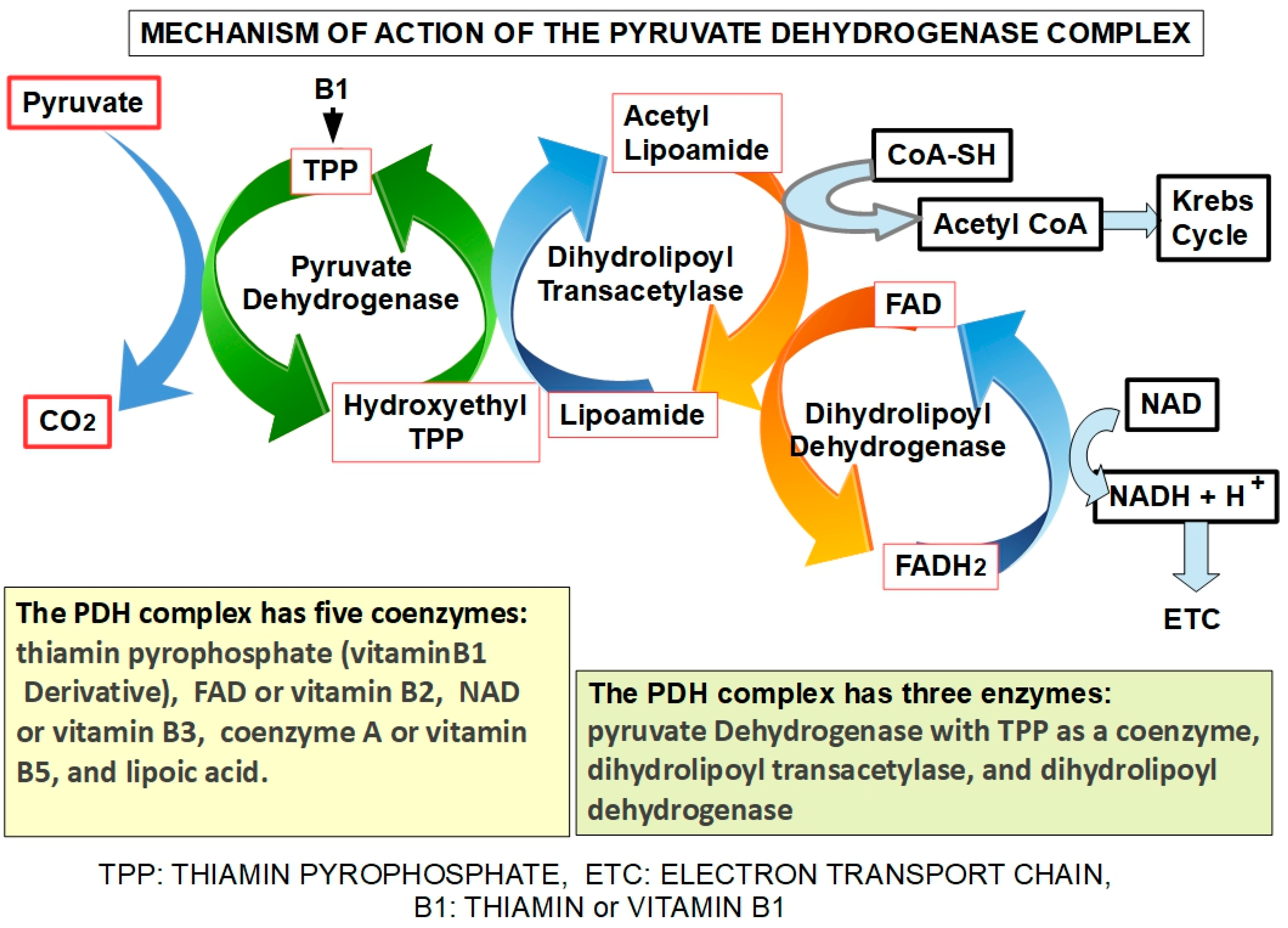

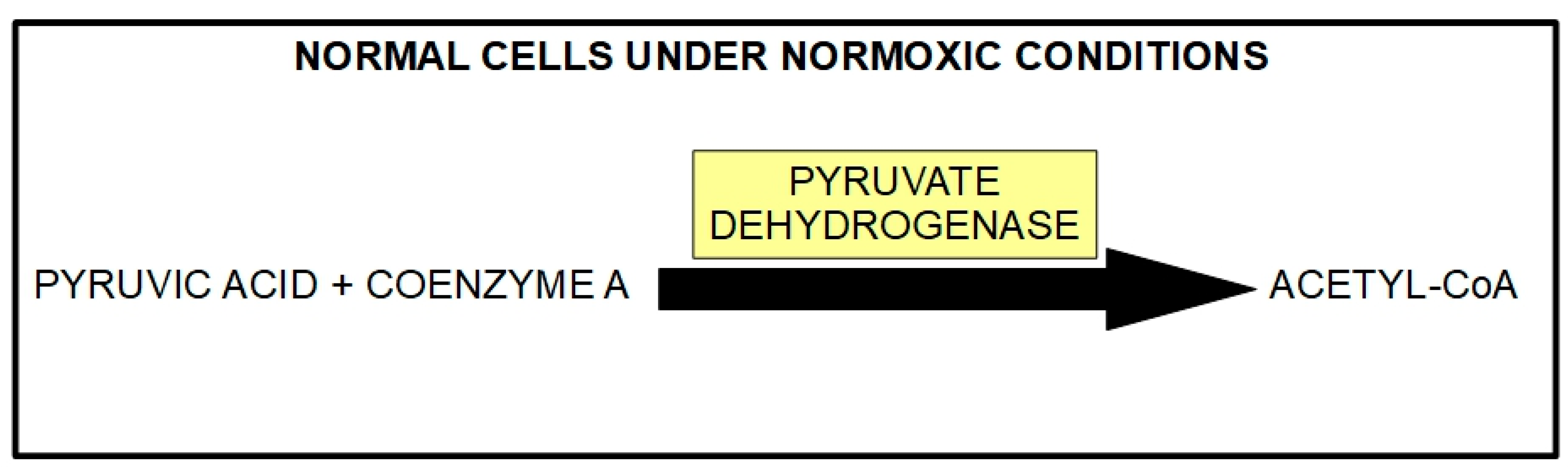
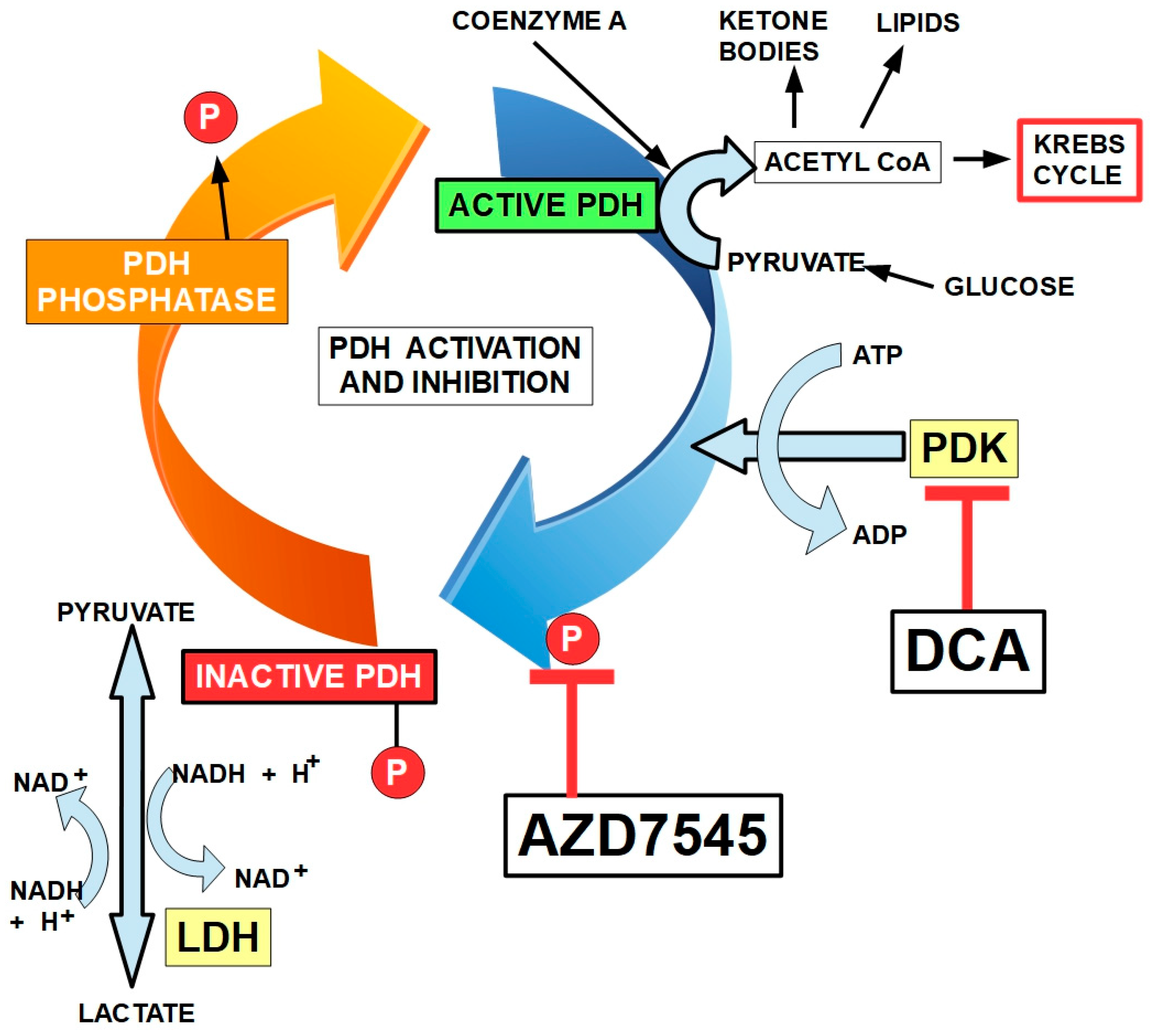
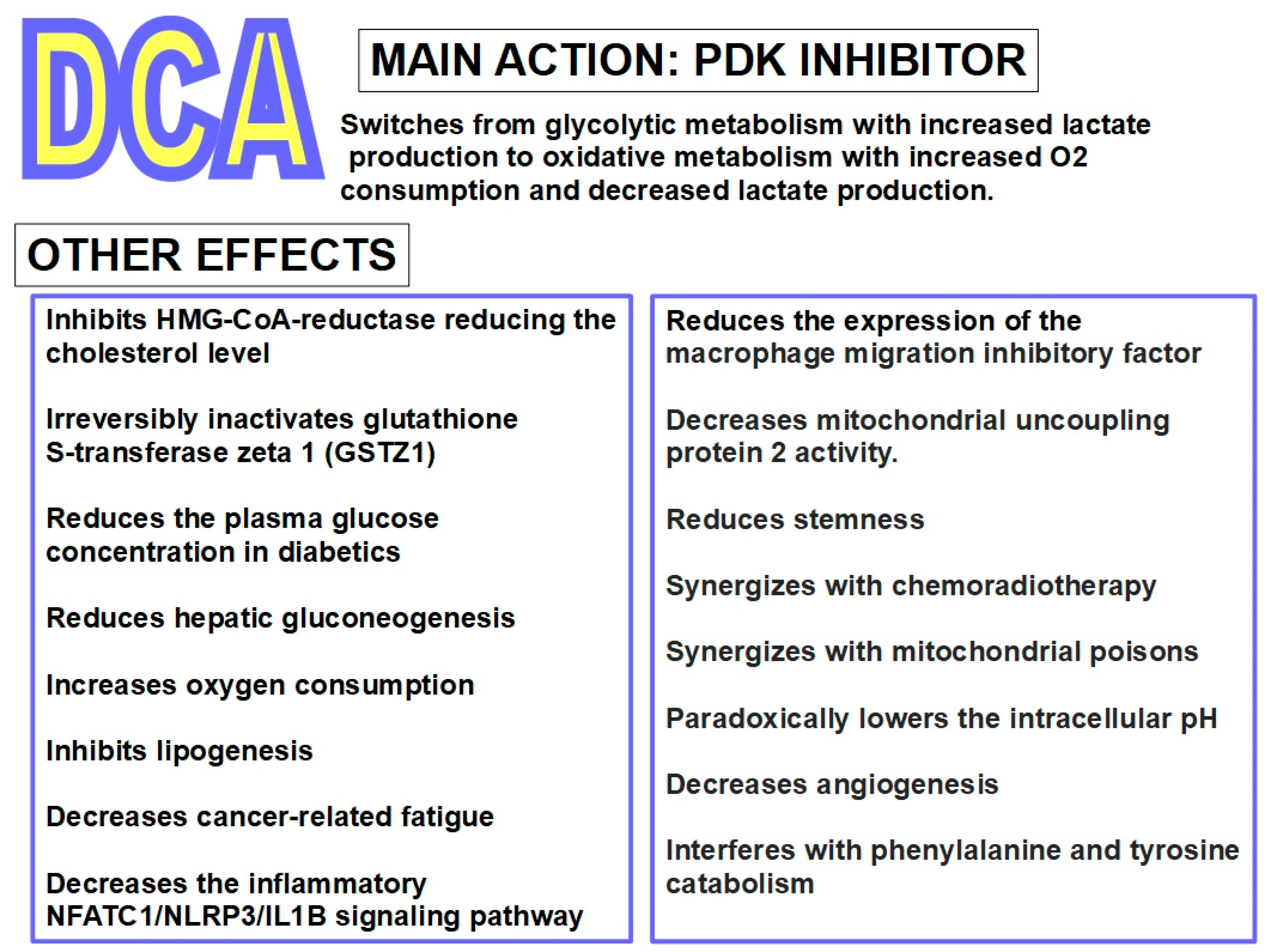
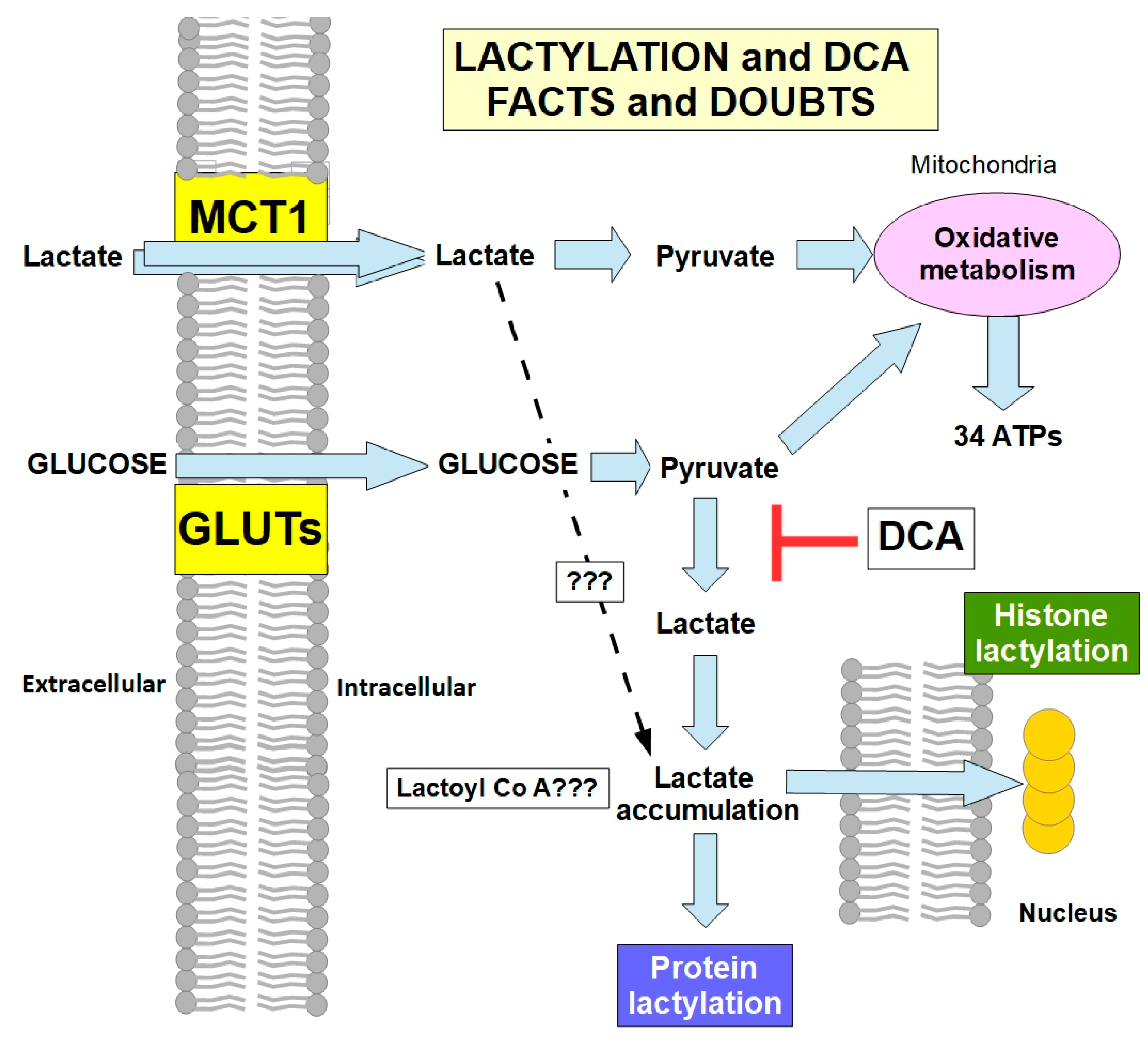
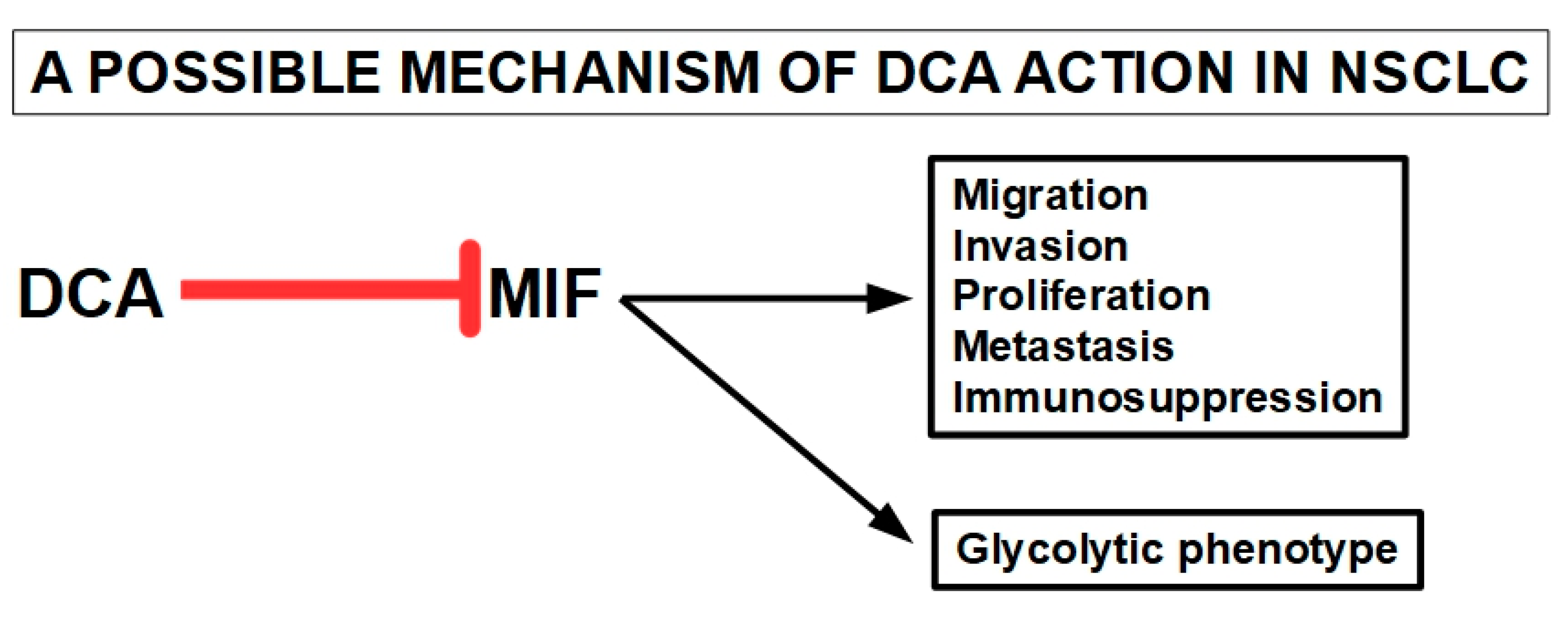

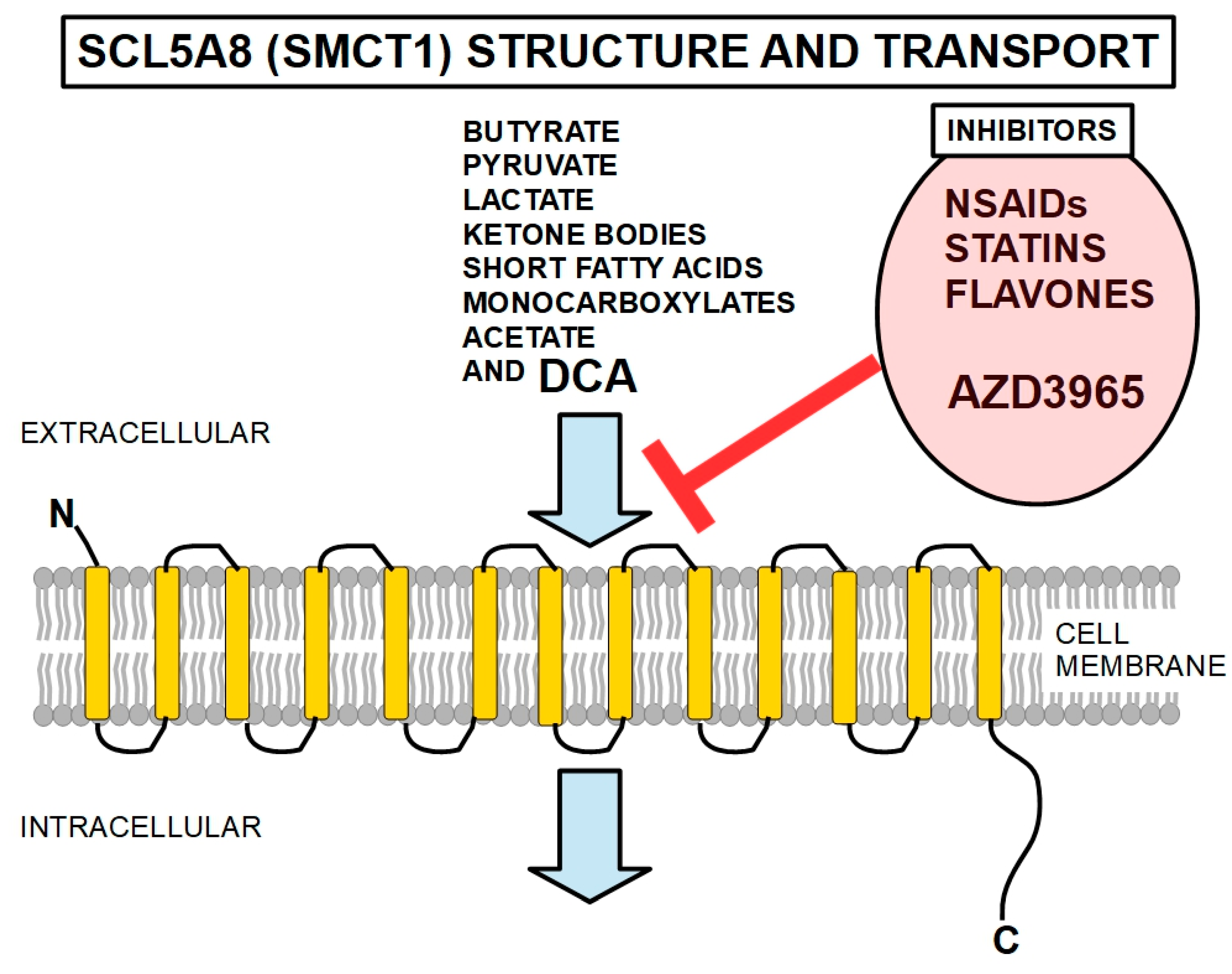
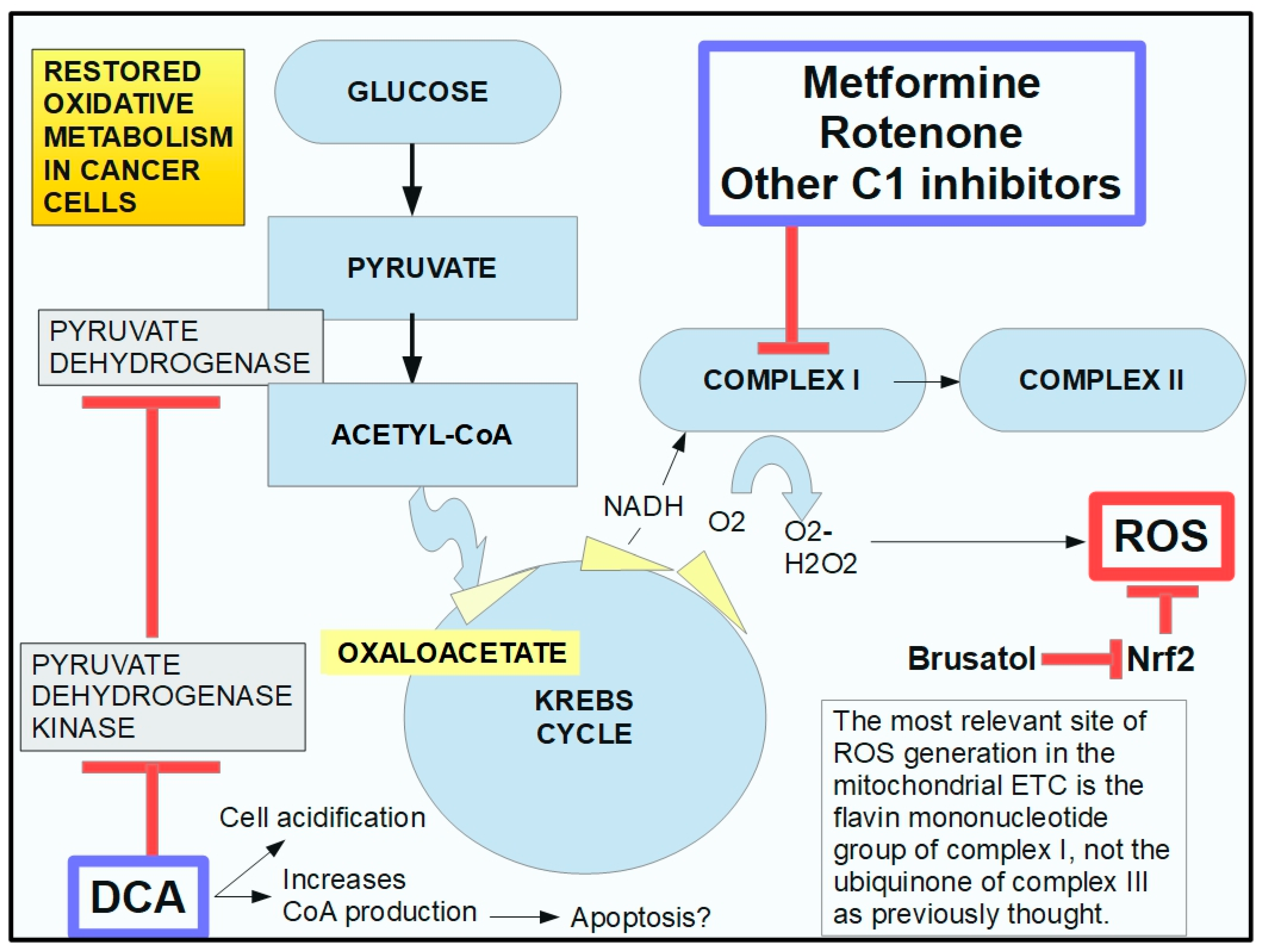

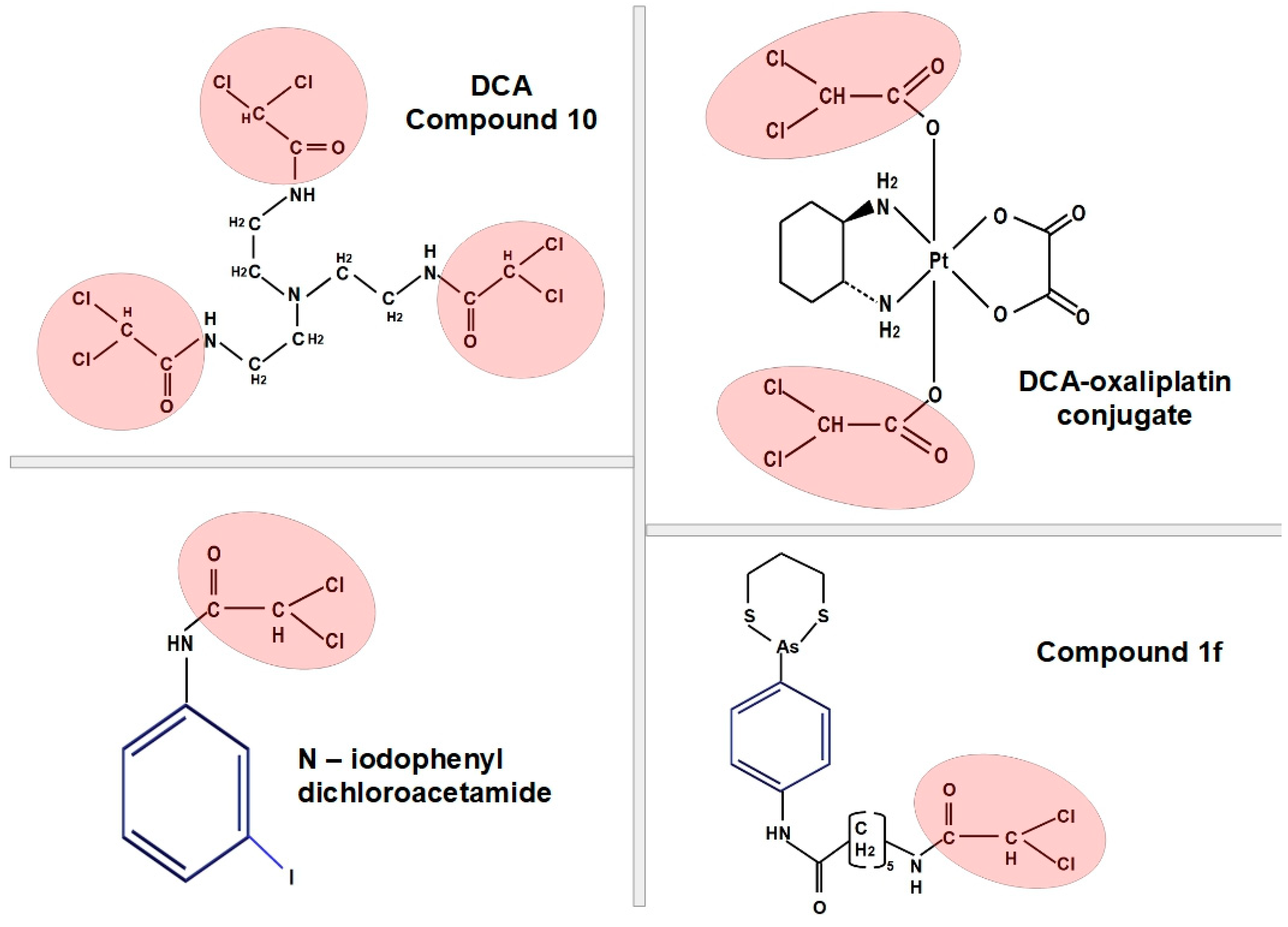


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Findings |
|---|---|
| Michelakis ED et al., 2010 [209] | The authors tested whether DCA can reverse cancer-specific metabolic and mitochondrial remodeling in glioblastoma. Freshly isolated glioblastomas from 49 patients showed mitochondrial hyperpolarization, which was rapidly reversed by DCA. A separate small experiment treated five patients with glioblastoma with oral DCA for up to 15 months. Three patients showed promising evidence of tumor regression; however, any conclusiuons need repetition with large-scale studies. DCA depolarized mitochondria, increased mitochondrial reactive oxygen species levels, and induced apoptosis in GBM cells, as well as in putative GBM stem cells. DCA therapy also inhibited the hypoxia-inducible factor–1α, promoted p53 activation, and suppressed angiogenesis both in vivo and in vitro. |
| Duan Y et al., 2013 [210] | DCA inhibited cell proliferation, induced apoptosis, and arrested C6 glioma cells in S phase. In vivo antitumor testing indicated that DCA markedly inhibited the growth of glioma tumors in brain tumor-bearing rats and tumor-bearing nude mice. DCA significantly induced ROS production and decreased the mitochondrial membrane potential in tumor tissues. Antiangiogenic effects were also found. |
| Kumar K et al., 2013 [211] | DCA synergistically enhanced the results of bevacizumab antiangiogenic treatment in a xenotransplanted mouse model of glioblastoma. |
| Morfouace M. et al., 2014 [212] | Oct4 is a major regulator of cell pluripotency. DCA increased the amount of Oct4–pyruvate kinase 2 complexes, which inhibited Oct4-dependent gene expression, inducing the differentiation of glioma stem cells. |
| Kolesnik DL et al., 2014 [213] | Hypoxia enhanced the cytotoxic effects of DCA on glioblastoma cells, inducing necrosis of tumor cells. |
| Vella et al., 2012 [214] | DCA had anticancer effects on NB (neuroblastoma) tumor cells, which were selectively directed to very malignant NB cells. More differentiated/less malignant NB cells were refractory to DCA treatment. |
| Sradhanjali S et al., 2017 [215] | DCA reduced retinoblastoma cell line and retinoblastoma explant growth and potentiated carboplatin-mediated inhibition of retinoblastoma cell growth. |
| Park JM et al., 2013 [216] | C6 glioma was transplanted into rat brains. Magnetic resonance imaging in vivo showed that DCA modified PDH flux much more in gliomas than in normal brains. |
| Fedorchuk AG et al., 2016 [217] | Experiments in rats with transplanted C6 glioma cells showed that different administration schedules of DCA could cause ambiguous effects: inhibition or stimulation of tumor growth. Prolonged daily administration showed the best antitumor effects. The anticancer efficacy of DCA was significantly increased under hypoxic conditions. |
| Wicks RT et al., 2014 [218] | Local delivery (wafers) of DCA to experimental brain tumors in rats caused significantly increased survival compared with controls and after oral administration of the drug. |
| Korsakova et al., 2021 [219] | The authors found synergistic apoptotic effects of metformin and DCA on glioblastomas cells in vitro and in vivo with dose-dependent cytotoxicity. Cytotoxic activity required DCA concentrations above 10 mM and metformin concentrations of 5 mM. There were no effects with concentrations of 5 mM DCA and 2.5 mM metformin. |
| Shen H et al., 2015 [220] | DCA increased radiosensitivity in orthotopic glioblastoma-bearing mice and of high-grade gliomas, also reviewed in Cook, 2021 [221]. |
| Jiang W et al., 2016 [222] | DCA increased cytotoxic activity of phenformin in glioblastoma stem cells. |
| Prokorhova IV et al., 2018 [223] | Co-administration of metformin with DCA improved anemia and thrombocytopenia produced by glioma C6 growth. |
| Kolesnik DL et al., 2019 [224] | It was found that metformin increased the cytotoxic activity of DCA against C6 glioma cells in vitro and in vivo. |
| Shen H et al., 2021 [225] | DCA associated with metformin and radiotherapy increased apoptosis in pediatric glioma cells in vitro and in vivo. The triple combination of DCA, metformin, and radiation therapy had more potent effects. |
| Yang Z et al., 2021 [226] | Phosphorylated PDH-A1 mediated tumor necrosis factor-α (TNF-α)-induced glioma cell migration. DCA decreased phosphorylated PDH-A1 levels and reduced glioma cell migration and invasion. |
| References | Organ/Cell | Findings |
|---|---|---|
| Ohashi T et al., 2013 [314] | Macrophages | DCA targets macrophages to suppress the activation of the IL-23/IL-17 pathway and prevents arginase 1 (ARG1) expression induced by lactic acid. Lactic acid-pretreated macrophages inhibited CD8+T-cell proliferation, but CD8+T-cell proliferation was restored when macrophages were pretreated with lactic acid and DCA. Although DCA treatment alone did not suppress tumor growth, it increased the antitumor immunotherapeutic activity. |
| Rooke M et al., 2014 [315] | Sarcoma models in vitro and in vivo | Three types of cells (mouse fibrosarcoma S180 cells, mouse osteosarcoma K7M2 cells and human fibrosarcoma HT1080-luc2 cells) were tested with DCA with and without doxorubicin. DCA alone significantly decreased viability at a concentration of 5 mM. At 0.5 mM, viability only decreased 15%. Importantly, there were no signs of apoptosis; therefore, DCA inhibited proliferation but did not induce apoptosis. DCA had additive effects with low concentrations of doxorubicin. |
| Ishiguro T et al., 2012 [316] | Fibrosarcoma and colon cancer cells, normal human fibroblasts | The combination of DCA and omeprazol exhibited more potent antitumor activity than DCA alone in tumor cells and did not affect the proliferation of normal cells. Caspase-dependent apoptosis through superoxide production was the suggested mechanism of action. |
| Sutendra G, et al., 2013 [317] | NSCLC and 2335 mammary cells | In a rat xenotransplanted model, DCA reduced NSCLC and breast cancer tumor vascularity in vivo. DCA also inhibited HIF-1 α in tumor cell lines. |
| El Sayed SM et al., 2019 [318] | Cancer cells | The authors propose that DCA is an antagonist of acetate, competing for enzymes, and thus its therapeutic action would be caused by acetate deprivation. |
| Khyzhnyak SV. et al., 2014 [319] | Mouse sarcoma | An examination of sarcoma mitochondria from mice under DCA treatment showed that they had a decrease in lactic acid levels and an increase in PDH activity with a decrease in the electron transport chain activity and an increase in ROS levels. |
| Choi YW et al. 2014 [320] | HeLa cells | DCA and metformin acted synergistically, enhancing cytotoxicity to cancer cells. |
| Xuan Y et al., 2014 [321] | Gastric cancer cells | DCA decreased resistance to 5-fluorouracil in highly hypoxic gastric cancer cells. |
| Badr MM et al., 2014 [322] | Fibrosarcoma | DCA showed immunomodulatory effects through the interleukin 12–interferon γ pathway. |
| Jin J. et al., 2022 [323] | Osteosarcoma cell lines | DCA mitochondria-targeting micelles induced pyroptosis, increasing the effects of immunotherapy. |
| Lam SK, et al., 2022 [324] | Mesothelioma | The combination of DCA with niclosamide blocked the growth and proliferation of different mesothelioma cells in vitro and in vivo. |
| Qin H et al., 2023 [325] | Cholangiocarcinoma | DCA increased sensitivity to cisplatin, changing glycolytic metabolism to oxidative metabolism and increasing ROS levels. Chloroquine further increased this sensitivity. |
| Dose in Humans | Concentration Range | Peak Concentration | Half-Life | |
|---|---|---|---|---|
| 0 | 19.9 µg/mL | 24.7 µg/mL | 0.16 mM | 20 min |
| IV 20 mg/kg | 57.3 µg/mL | 74.9 µg/mL | 0.49 mM | 36 min |
| Oral 25 mg/kg | 41 µg/mL (0.27 mM) (slow metabolizer) | |||
| Oral 25 mg/kg | 30 µg/mL (0.20 mM) (quick metabolizer) | |||
| Other sources | ||||
| IV 25 mg/kg | 130 µg/mL (0.86 mM) | |||
| After 5 infusions | 163 µg/mL (1.08 mM) | |||
| DOSE IN RATS | ||||
| 100 mg/Kg | 120 µg/mL | 164 µg/mL | 1.08 mM | 4 h |
| DOSE IN DOGS | ||||
| 100 mg/Kg | 447 µg/mL | 508 µg/mL | 3.36 mM | 24 h |
| DCA ALONE | |||
|---|---|---|---|
| Reference | Authors | DCA concentration | |
| [158] | Sun et al. | 5 mM | |
| [159] | Gang et al. | 5 mM | |
| [160] | Harting et al. | 10 mM | |
| [161] | De Preter et al. | 5 mM | |
| [170] | Xintaropoulou et al. | no response at 1 mM, response above 5 mM | |
| [178] | Cao et al. | 0.5 and 1 mM | |
| [180] | Harting et al. | 10 mM | |
| [184] | Lai et al. | 40 mM | |
| [187] | Madhok et al. | 20 mM | |
| [188] | Lin et al. | 100, 80, and 75 mM | |
| [189] | Delaney et al. | 20 and 50 mM | |
| [198] | Franco Molina et al. | 75 mM | |
| [209] | Michelakis et al. | 0.5 mM | |
| [210] | Duan et al. | 0 to 128 mM, response at 10 and 25 mM | |
| [214] | Vella et al. | 5 and 50 mM | |
| DCA CO-ADMINISTERED WITH OTHER DRUGS | |||
| Reference | Authors | DCA concentration | Association |
| [164] | Woo et al. | 20 mM | tamoxifen |
| [165] | Haugrud et al. | 0.5–5 mM | metformin |
| [166] | Sun et al. | 5 mM | arsenite trioxide |
| [167] | Verma et al. | 0.038 mM | arginase |
| [172] | DE Mey et al. | 30, 45, and 60 mM | radiosensitization |
| [181] | Zeng et al. | 5 mM | cisplatin |
| [182] | Olszewski et al. | 10 mM | cisplatin |
| [191] | Tong et al. | 0 to 90 mM | 5-FU |
| [192] | Liang et al. | 15 and 20 mM | 5-FU |
| [193] | Liang et al. | 15 and 20 mM | oxaliplatin |
| [201] | Abdilgaard et al. | 10 mM | BRAF inhibitor |
| [211] | Kumar et al. | 10 mM | bevacizumab |
| [213] | Kolesnik et al. | 35.8–42.3 mM | hypoxia |
| [219] | Korsakova et al. | 2.5, 5, 10, and 20 mM | metformin |
| [222] | Jiang et al. | 20 mM | phenformin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koltai, T.; Fliegel, L. Dichloroacetate for Cancer Treatment: Some Facts and Many Doubts. Pharmaceuticals 2024, 17, 744. https://doi.org/10.3390/ph17060744
Koltai T, Fliegel L. Dichloroacetate for Cancer Treatment: Some Facts and Many Doubts. Pharmaceuticals. 2024; 17(6):744. https://doi.org/10.3390/ph17060744
Chicago/Turabian StyleKoltai, Tomas, and Larry Fliegel. 2024. "Dichloroacetate for Cancer Treatment: Some Facts and Many Doubts" Pharmaceuticals 17, no. 6: 744. https://doi.org/10.3390/ph17060744
APA StyleKoltai, T., & Fliegel, L. (2024). Dichloroacetate for Cancer Treatment: Some Facts and Many Doubts. Pharmaceuticals, 17(6), 744. https://doi.org/10.3390/ph17060744