
Pharmacokinetics, Dose-Proportionality, and Tolerability of Intravenous Tanespimycin (17-AAG) in Single and Multiple Doses in Dogs: A Potential Novel Treatment for Canine Visceral Leishmaniasis

 ,
,  , , ,
, , ,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Study A—Dose-Escalation Protocol
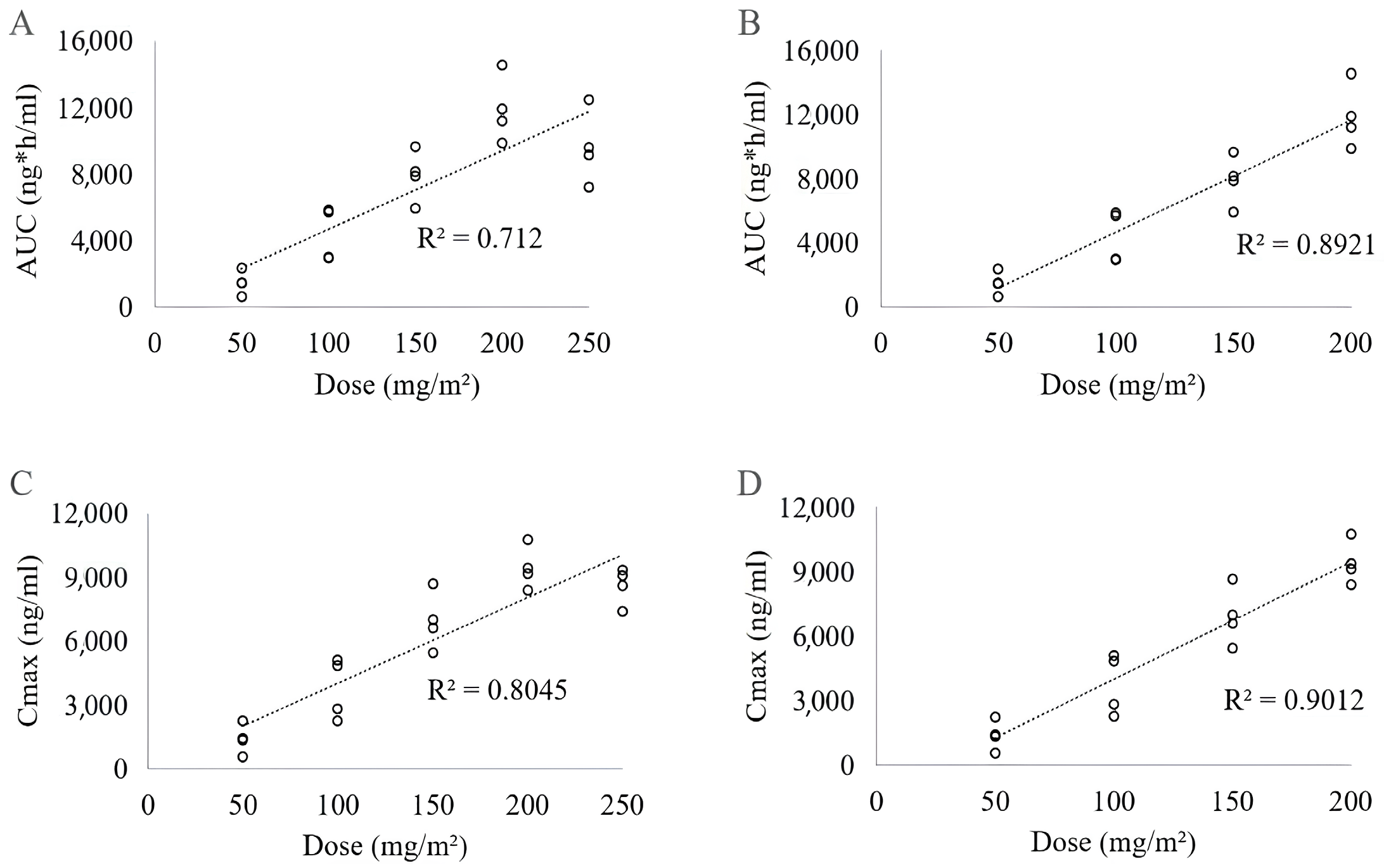
2.1.1. Evaluation of 17-AAG Pharmacokinetics in Plasma
2.1.2. Assessments of 17-AAG Tolerability
2.2. Study B–Multiple Administrations at a Single Dose of 150 mg/m2
2.2.1. Analysis of the Pharmacokinetics of 17-AAG in Plasma and Subcutaneous Interstitial Fluid
2.2.2. Assessments of 17-AAG Safety and Toxicity
2.2.3. Histopathology Analysis of Different Tissues of Dogs after Treatment
3. Discussion
4. Materials and Methods
4.1. Preparation of 17-AAG Formulation for Systemic Administration
4.2. Animal Selection and Care Procedures
4.3. Experiment Design of Pharmacological Studies
4.4. 17-AAG Pharmacokinetics Evaluation
4.4.1. Peripheral Blood Sample Collection
4.4.2. Collection of Subcutaneous Interstitial Liquid
4.4.3. Determination of 17-AAG Concentration by HPLC
4.4.4. Determination of 17-AAG Concentration by HPLC
4.5. 17-AAG Tolerability and Toxicity Assessments
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Travi, B.L.; Cordeiro-da-Silva, A.; Dantas-Torres, F.; Miró, G. Canine visceral leishmaniasis: Diagnosis and management of the reservoir living among us. PLoS Negl. Trop. Dis. 2018, 12, e0006082. [Google Scholar] [CrossRef] [PubMed]
- Teixeira-Neto, R.G.; Da Silva, E.S.; Nascimento, R.A.; Belo, V.S.; De Oliveira, C.D.L.; Pinheiro, L.C.; Gontijo, C.M.F. Canine visceral leishmaniasis in an urban setting of Southeastern Brazil: An ecological study involving spatial analysis. Parasit. Vectors 2014, 7, 485. [Google Scholar] [CrossRef] [PubMed]
- França-Silva, J.C.; Barata, R.A.; Costa, R.T.; Monteiro, E.M.; Machado-Coelho, G.L.L.; Vieira, E.P.; Prata, A.; Mayrink, W.; Nascimento, E.; Fortes-Dias, C.L.; et al. Importance of Lutzomyia longipalpis in the dynamics of transmission of canine visceral leishmaniasis in the endemic area of Porteirinha Municipality, Minas Gerais, Brazil. Vet. Parasitol. 2005, 131, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.S.; Macedo, L.O.; Oliveira, J.C.P.; Alves, L.C.; Carvalho, G.A.; Ramos, R.A.N. Canine visceral leishmaniasis: Risk factors and spatial analysis in an endemic area of Northeastern Brazil. Rev. Bras. Parasitol. Vet. 2023, 32, e003223. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.E.; Langton, D.A.; Hillman, T.J. Canine leishmaniosis in the United Kingdom: A zoonotic disease waiting for a vector? Vet. Parasitol. 2009, 163, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Morales-Yuste, M.; Martín-Sánchez, J.; Corpas-Lopez, V. Canine leishmaniasis: Update on epidemiology, diagnosis, treatment, and prevention. Vet. Sci. 2022, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Gontijo, C.M.F.; Melo, M.N. Visceral Leishmaniasis in Brazil: Current status, challenges and prospects. Rev. Bras. Epidemiol. 2004, 7, 338–349. [Google Scholar] [CrossRef]
- Alexander, J.; Satoskar, A.R.; Russell, D.G. Leishmania species: Models of intracellular parasitism. J. Cell Sci. 1999, 18, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Marzochi, M.C.; Coutinho, S.G.; De Souza, W.J.; De Toledo, L.M.; Grimaldi Júnior, G.; Momen, H.; Pacheco, R.S.; Sabroza, P.C.; Souza, M.A.; Rangel Júnior, F.B.; et al. Canine visceral leishmaniasis in Rio de Janeiro, Brazil. Clinical, parasitological, therapeutical and epidemiological findings (1977–1983). Mem. Inst. Oswaldo Cruz 1985, 80, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martínez, A.F.; Newman, S.; Ramanan, P.; Suarez, J.A. A review of leishmaniasis: Current knowledge and future directions. Curr. Trop. Med. Rep. 2021, 8, 121–132. [Google Scholar] [CrossRef]
- Azevedo, M.A.A.; Dias, A.K.K.; Paula, H.B.; Perri, S.H.V.; Nunes, C.M. Canine visceral leishmaniasis evaluation in Poxoréo, Mato Grosso State, Brazil. Rev. Bras. Parasitol. Vet. 2008, 17, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, P.H.P.; Santos, S.O.; Pinheiro, A.A.; Bittencourt, D.V.V.; Costa, R.L.G.; Julião, F.S.; Santos, W.L.C.; Barrouin-Melo, S.M. Clinical profile of naturally infected dogs from an endemic area for “Leishmania chagasi” (infantum) in Bahia State, Brazil. Rev. Bras. Saude Prod. An. 2007, 8, 283–294. [Google Scholar]
- Koutinas, A.F.; Polizopoulou, Z.S.; Saridomichelakis, M.N.; Argyriadis, D.; Fytianou, A.; Plevraki, K.G. Clinical considerations on canine visceral leishmaniasis in Greece: A retrospective study of 158 cases (1989–1996). J. Am. Anim. Hosp. Assoc. 1999, 35, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Ciaramella, P.; Oliva, G.; Luna, R.D.; Ambrosio, R.; Cortese, L.; Scalone, A.; Persechino, A. A retrospective clinical study of canine leishmaniasis in 150 dogs naturally infected by Leishmania infantum. Vet. Rec. 1997, 141, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Ivănescu, L.; Andronic, B.L.; Grigore-Hristodorescu, S.; Martinescu, G.V.; Mîndru, R.; Miron, L. The immune response in canine and human leishmaniasis and how this influences the diagnosis- a review and assessment of recent research. Front. Cell Infect. Microbiol. 2023, 13, 1326521. [Google Scholar] [CrossRef] [PubMed]
- Reguera, R.M.; Morán, M.; Pérez-Pertejo, Y.; García-Estrada, C.; Balaña-Fouce, R. Current status on prevention and treatment of canine leishmaniasis. Vet. Parasitol. 2016, 227, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.A.N.; Giannelli, A.; Fasquelle, F.; Scuotto, A.; Betbeder, D. Effective immuno-therapeutic treatment of canine leishmaniasis. PLoS Negl. Trop. Dis. 2023, 17, e0011360. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, F.S.; Avino, V.C.; Galvis-Ovallos, F.; Pereira-Chioccola, V.L.; Moreira, M.A.B.; Romariz, A.P.P.L.; Molla, L.M.; Menz, I. Use of miltefosine to treat canine visceral leishmaniasis caused by Leishmania infantum in Brazil. Parasit. Vectors 2019, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Yasur-Landau, D.; Jaffe, C.L.; David, L.; Baneth, G. Allopurinol resistance in Leishmania infantum from dogs with disease relapse. PLoS Negl. Trop. Dis. 2016, 10, e0004341. [Google Scholar] [CrossRef] [PubMed]
- Alvar, J.; Cañavate, C.; Molina, R.; Moreno, J.; Nieto, J. Canine leishmaniasis. Adv. Parasitol. 2004, 57, 1–88. [Google Scholar] [PubMed]
- Li, Z.N.; Luo, Y. HSP90 inhibitors and cancer: Prospects for use in targeted therapies (Review). Oncol. Rep. 2023, 49, 6. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Berman, D.; Murthy, B.; Jones, S. Tanespimycin pharmacokinetics: A randomized dose-escalation crossover phase 1 study of two formulations. Cancer Chemother. Pharmacol. 2011, 67, 1045–1054. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Egorin, M.J.; Eiseman, J.L.; Ramalingam, S.; Friedland, D.; Agarwala, S.S.; Ivy, S.P.; Potter, D.M.; Chatta, G.; Zuhowski, E.G.; et al. Phase I and pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with refractory advanced cancers. Clin. Cancer Res. 2007, 13, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Mitsiades, C.S.; Anderson, K.C.; Richardson, P.G. Tanespimycin as antitumor therapy. Clin. Lymphoma Myeloma Leuk. 2011, 11, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.; Abdallah, J.; Abou-Antoun, T. A novel mechanism of 17-AAG therapeutic efficacy on HSP90 inhibition in MYCN-amplified neuroblastoma cells. Front. Oncol. 2020, 10, 624560. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The Therapeutic Target Hsp90 and Cancer Hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.; Bona, R.; Li, Z. 17 AAG for HSP90 Inhibition in Cancer—From Bench to Bedside. Curr. Mol. Med. 2009, 9, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 1998, 42, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.P.; Ghosh, A. Hsp90 in human diseases: Molecular mechanisms to therapeutic approaches. Cells 2022, 11, 976. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Clos, J. No stress--Hsp90 and signal transduction in Leishmania. Parasitology 2014, 141, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.L.O.A.; Cull, B.; Dias, B.R.S.; Palma, L.C.; Luz, Y.S.; Menezes, J.P.B.; Mottram, J.C.; Veras, P.S.T. 17-AAG-induced activation of the autophagic pathway in leishmania is associated with parasite death. Microorganisms 2021, 9, 1089. [Google Scholar] [CrossRef] [PubMed]
- Palma, L.C.; Ferreira, L.F.G.R.; Petersen, A.L.O.A.; Dias, B.R.S.; Menezes, J.P.B.; Moreira, D.R.M.; Hernandes, M.Z.; Veras, P.S.T. A docking-based structural analysis of geldanamycin-derived inhibitor binding to human or Leishmania Hsp90. Sci. Rep. 2019, 9, 14756. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.L.O.A.; Guedes, C.E.S.; Versoza, C.L.; Lima, J.G.B.; Freitas, L.A.R.; Borges, V.M.; Veras, P.S.T. 17-AAG Kills Intracellular Leishmania amazonensis while Reducing Inflammatory Responses in Infected Macrophages. PLoS ONE 2012, 7, e49496. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ahmad, L.; Raza, F.; Khan, S.A.; Ali, T. The practical implication of clinical pharmacokinetics in drug development, pharmaceutical analysis, and clinical research. Front. Pharmacol. 2023, 14, 1252030. [Google Scholar] [CrossRef] [PubMed]
- Shawahna, R.; Shraim, N.; Aqel, R. Views, knowledge, and practices of hospital pharmacists about using clinical pharmacokinetics to optimize pharmaceutical care services: A cross-sectional study. BMC Health Serv. Res. 2022, 22, 411. [Google Scholar] [CrossRef]
- Usman, M.; Khadka, S.; Saleem, M.; Rasheed, H.; Kunwar, B.; Ali, M. Pharmacometrics: A new era of pharmacotherapy and drug development in low- and middle-income countries. Adv. Pharmacol. Pharm. Sci. 2023, 2023, 3081422. [Google Scholar] [CrossRef] [PubMed]
- Menon, G.; Okeke, C.; Krishnan, J. Modelling compartmentalization towards elucidation and engineering of spatial organization in biochemical pathways. Sci. Rep. 2017, 7, 12057. [Google Scholar] [CrossRef] [PubMed]
- Verrest, L.; Dorlo, T.P.C. Lack of clinical pharmacokinetic studies to optimize the treatment of neglected tropical diseases: A systematic review. Clin. Pharmacokinet. 2017, 56, 583–606. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Qiang, W.; Wen, Z.; Li, L.; Wang, L.; Chenga, Z. A new model to describe the single-dose pharmacokinetics of bevacizumab and predict its multiple-dose pharmacokinetics in beagle dogs. Iran J. Pharm. Res. 2019, 18, 1147–1155. [Google Scholar] [PubMed]
- Fan, J.; Lannoy, I.A.M. Pharmacokinetics. Biochem. Pharmacol. 2014, 87, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Dansirikul, C.; Choi, M.; Duffull, S.B. Estimation of pharmacokinetic parameters from non-compartmental variables using Microsoft Excel. Comput. Biol. Med. 2005, 35, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.Y.; Baverel, P.G.; Lythe, G.D.; Vicini, P.; Yates, J.W.T.; Dubois, V.F.S. Step-by-step comparison of ordinary differential equation and agent-based approaches to pharmacokinetic-pharmacodynamic models. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Samant, R.S.; Batista, S.; Larance, M.; Ozer, B.; Milton, C.I.; Bludau, I.; Wu, E.; Biggins, L.; Andrews, S.; Hervieu, A.; et al. Native size-exclusion chromatography–based mass spectrometry reveals new components of the early heat shock protein 90 inhibition response among limited global changes. Mol. Cell Proteom. 2023, 22, 100485. [Google Scholar] [CrossRef] [PubMed]
- Katragadda, U.; Fan, W.; Wang, Y.; Teng, Q.; Tan, C. Combined delivery of paclitaxel and tanespimycin via micellar nanocarriers: Pharmacokinetics, efficacy and metabolomic analysis. PLoS ONE 2013, 8, e58619. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.C.; Cho, H.; Lai, T.C.; Kozak, K.R.; Kolesar, J.M.; Kwon, G.S. Pharmacokinetic study of 3-in-1 poly(ethylene glycol)-block-poly(D, L-lactic acid) micelles carrying paclitaxel, 17-allylamino-17-demethoxygeldanamycin, and rapamycin. J. Control. Release 2012, 163, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.S.; Kim, G.P.; Foster, N.R.; Wang-Gillam, A.; Erlichman, C.; McWilliams, R.R. Phase II trial of gemcitabine and tanespimycin (17AAG) in metastatic pancreatic cancer: A mayo clinic phase II consortium study. Investig. New Drugs 2015, 33, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Erlichman, C. Tanespimycin: The opportunities and challenges of targeting heat shock protein 90. Expert Opin. Investig. Drugs 2009, 18, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Iyer, G.; Morris, M.J.; Rathkopf, D.; Slovin, S.F.; Steers, M.; Larson, S.M.; Schwartz, L.H.; Curley, T.; DeLaCruz, A.; Ye, Q.; et al. A phase I trial of docetaxel and pulse-dose 17-allylamino-17-demethoxygeldanamycin in adult patients with solid tumors. Cancer Chemother. Pharmacol. 2012, 69, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Chanan-Khan, A.A.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Mitsiades, C.S.; Anderson, K.C. Tanespimycin monotherapy in relapsed multiple myeloma: Results of a phase 1 dose-escalation study. Br. J. Haematol. 2010, 150, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Kenward, H.; Pelligand, L.; Savary-Bataille, K.; Elliott, J. Nausea: Current knowledge of mechanisms, measurement and clinical impact. Vet J. 2015, 203, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.S.; Silva, J.S.; Almeida, V.A.; Leal Junior, F.G.; Souza, P.A.N.; Larangeira, D.F.; Moura-Neto, J.P.; Fraga, D.B.M.; Freitas, L.A.R.; Santos, W.L.C. Severe clinical presentation of visceral leishmaniasis in naturally infected dogs with disruption of the splenic white pulp. PLoS ONE 2014, 9, e87742. [Google Scholar] [CrossRef] [PubMed]
- Egorin, M.J.; Zuhowski, E.G.; Rosen, D.M.; Sentz, D.L.; Covey, J.M.; Eiseman, J.L. Plasma pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) in CD2F1 mice1. Cancer Chemother. Pharmacol. 2001, 47, 291–302. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tanespimycin#section=Computed-Properties (accessed on 14 February 2024).
- Wolfsegger, M.J.; Bauer, A.; Labes, D.; Schütz, H.; Vonk, R.; Lang, B.; Lehr, S.; Jaki, T.F.; Engl, W.; Hale, M.D. Assessing goodness-of-fit for evaluation of dose-proportionality. Pharm. Stat. 2020, 20, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Trump, D.L.; Eiseman, J.L.; Belani, C.P.; Agarwala, S.S.; Zuhowski, E.G.; Lan, J.; Potter, D.M.; Ivy, S.P.; Ramalingam, S.; et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin. Cancer Res. 2005, 11, 3385–3391. [Google Scholar] [CrossRef] [PubMed]
- Nation, R.L.; Theuretzbacher, U.; Tsuji, B.T. Concentration-dependent plasma protein binding: Expect the unexpected. Eur. J. Pharm. Sci. 2018, 122, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Klotz, U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab. Rev. 2009, 41, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Silva-O’Hare, J.; Oliveira, I.S.; Klevorn, T.; Almeida, V.A.; Oliveira, G.G.S.; Atta, A.M.; Freitas, L.A.R.; Santos, W.L.C. Disruption of splenic lymphoid tissue and plasmacytosis in canine visceral leishmaniasis: Changes in homing and survival of plasma cells. PLoS ONE 2016, 11, e0156733. [Google Scholar] [CrossRef] [PubMed]
- Pelligand, L.; King, J.N.; Toutain, P.L.; Elliott, J.; Lees, P. Pharmacokinetic/pharmacodynamic modelling of robenacoxib in a feline tissue cage model of inflammation. J. Vet. Pharmacol. Ther. 2012, 35, 19–32. [Google Scholar] [CrossRef]
- Pelligand, L.; King, J.N.; Hormazabal, V.; Toutain, P.L.; Elliott, J.; Lees, P. Differential Pharmacokinetics and pharmacokinetic/pharmacodynamic modelling of robenacoxib and ketoprofen in a feline model of inflammation. J. Vet. Pharmacol. Ther. 2014, 37, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Stegemann, M.R.; Sherington, J.; Coati, N.; Brown, S.A.; Blanchflower, S. Pharmacokinetics of cefovecin in cats. J. Vet. Pharmacol. Ther. 2006, 29, 513–524. [Google Scholar] [CrossRef]
- Stegemann, M.R.; Sherington, J.; Blanchflower, S. Pharmacokinetics and pharmacodynamics of cefovecin in dogs. J. Vet. Pharmacol. Ther. 2006, 29, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef]
- Verscheijden, L.F.M.; Van der Zanden, T.M.; Van Bussel, L.P.M.; Hoop-Sommen, M.; Russel, F.G.M.; Johnson, T.N.; Wildt, S.N. Chloroquine dosing recommendations for pediatric COVID-19 supported by modeling and simulation. Clin. Pharmacol. Ther. 2020, 108, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Cleary, Y.; Parrott, N.; Reigner, B.; Smith, J.R.; Toovey, S. Development of a physiologically based pharmacokinetic model for mefloquine and its application alongside a clinical effectiveness model to select an optimal dose for prevention of malaria in young Caucasian children. Br. J. Clin. Pharmacol. 2019, 85, 100–113. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, X.; Huang, Z.; Kang, Z.; Chen, Y.; Shen, X.; Cai, Q.; Ding, H. Pharmacokinetic/pharmacodynamic integration of cefquinome against Pasteurella Multocida in a piglet tissue cage model. J. Vet. Pharmacol. Ther. 2019, 42, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Qu, Y.; Sun, M.; Qiu, Z.; Huang, X.; Huai, B.; Lu, Y.; Zeng, Z. In vivo antimicrobial activity of marbofloxacin against Pasteurella multocida in a tissue cage model in calves. Front. Microbiol. 2015, 6, 759. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Wang, J.; Yang, F.; Ding, H.; Liang, C.; Lv, Z.; Li, Z.; Zeng, Z. Pharmacokinetic/pharmacodynamic relationship of marbofloxacin against Pasteurella multocida in a tissue-cage model in yellow cattle. J. Vet. Pharmacol. Ther. 2014, 37, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Dorlo, T.P.C.; Kip, A.E.; Younis, B.M.; Ellis, S.J.; Alves, F.; Beijnen, J.H.; Njenga, S.; Kirigi, G.; Hailu, A.; Olobo, J.; et al. Visceral leishmaniasis relapse hazard is linked to reduced miltefosine exposure in patients from Eastern Africa: A population pharmacokinetic/pharmacodynamic study. J. Antimicrob. Chemother. 2017, 72, 3131–3140. [Google Scholar] [CrossRef]
- Fortin, A.; Dorlo, T.P.C.; Hendrickx, S.; Maes, L. Pharmacokinetics and pharmacodynamics of oleylphosphocholine in a hamster model of visceral leishmaniasis. J. Antimicrob. Chemother. 2016, 71, 1892–1898. [Google Scholar] [CrossRef]
- Dorlo, T.P.C.; Rijal, S.; Ostyn, B.; Vries, P.J.; Singh, R.; Bhattarai, N.; Uranw, S.; Dujardin, J.C.; Boelaert, M.; Beijnen, J.H.; et al. Failure of miltefosine in visceral leishmaniasis is associated with low drug exposure. J. Infect. Dis. Adv. 2014, 210, 146–153. [Google Scholar] [CrossRef]
- Dorlo, T.P.C.; Huitema, A.D.R.; Beijnen, J.H.; De Vries, P.J. Optimal dosing of miltefosine in children and adults with visceral leishmaniasis. Antimicrob. Agents Chemother. 2012, 56, 3864–3872. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.L.O.A.; Campos, T.A.; Dantas, D.A.S.; Rebouças, J.S.; Silva, J.C.; Menezes, J.P.B.; Formiga, F.R.; Melo, J.V.; Machado, G.; Veras, P.S.T. Encapsulation of the HSP-90 chaperone inhibitor 17-AAG in stable liposome allow increasing the therapeutic index as assessed, in vitro, on Leishmania (L) amazonensis amastigotes-hosted in mouse CBA macrophages. Front. Cell. Infect. Microbiol. 2018, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.; Tomaszewski, J.; Ivy, P. Clinical Development of 17-Allylamino, 17-Demethoxygeldanamycin. Curr. Cancer Drug Targets 2005, 3, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Page, J.G.; Noker, P.E.; Tomaszewski, J.E.; Smith, A.C. Lack of schedule dependent toxicity of 17-allylaminogeldanamycin (17-AAG, NSC 330507) in rats. Proc. Am. Assoc. Cancer Res. 1999, 40, 121. [Google Scholar]
- Weber, H.; Valbuena, J.R.; Barbhuiya, M.A.; Stein, S.; Kunkel, H.; García, P.; Bizama, C.; Riquelme, I.; Espinoza, J.A.; Kurtz, S.E.; et al. Small molecule inhibitor screening identifified HSP90 inhibitor 17-AAG as potential therapeutic agent for gallbladder cancer. Oncotarget 2017, 8, 26169–26184. [Google Scholar] [CrossRef]
- Saif, M.W.; Erlichman, C.; Dragovich, T.; Mendelson, D.; Toft, D.; Burrows, F.; Storgard, C.; Von Hoff, D. Open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study of intravenously administered CNF1010 (17-(allylamino)-17- demethoxygeldanamycin [17-AAG]) in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1345–1355. [Google Scholar] [CrossRef]
- Solit, D.B.; Ivy, S.P.; Kopil, C.; Sikorski, R.; Morris, M.J.; Slovin, F.; Kelly, W.K.; DeLaCruz, A.; Curley, T.; Heller, G.; et al. Phase I Trial of 17-Allylamino-17-Demethoxygeldanamycin in Patients with Advanced Cancer. Clin. Cancer Res. 2007, 13, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Schenk, E.; Hendrickson, A.E.W.; Northfelt, D.; Toft, D.O.; Ames, M.M.; Menefee, M.; Satele, D.; Qin, R.; Erlichman, C. Phase I study of tanespimycin in combination with bortezomib in patients with advanced solid malignancies. Investig. New Drugs 2013, 31, 1251–1256. [Google Scholar] [CrossRef]
- Ho, S.W.; Tsui, Y.T.C.; Wong, T.T.; Cheung, S.K.K.; Goggins, W.B.; Yi, L.M.; Cheng, K.K.; Baum, L. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl. Neurodegener. 2013, 2, 24. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.T.; Gordon, M.S.; Mendelson, D.; Solit, D.B.; Bagatell, R.; Ma, W.; Wheler, J.; Rosen, N.; Norton, L.; et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2-overexpressing breast cancer: A phase I dose-escalation study. J. Clin. Oncol. 2007, 25, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Glaze, E.R.; Lambert, A.L.; Smith, A.C.; Page, J.G.; Johnson, W.D.; McCormick, D.L.; Brown, A.P.; Levine, B.S.; Covey, J.M.; Egorin, M.J.; et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: Potential clinical relevance. Cancer Chemother. Pharmacol. 2005, 56, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Page, J.; Heath, J.; Fulton, R.; Yalkowsky, E.; Tabibi, E.; Tomzszewski, J.; Smith, A.; Rodman, L. Comparison of geldanamycin (NSC-122750) and 17-allylaminogeldanamycin (NSC-330507D) toxicity in rats. Proc. Am. Assoc. Cancer Res. 1997, 38, 2067. [Google Scholar]
- Goetz, M.P.; Toft, D.; Reid, J.; Ames, M.; Stensgard, B.; Safgren, S.; Adjei, A.A.; Sloan, J.; Atherton, P.; Vasile, V.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.; Ip, C.; Jimenez, L.; Tyson, C.; Behrsing, H. In vitro detection of differential and cell-specific hepatobiliary toxicity induced by geldanamycin and 17-allylaminogeldanamycin using dog liver slices. Toxicol. Sci. 2005, 87, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.S.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren III, J.; Modi, S.; et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin. Investig. Drug. 2014, 23, 611–628. [Google Scholar] [CrossRef]
- Samuni, Y.; Ishii, H.; Hyodo, F.; Samuni, U.; Krishna, M.C.; Goldstein, S.; Mitchell, J.B. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic. Biol. Med. 2010, 48, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Menezes, J.P.B.; Guedes, C.E.S.; Petersen, A.L.O.A.; Fraga, D.B.M.; Veras, P.S.T. Advances in development of new treatment for Leishmaniasis. BioMed Res. Int. 2015, 2015, 815023. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.C.; Morais-Teixeira, E.; Reis, P.G.; Silva-Barcellos, N.M.; Salaün, P.; Campos, P.P.; Corrêa-Junior, J.D.; Rabello, A.; Demicheli, C.; Frézard, F. Hepatotoxicity of pentavalent antimonial drug: Possible role of residual Sb(III) and protective effect of ascorbic acid. Antimicrob. Agents Chemother. 2014, 58, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.P.; Crank, C.W.; Leikin, J.B. An evaluation of hepatotoxicity and nephrotoxicity of liposomal amphotericin B (L-AMB). J. Med. Toxicol. 2011, 7, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Soto, P. Miltefosine: Oral treatment of leishmaniasis. Expert Rev. Anti-Infect. Ther. 2006, 4, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Talaei, S.; Mellatyar, H.; Asadi, A.; Akbarzadeh, A.; Sheervalilou, R.; Zarghami, N. Spotlight on 17-AAG as an Hsp90 inhibitor for molecular targeted cancer treatment. Chem. Biol. Drug Des. 2019, 93, 760–786. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.Q.; Liu, Y.; Zhang, D.; Wang, Z.; Dong, S.D.; Carreras, C.W.; Zhou, Y.; Rastelli, G.; Santi, D.V.; Myles, D.C. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg. Med. Chem. 2004, 12, 5317–5329. [Google Scholar] [CrossRef]
- Hollingshead, M.; Alley, M.; Burger, A.M.; Borgel, S.; Pacula-Cox, C.; Fiebig, H.H.; Sausville, E.A. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother. Pharmacol. 2005, 56, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Egorin, M.J.; Lagattuta, T.F.; Hamburger, D.R.; Covey, J.M.; White, K.D.; Musser, S.M.; Eiseman, J.L. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer Chemother. Pharmacol. 2002, 49, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Suna, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updat. 2009, 12, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, G.S.; McCollum, A.K.; Ames, M.M.; Mandrekar, S.J.; Reid, J.M.; Adjei, A.A.; Toft, D.O.; Safgren, S.L.; Erlichmanet, C. A phase I trial of twice-weekly 17-allylamino-demethoxy-geldanamycin in patients with advanced cancer. Clin. Cancer Res. 2006, 12, 6087–6093. [Google Scholar] [CrossRef] [PubMed]
- Cruz, K.P.; Patricio, B.F.C.; Pires, V.C.; Amorim, M.F.; Pinho, A.G.S.F.; Quadros, H.C.; Dantas, D.A.S.; Chaves, M.H.C.; Formiga, F.R.; Roch, H.V.A.; et al. Development and characterization of PLGA nanoparticles containing 17-DMAG, an Hsp90 inhibitor. Front. Chem. 2021, 13, 644827. [Google Scholar] [CrossRef]
- Xiong, M.P.; Yáñez, J.A.; Kwon, G.S.; Davies, N.M.; Forrest, M.L. A Cremophor-Free formulation for Tanespimycin (17-AAG) using PEO-b-PDLLA micelles: Characterization and pharmacokinetics in rats. J. Pharm. Sci. 2009, 98, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Katragadda, U.; Teng, Q.; Rayaprolu, B.M.; Chandran, T.; Tan, C. Multi-drug delivery to tumor cells via micellar nanocarriers. Int. J. Pharm. 2011, 419, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Wasan, K.M.; Wasan, E.K.; Hnik, P. Assessing the safety, tolerability, pharmacokinetics, and biodistribution of novel oral formulations of Amphotericin B following single- and multiple-dose administration to beagle dogs. Antimicrob. Agents Chemother. 2020, 64, e01111-20. [Google Scholar] [CrossRef] [PubMed]
- Griensven, J.V.; Dorlo, T.P.C.; Diro, E.; Costa, C.; Burza, S. The status of combination therapy for visceral leishmaniasis: An updated review. Lancet Infect. Dis. 2024, 24, e36–e46. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.; López, M.; Gallego-Yerga, L.; Álvarez, R.; Peláez, R. Experimental structure based drug design (SBDD) applications for anti-leishmanial drugs: A paradigm shift? Med. Res. Rev. 2024, 44, 1055–1120. [Google Scholar] [CrossRef] [PubMed]
- Khandibharad, S.; Singh, S. Synthetic biology for combating leishmaniasis. Front. Microbiol. 2024, 15, 1338749. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bies, R.R.; Ramanathan, R.K.; Zuhowski, E.G.; Trump, D.L.; Egorin, M.J. Population pharmacokinetic analysis of 17-(allylamino)-17-demethoxygeldanamycin (17AAG) in adult patients with advanced malignancies. Cancer Chemother. Pharmacol. 2005, 55, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Agnew, E.B.; Wilson, R.H.; Grem, J.L.; Neckers, L.; Bi, D.; Takimoto, C.H. Measurement of the novel antitumor agent 17-(allylamino)-17-demethoxygeldanamycin in human plasma by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2001, 755, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.S.; Phelps, M.A.; Blum, K.A.; Blum, W.; Grever, M.R.; Farley, K.L.; Dalton, J.T. Development and validation of a rapid and sensitive high-performance liquid chromatography-mass spectroscopy assay for determination of 17-(allylamino)-17-demethoxygeldanamycin and 17-(amino)-17-demethoxygeldanamycin in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 15–21. [Google Scholar] [CrossRef] [PubMed]
- ICH. Validation of Analytical Procedures: Text and Methodology Q2 (R1). International Conference on Harmonisation. 2005. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 1 May 2024).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (mg/m2) | AUC (ng × h/mL) | Cmax (ng/mL) | t½ (h) | Clearance (L/m2/h) | DV (L/m2) |
|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |
| 50 | 1483.26 ± 694.52 | 1405.97 ± 686.71 | 0.54 ± 0.03 | 0.04 ± 0.02 | 0.03 ± 0.02 |
| 100 | 4380.35 ± 1626.22 | 3756.41 ± 1422.52 | 0.61 ± 0.08 | 0.03 ± 0.01 | 0.02 ± 0.01 |
| 150 | 7927.84 ± 1548.51 | 6938.99 ± 1342.39 | 0.59 ± 0.04 | 0.02 ± 0.00 | 0.02 ± 0.00 |
| 200 | 11,902.75 ± 1962.12 | 9439.70 ± 991.11 | 0.67 ± 0.05 | 0.02 ± 0.00 | 0.02 ± 0.00 |
| 250 | 9632.29 ± 2667.12 | 8611.51 ± 1062.88 | 0.57 ± 0.21 | 0.03 ± 0.01 | 0.02 ± 0.00 |
| Alterations | Number of Animals Evaluated (n = 4) N (%) | ||||
|---|---|---|---|---|---|
| 50 mg/m2 | 100 mg/m2 | 150 mg/m2 | 200 mg/m2 | 250 mg/m2 | |
| Biochemical | |||||
| ALT | 1 (25%) | 2 (50%) | 3 (75%) | 3 (75%) | 4 (100%) |
| AST | 0 | 2 (50%) | 2 (50%) | 4 (100%) | 4 (100%) |
| Gastrointestinal | |||||
| Bloody diarrhea | 0 | 0 | 0 | 3 (75%) | 4 (100%) |
| Diarrhea (pasty feces) | 0 | 1 (25%) | 1 (25%) | 1 (25%) | 1 (25%) |
| Nausea | 0 | 0 | 0 | 4 (100%) | 4 (100%) |
| Vomit | 0 | 0 | 0 | 1 (25%) | 1 (25%) |
| Other | |||||
| Hyperthermia | 0 | 0 | 1 (25%) | 0 | 1 (25%) |
| Pruritus | 0 | 0 | 1 (25%) | 3 (75%) | 4 (100%) |
| Erythema | 0 | 0 | 1 (25%) | 2 (50%) | 4 (100%) |
| Administration | t ½ (h) | Cmax (μg/mL) | Clearance (L/m2/h) | DV (L/m2) | AUC (μg/mL × h) 0–8 h |
|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |
| 1st | 0.69 ± 0.22 | 6309 ± 2204 | 0.023 ± 0.002 | 0.029 ± 0.003 | 6353 ± 432 |
| 2nd | 0.69 ± 0.24 | 5912 ± 1127 | 0.021 ± 0.001 | 0.030 ± 0.002 | 7054 ± 382 |
| 3rd | 0.81 ± 0.35 | 5254 ± 2784 | 0.022 ± 0.002 | 0.033 ± 0.003 | 6850 ± 69 |
| Administration | t ½ (h) | Cmax (μg/mL) | AUC (μg/mL × h) | ||||
|---|---|---|---|---|---|---|---|
| 0–4 h | 0–6 h | 0–8 h | 0–24 h | 0–47.5 h | |||
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |
| 1st | 38 ± 16 | 704 ± 332 | 1820 ± 430 | 2301 ± 409 | 2659 ± 437 | 4765 ± 333 | 7382 ± 1357 |
| 2nd | 28 ± 9 | 736 ± 294 | 1943 ± 618 | 2553 ± 638 | 2913 ± 686 | 4070 ± 700 | 7054 ± 2189 |
| 3rd | N/A | 726 ± 401 | 895 ± 408 | 2040 ± 1097 | 2734 ± 1442 | 3505 ± 2417 | N/A |
| Alterations | Animals Presenting Adverse Reactions n (%) |
|---|---|
| Biochemistry | |
| ALT | 3 (33%) |
| AST | 3 (33%) |
| Gastrointestinal | |
| Diarrhea (liquid feces) | 3 (33%) |
| Pasty feces | 9 (100%) |
| Bloody diarrhea | 1 (11%) |
| Nausea | 0 |
| Vomit | 0 |
| Others | |
| Hyperthermia | 0 |
| Skin reaction | 1 (11%) |
| Erythema | 0 |
| Pruritus | 0 |
| Histopathological Alterations | Animals Presenting Alterations n (%) |
|---|---|
| Kidney | |
| Vacuolar degeneration of the renal tubular epithelium | 9 (100%) |
| Liver | |
| Mobilization of Kupffer cells | 9 (100%) |
| Spleen | |
| Congestion | 9 (100%) |
| Immunoblastic hyperplasia | 9 (100%) |
| Spleen disorganization in type II/III | 9 (100%) |
| Small Intestine | |
| Mucosal calcification foci | 2 (22%) |
| Thick Intestine | |
| Mucosal calcification foci | 1 (11%) 5 (55%) |
| Colitis |
| Reagent | Quantity |
|---|---|
| 17-AAG or Tanespimycin (LC-Laboratories®, Woburn, MA, USA) | 0.1 g |
| Cremophor (BASF®, Cincinnati, OH, USA) | 2.1 g |
| Propylene glycol (Palmar®, Salvador, BA, Brazil) | 3.0 g |
| Methylparaben (Dinamica®, Recreio Campestre Jóia, Indaiatuba, SP, Brazil) | 0.002 g |
| Propylparaben (Dinamica®) | 0.0002 g |
| Ethyl alcohol (Merck®, Darmstadt, Germany) | 4.9 mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrante, M.; Leite, B.M.M.; Fontes, L.B.C.; Santos Moreira, A.; Nascimento de Almeida, É.M.; Brodskyn, C.I.; Lima, I.d.S.; dos Santos, W.L.C.; Pacheco, L.V.; Cardoso da Silva, V.; et al. Pharmacokinetics, Dose-Proportionality, and Tolerability of Intravenous Tanespimycin (17-AAG) in Single and Multiple Doses in Dogs: A Potential Novel Treatment for Canine Visceral Leishmaniasis. Pharmaceuticals 2024, 17, 767. https://doi.org/10.3390/ph17060767
Ferrante M, Leite BMM, Fontes LBC, Santos Moreira A, Nascimento de Almeida ÉM, Brodskyn CI, Lima IdS, dos Santos WLC, Pacheco LV, Cardoso da Silva V, et al. Pharmacokinetics, Dose-Proportionality, and Tolerability of Intravenous Tanespimycin (17-AAG) in Single and Multiple Doses in Dogs: A Potential Novel Treatment for Canine Visceral Leishmaniasis. Pharmaceuticals. 2024; 17(6):767. https://doi.org/10.3390/ph17060767
Chicago/Turabian StyleFerrante, Marcos, Bruna Martins Macedo Leite, Lívia Brito Coelho Fontes, Alice Santos Moreira, Élder Muller Nascimento de Almeida, Claudia Ida Brodskyn, Isadora dos Santos Lima, Washington Luís Conrado dos Santos, Luciano Vasconcellos Pacheco, Vagner Cardoso da Silva, and et al. 2024. "Pharmacokinetics, Dose-Proportionality, and Tolerability of Intravenous Tanespimycin (17-AAG) in Single and Multiple Doses in Dogs: A Potential Novel Treatment for Canine Visceral Leishmaniasis" Pharmaceuticals 17, no. 6: 767. https://doi.org/10.3390/ph17060767
APA StyleFerrante, M., Leite, B. M. M., Fontes, L. B. C., Santos Moreira, A., Nascimento de Almeida, É. M., Brodskyn, C. I., Lima, I. d. S., dos Santos, W. L. C., Pacheco, L. V., Cardoso da Silva, V., dos Anjos, J. P., Guarieiro, L. L. N., Landoni, F., de Menezes, J. P. B., Fraga, D. B. M., Santos Júnior, A. d. F., & Veras, P. S. T. (2024). Pharmacokinetics, Dose-Proportionality, and Tolerability of Intravenous Tanespimycin (17-AAG) in Single and Multiple Doses in Dogs: A Potential Novel Treatment for Canine Visceral Leishmaniasis. Pharmaceuticals, 17(6), 767. https://doi.org/10.3390/ph17060767