
Vorinostat Treatment of Gastric Cancer Cells Leads to ROS-Induced Cell Inhibition and a Complex Pattern of Molecular Alterations in Nrf2-Dependent Genes

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Vorinostat Induces Reactive Oxygen Species (ROS) Increase in Gastric Carcinoma Cells
2.2. Tumor Cell Inhibition upon Vorinostat-Mediated ROS Increase
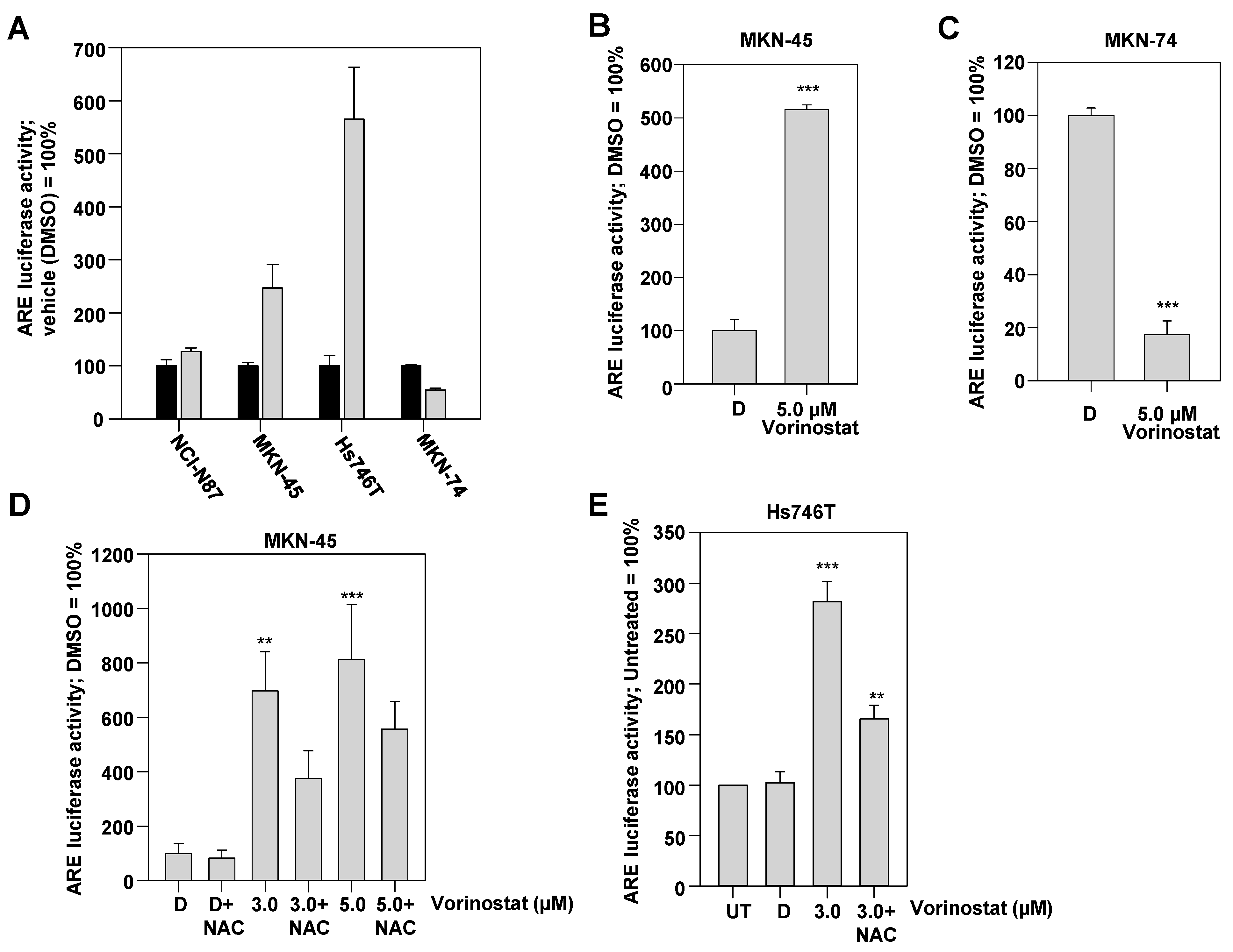
2.3. ROS-Dependent Induction of Antioxidant Response Element (ARE)-Regulated Luciferase Reporter upon Vorinostat Treatment
2.4. Nrf2 Knockdown Leads to a Cell Line-Dependent Enhancement of Tumor Cell Inhibition upon Vorinostat Treatment
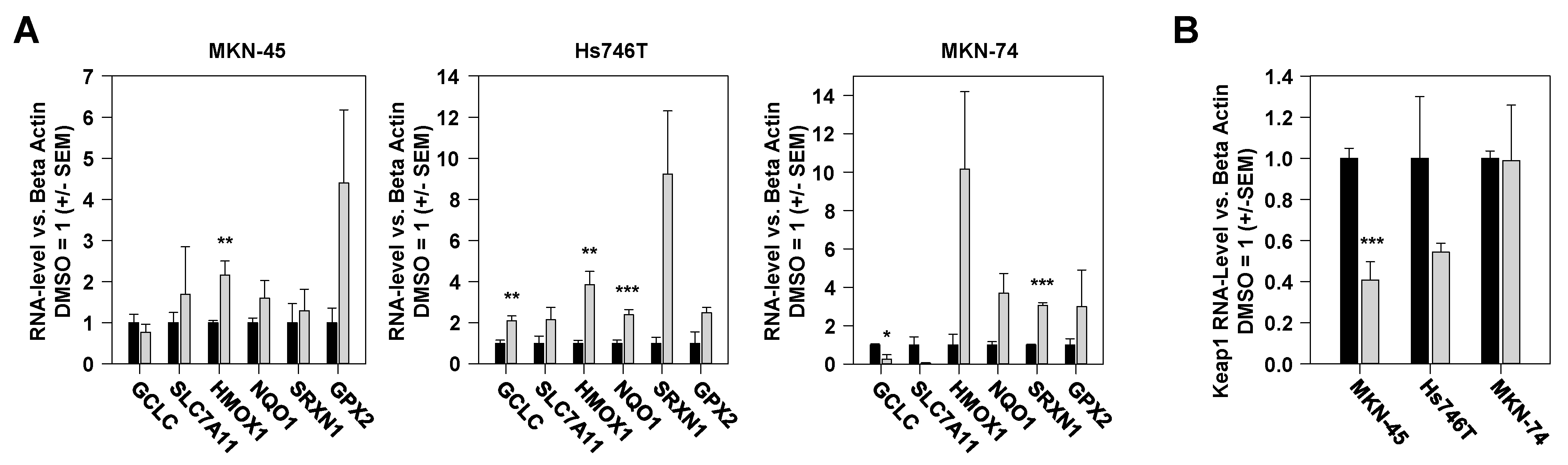
2.5. Vorinostat-Mediated Alterations in Expression Levels of Nrf2-Dependent Genes
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Treatment and Transfection
4.4. Fluorescence-Based Determination of Reactive Oxygen Species (ROS)
4.5. ARE Luciferase Reporter Assay
4.6. RNA Isolation and RT-qPCR Analyses
4.7. Proteomic Analyses of Nrf2 Target Gene Expression
4.8. Proliferation Assay
4.9. Colony-Forming Assay
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Khan, M.A.; Siddique, H.R. Role of epigenetic drugs in sensitizing cancers to anticancer therapies: Emerging trends and clinical advancements. Epigenomics 2023, 15, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Christodoulidis, G.; Koumarelas, K.-E.; Kouliou, M.-N.; Thodou, E.; Samara, M. Gastric Cancer in the Era of Epigenetics. Int. J. Mol. Sci. 2024, 25, 3381. [Google Scholar] [CrossRef] [PubMed]
- Badie, A.; Gaiddon, C.; Mellitzer, G. Histone Deacetylase Functions in Gastric Cancer: Therapeutic Target? Cancers 2022, 14, 5472. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Liu, S.; Zhang, J.; Pei, M.; Xiao, Y.; Li, J.; Hong, L.; Lin, J.; Wang, J.; Wu, X.; et al. Vorinostat triggers miR-769-5p/3p-mediated suppression of proliferation and induces apoptosis via the STAT3-IGF1R-HDAC3 complex in human gastric cancer. Cancer Lett. 2021, 521, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Seah, K.S.; Loh, J.Y.; Nguyen, T.T.T.; Tan, H.L.; Hutchinson, P.E.; Lim, K.K.; Dymock, B.W.; Long, Y.C.; Lee, E.J.D.; Shen, H.-M.; et al. SAHA and cisplatin sensitize gastric cancer cells to doxorubicin by induction of DNA damage, apoptosis and perturbation of AMPK-mTOR signalling. Exp. Cell Res. 2018, 370, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.R.; Kim, D.Y.; Jin, H.; Meng, R.; Chae, O.H.; Kim, S.H.; Park, B.H.; Kim, S.M. Inactivation of the Akt/FOXM1 signaling pathway by panobinostat suppresses the proliferation and metastasis of gastric cancer cells. Int. J. Mol. Sci. 2021, 22, 5955. [Google Scholar] [CrossRef]
- Yoo, C.; Ryu, M.-H.; Na, Y.-S.; Ryoo, B.-Y.; Lee, C.-W.; Kang, Y.-K. Vorinostat in combination with capecitabine plus cisplatin as a first-line chemotherapy for patients with metastatic or unresectable gastric cancer: Phase II study and biomarker analysis. Br. J. Cancer 2016, 114, 1185–1190. [Google Scholar] [CrossRef]
- Fushida, S.; Kinoshita, J.; Kaji, M.; Oyama, K.; Hirono, Y.; Tsukada, T.; Fujimura, T.; Ohta, T. Paclitaxel plus valproic acid versus paclitaxel alone as second- or third-line therapy for advanced gastric cancer: A randomized Phase II trial. Drug Des. Dev. Ther. 2016, 10, 2353–2358. [Google Scholar] [CrossRef]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- Jenke, R.; Oliinyk, D.; Zenz, T.; Körfer, J.; Schäker-Hübner, L.; Hansen, F.K.; Lordick, F.; Meier-Rosar, F.; Aigner, A.; Büch, T. HDAC inhibitors activate lipid peroxidation and ferroptosis in gastric cancer. Biochem. Pharmacol. 2024, 225, 116257. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Pourbagher-Shahri, A.M.; Azimi-Nezhad, M.; Forouzanfar, F.; Brockmueller, A.; Ashrafizadeh, M.; Talebi, M.; Shakibaei, M.; Samarghandian, S. Roles of Nrf2 in Gastric Cancer: Targeting for Therapeutic Strategies. Molecules 2021, 26, 3157. [Google Scholar] [CrossRef]
- Telkoparan-Akillilar, P.; Panieri, E.; Cevik, D.; Suzen, S.; Saso, L. Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer. Molecules 2021, 26, 1417. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Campbell, K.H.; MacLeod, A.K.; McLaughlin, L.A.; Henderson, C.J.; Wolf, C.R. HDAC inhibitors increase NRF2-signaling in tumour cells and blunt the efficacy of co-adminstered cytotoxic agents. PLoS ONE 2014, 9, e114055. [Google Scholar] [CrossRef]
- Yang, M.; He, J.; Xia, S.; Wang, Y.; Xiong, J.; Liao, C.; Li, N.; Qu, S.; Shen, C. Investigation of the mixed origins of the MGC-803 cell line reveals that it is a hybrid cell line derived from HeLa. Hum. Cell 2024, 37, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.R.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.E.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, X.Q.; Delaidelli, A.; Rafn, B.; Cran, J.; et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Southam, A.D.; Trova, S.; Beke, F.; Alhazmi, B.; Francis, T.; Radotra, A.; Di Maio, A.; Drayson, M.T.; Bunce, C.M.; et al. Valproic acid disables the Nrf2 anti-oxidant response in acute myeloid leukaemia cells enhancing reactive oxygen species-mediated killing. Br. J. Cancer 2022, 126, 275–286. [Google Scholar] [CrossRef]
- Tanaka, S.; Hosokawa, M.; Tatsumi, A.; Asaumi, S.; Imai, R.; Ogawara, K.-I. Improvement of resistance to oxaliplatin by vorinostat in human colorectal cancer cells through inhibition of Nrf2 nuclear translocation. Biochem. Biophys. Res. Commun. 2022, 607, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, M.J.; Han, T.H.; Lee, J.Y.; Kim, S.; Kim, H.; Oh, K.J.; Kim, W.K.; Han, B.S.; Bae, K.H.; et al. FSP1 confers ferroptosis resistance in KEAP1 mutant non-small cell lung carcinoma in NRF2-dependent and -independent manner. Cell Death Dis. 2023, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Consoli, V.; Sorrenti, V.; Salvo Grosso, S.; Vanella, L. Heme oxygenase-1 signaling and redox homeostasis in physiopathological conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Kusano, Y.; Okazaki, K.; Akaike, T.; Motohashi, H. NRF2 signalling in cytoprotection and metabolism. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Hammad, A.; Namani, A.; Elshaer, M.; Wang, X.J.; Tang, X. “NRF2 addiction” in lung cancer cells and its impact on cancer therapy. Cancer Lett. 2019, 467, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Shi, Y.; Wu, L.; Tan, Y.; Li, T.; Chen, Y.; Xia, J.; Hu, R. FAM117B promotes gastric cancer growth and drug resistance by targeting the KEAP1/NRF2 signaling pathway. J. Clin. Investig. 2023, 133, e158705. [Google Scholar] [CrossRef]
- Storz, L.; Walther, P.; Chemnitzer, O.; Lyros, O.; Niebisch, S.; Mehdorn, M.; Jansen-Winkeln, B.; Moulla, Y.; Büch, T.; Gockel, I.; et al. Nrf2/Keap1-Pathway Activation and Reduced Susceptibility to Chemotherapy Treatment by Acidification in Esophageal Adenocarcinoma Cells. Cancers 2021, 13, 2806. [Google Scholar] [CrossRef]
- Paik, J.Y.; Lee, H.G.; Piao, J.-Y.; Kim, S.-J.; Kim, D.-H.; Na, H.-K.; Surh, Y.-J. Helicobacter pylori infection promotes autophagy through Nrf2-mediated heme oxygenase upregulation in human gastric cancer cells. Biochem. Pharmacol. 2019, 162, 89–97. [Google Scholar] [CrossRef]
- Bacon, S.; Seeneevassen, L.; Fratacci, A.; Rose, F.; Tiffon, C.; Sifré, E.; Haykal, M.M.; Moubarak, M.M.; Ducournau, A.; Bruhl, L.; et al. Nrf2 Downregulation Contributes to Epithelial-to-Mesenchymal Transition in Helicobacter pylori-Infected Cells. Cancers 2022, 14, 4316. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Qiao, Y.; Yang, H.; Nan, Q.; Qu, W.; Feng, F.; Liu, W.; Chen, Y.; Sun, H. Small molecular Nrf2 inhibitors as chemosen-sitizers for cancer therapy. Future Med. Chem. 2020, 12, 243–267. [Google Scholar] [CrossRef]
- Srivastava, R.; Fernández-Ginés, R.; Encinar, J.A.; Cuadrado, A.; Wells, G. The current status and future prospects for therapeutic targeting of KEAP1-NRF2 and β-TrCP-NRF2 interactions in cancer chemoresistance. Free Radic. Biol. Med. 2022, 192, 246–260. [Google Scholar] [CrossRef]
- Friedrich, M.; Aigner, A. Therapeutic siRNA: State-of-the-art and future perspectives. BioDrugs 2022, 36, 549–571. [Google Scholar] [CrossRef]
- Song, M.Y.; Lee, D.Y.; Chun, K.S.; Kim, E.H. The role of NRF2/KEAP1 signaling pathway in cancer metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Gulati, R.; Fleifil, Y.; Jennings, K.; Bondoc, A.; Tiao, G.; Geller, J.; Timchenko, L.; Timchenko, N. Inhibition of histone deacetylase activity increases cisplatin efficacy to eliminate metastatic cells in pediatric liver cancers. Cancers 2024, 16, 2300. [Google Scholar] [CrossRef]
- Lim, J.S.; Kyung, S.Y.; Jeon, Y.; Kim, I.S.; Kwak, J.H.; Kim, H.S. Anticancer effects of the HDAC inhibitor, 3β,6β-dihydroxyurs-12-en-27-oic acid, in MCF-7 breast cancer cells via the inhibition of Akt/mTOR pathways. Oncol. Rep. 2023, 49, 43. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Deng, X. Recent advances in organotypic tissue slice cultures for anticancer drug development. Int. J. Biol. Sci. 2022, 18, 5885–5896. [Google Scholar] [CrossRef] [PubMed]
- Leibold, J.; Tsanov, K.M.; Amor, C.; Ho, Y.J.; Sánchez-Rivera, F.J.; Feucht, J.; Baslan, T.; Chen, H.A.; Tian, S.; Simon, J.; et al. Somatic mouse models of gastric cancer reveal genotype-specific features of metastatic disease. Nat. Cancer 2024, 5, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kang, S.K.; Kwon, W.S.; Kim, T.S.; Jeong, I.; Jeung, H.C.; Kragh, M.; Horak, I.D.; Chung, H.C.; Rha, S.Y. Forty-nine gastric cancer cell lines with integrative genomic profiling for development of c-MET inhibitor. Int. J. Cancer 2018, 143, 151–159. [Google Scholar]
- Yoshioka, T.; Shien, K.; Namba, K.; Torigoe, H.; Sato, H.; Tomida, S.; Yamamoto, H.; Asano, H.; Soh, J.; Tsukuda, K.; et al. Antitumor activity of pan-HER inhibitors in HER2-positive gastric cancer. Cancer Sci. 2018, 109, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Schäker-Hübner, L.; Haschemi, R.; Büch, T.; Kraft, F.B.; Brumme, B.; Schöler, A.; Jenke, R.; Meiler, J.; Aigner, A.; Bendas, G.; et al. Balancing Histone Deacetylase (HDAC) Inhibition and Drug-likeness: Biological and Physicochemical Evaluation of Class I Selective HDAC Inhibitors. ChemMedChem 2022, 17, e202100755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.N.; Zhu, J.J.; Liu, X.X.; You, H.; Gong, M.Y.; Zou, M.; Cheng, W.H.; Zhu, J.H. N-acetylcysteine negatively regulates Notch3 and its malignant signaling. Oncotarget 2016, 7, 30855–30866. [Google Scholar] [CrossRef]
- Walther, M.; Jenke, R.; Aigner, A.; Ewe, A. Efficient polymeric nanoparticles for RNAi in macrophage reveal complex effects on polarization markers upon knockdown of STAT3/STAT6. Eur. J. Pharm. Biopharm. 2024, 197, 114232. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.-D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenz, L.; Zenz, T.; Oliinyk, D.; Meier-Rosar, F.; Jenke, R.; Aigner, A.; Büch, T. Vorinostat Treatment of Gastric Cancer Cells Leads to ROS-Induced Cell Inhibition and a Complex Pattern of Molecular Alterations in Nrf2-Dependent Genes. Pharmaceuticals 2024, 17, 1080. https://doi.org/10.3390/ph17081080
Lorenz L, Zenz T, Oliinyk D, Meier-Rosar F, Jenke R, Aigner A, Büch T. Vorinostat Treatment of Gastric Cancer Cells Leads to ROS-Induced Cell Inhibition and a Complex Pattern of Molecular Alterations in Nrf2-Dependent Genes. Pharmaceuticals. 2024; 17(8):1080. https://doi.org/10.3390/ph17081080
Chicago/Turabian StyleLorenz, Leoni, Tamara Zenz, Denys Oliinyk, Florian Meier-Rosar, Robert Jenke, Achim Aigner, and Thomas Büch. 2024. "Vorinostat Treatment of Gastric Cancer Cells Leads to ROS-Induced Cell Inhibition and a Complex Pattern of Molecular Alterations in Nrf2-Dependent Genes" Pharmaceuticals 17, no. 8: 1080. https://doi.org/10.3390/ph17081080
APA StyleLorenz, L., Zenz, T., Oliinyk, D., Meier-Rosar, F., Jenke, R., Aigner, A., & Büch, T. (2024). Vorinostat Treatment of Gastric Cancer Cells Leads to ROS-Induced Cell Inhibition and a Complex Pattern of Molecular Alterations in Nrf2-Dependent Genes. Pharmaceuticals, 17(8), 1080. https://doi.org/10.3390/ph17081080