

Discovery of Benzopyrone-Based Candidates as Potential Antimicrobial and Photochemotherapeutic Agents through Inhibition of DNA Gyrase Enzyme B: Design, Synthesis, In Vitro and In Silico Evaluation


 ,
,  , and
, and
Abstract
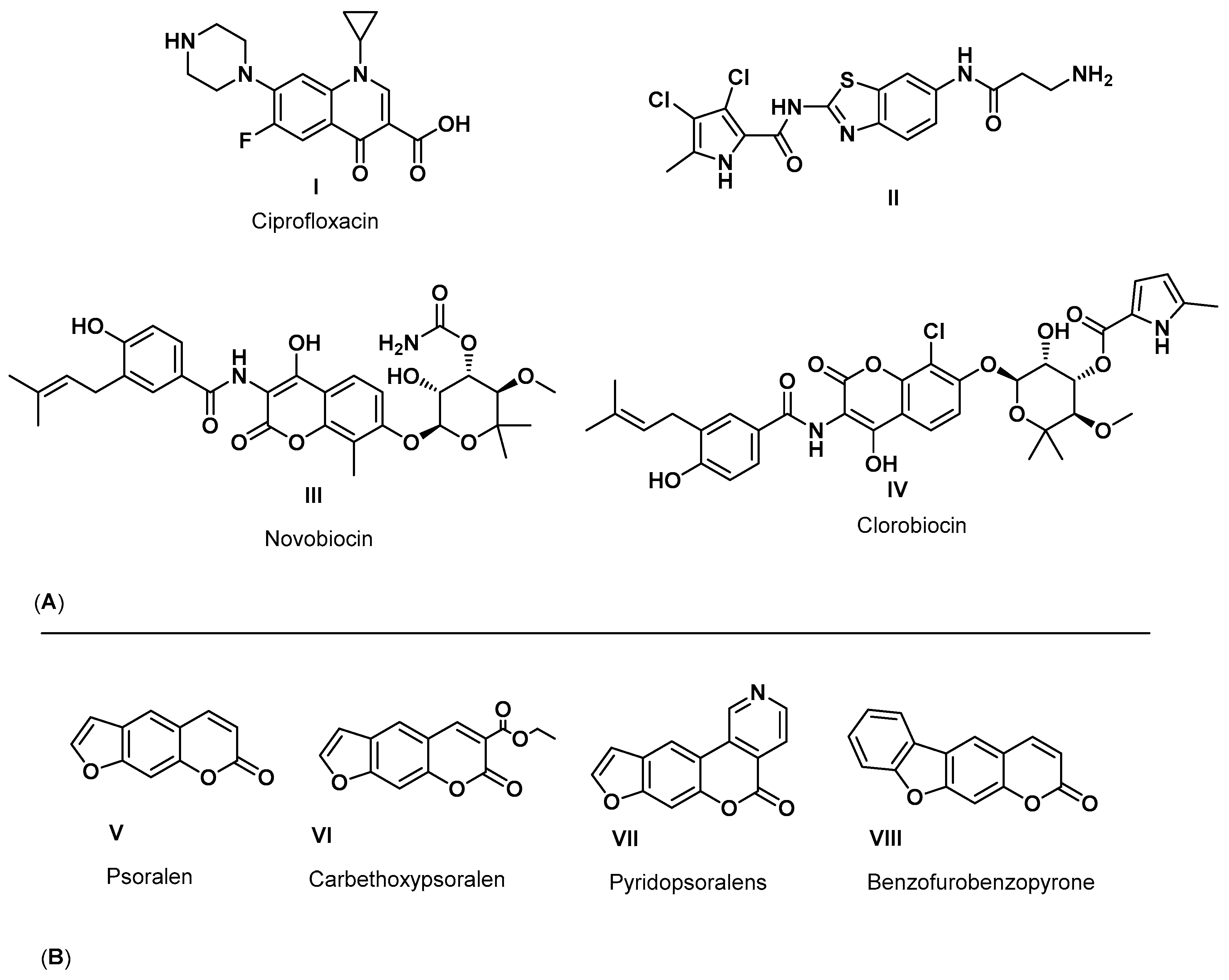
1. Introduction
2. Results and Discussion
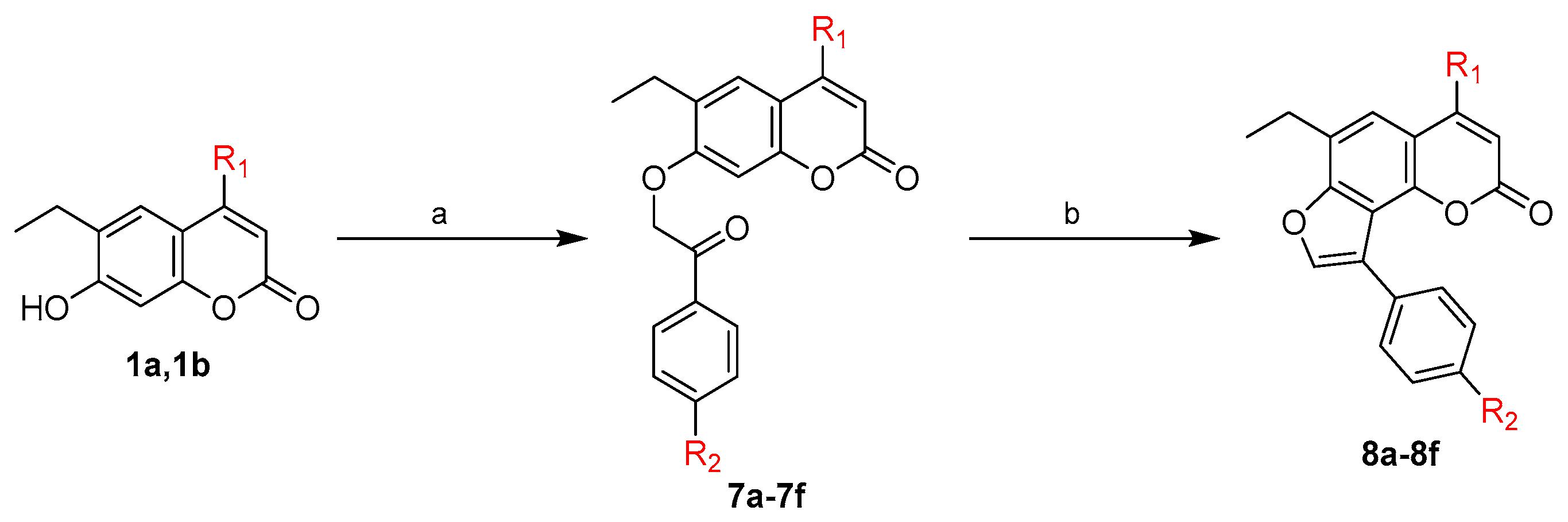
2.1. Chemistry
2.2. Antimicrobial Assay
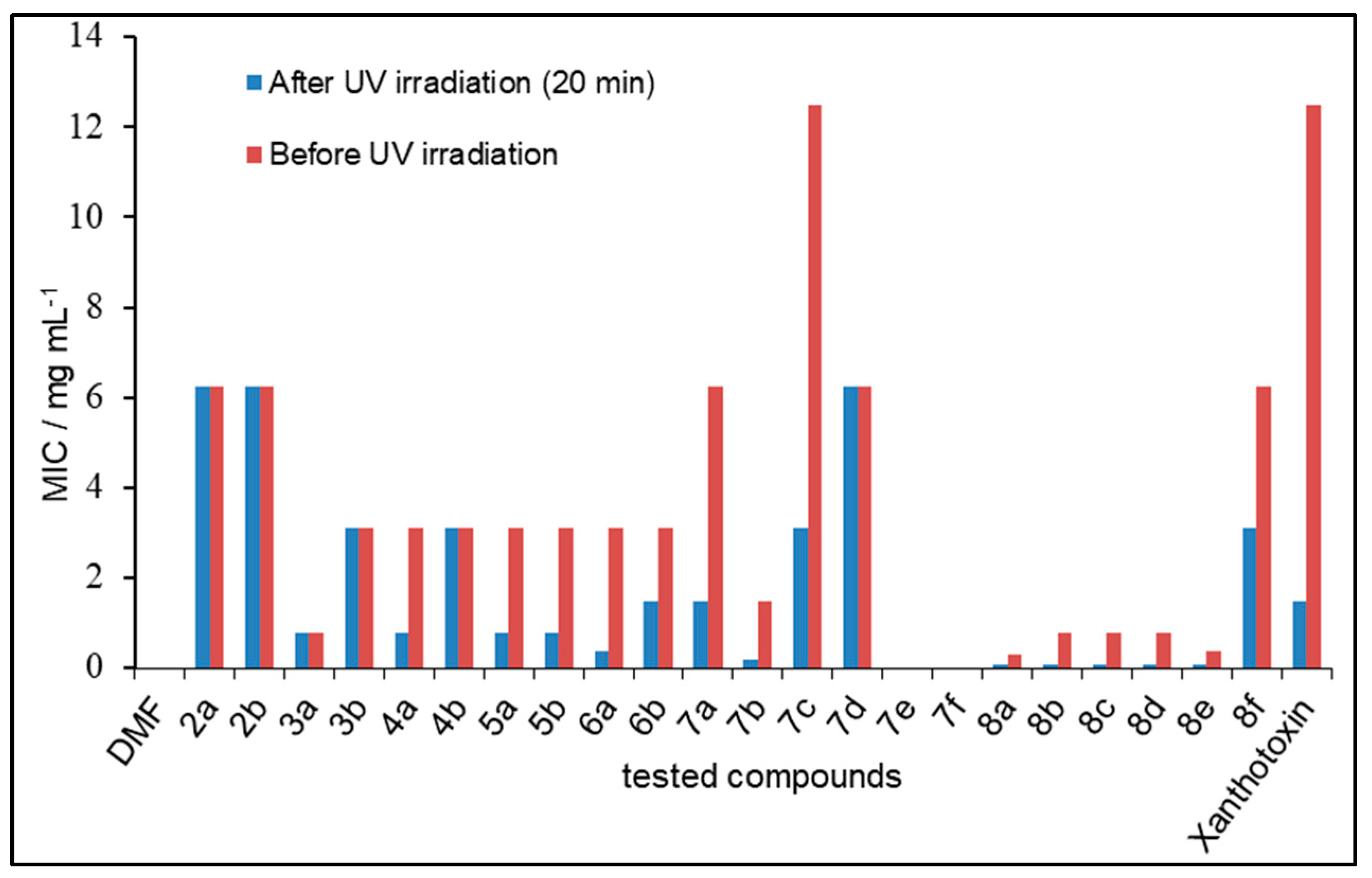
2.3. Photosensitizing Assay
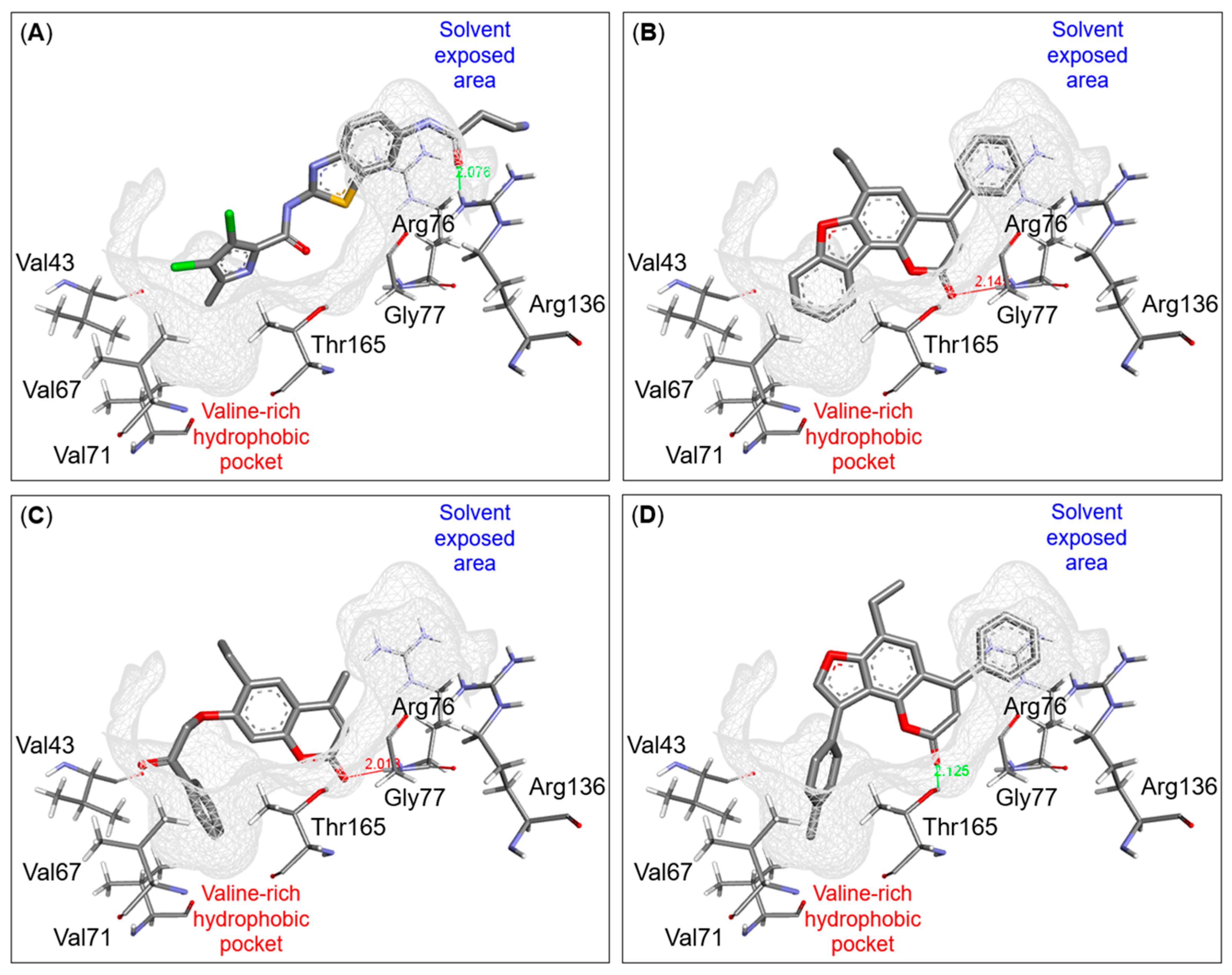
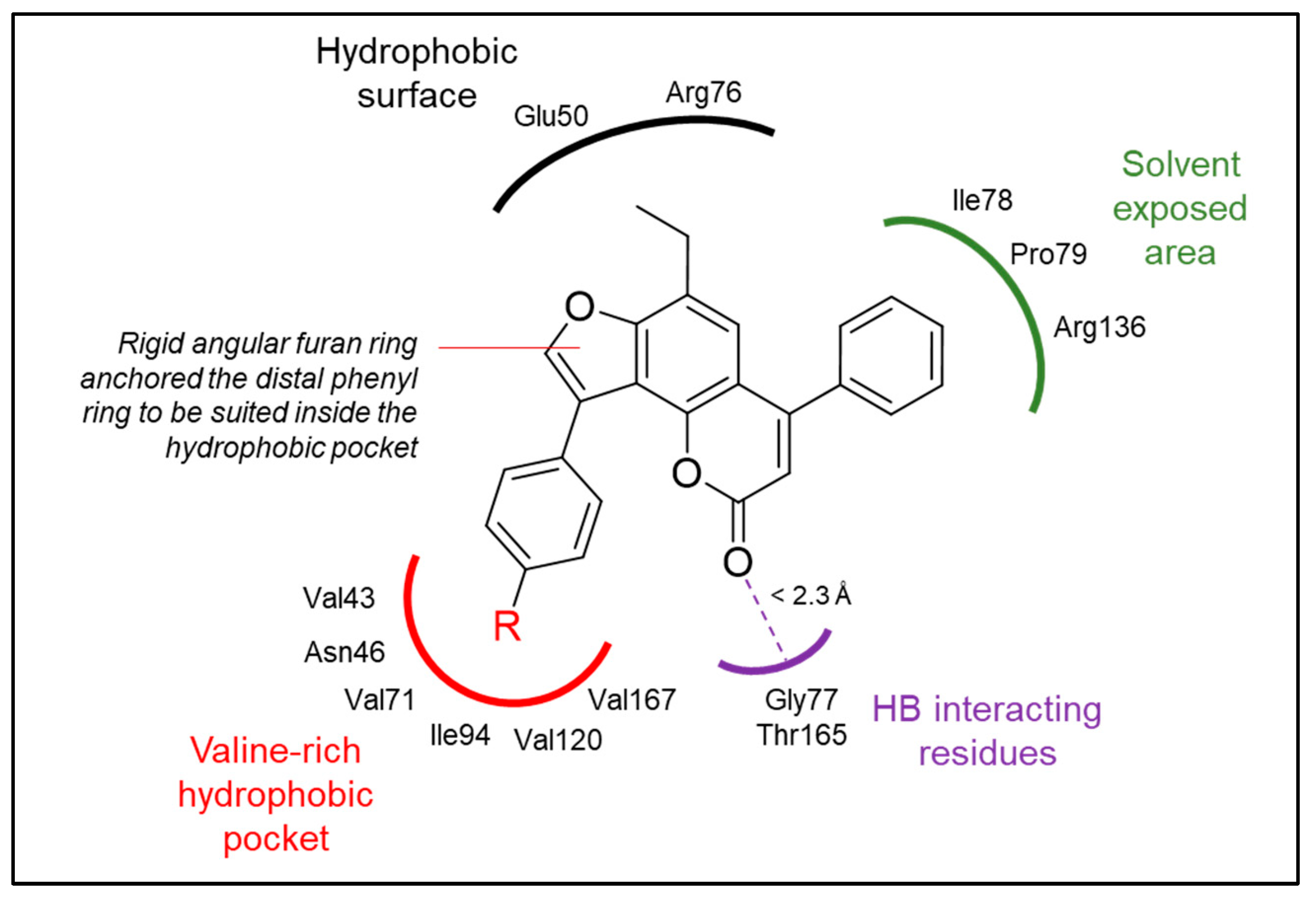
2.4. Binding Mode Investigation Using Molecular Docking
2.5. In Vitro DNA Gyrase Supercoiling Inhibitory Assay
2.6. In Silico ADME Prediction
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for Synthesis of 6-Ethyl-7-(3-oxobutan-2-yloxy)-4-substituted-2H-1-benzopyran-2-ones (2a,b)
3.1.3. General Procedure for Synthesis of 2,3-Dimethyl-9-ethyl-7-substituted-5H-furo[2,3-h]benzopyran-5-ones (3a,b)
3.1.4. General Procedure for Synthesis of 6-Ethyl-7-(2-oxocyclohexyloxy)-4-substituted-2H-1-benzopyran-2-ones (4a,b)
3.1.5. General Procedure for Synthesis of 6-Ethyl-4-substituted-8,9,10,11-tetrahydro-2H-benzofuro[2,3-h]benzopyran-2-ones (5a,b)
3.1.6. General Procedure for Synthesis of 6-Ethyl-4-substituted-2H-benzofuro[2,3-h]benzopyran-2-ones (6a,b)
3.1.7. General Procedure for Synthesis of 6-Ethyl-4-substituted-7-(un)substituted phenacyloxy-2H-1-benzopyran-2-ones (7a-f)
3.1.8. General Procedure for Synthesis of 9-Ethyl-7-substituted-3-(un)substituted phenyl-5H-furo[2,3-h]benzopyran-5-ones (8a-8f)
3.2. Antimicrobial Assay
3.2.1. General
3.2.2. Pre-Experimental Preparations
3.2.3. Cup-Plate Method
3.2.4. Statistical Analysis
3.3. Photosensitizing Assay
3.4. In Silico Molecular Docking
3.4.1. Ligand Preparation
3.4.2. Protein Preparation
3.4.3. Molecular Docking Protocol Validation
3.4.4. Molecular Docking Analysis
3.5. In Vitro DNA Gyrase Supercoiling Inhibitory Assay
3.6. In Silico ADME Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skok, Z.; Barancokova, M.; Benek, O.; Cruz, C.D.; Tammela, P.; Tomasic, T.; Zidar, N.; Masic, L.P.; Zega, A.; Stevenson, C.E. Exploring the chemical space of benzothiazole-based DNA gyrase B inhibitors. ACS Med. Chem. Lett. 2020, 11, 2433–2440. [Google Scholar] [CrossRef]
- Weiss, S.; Ben-Shmuel, A.; Chajanovsky, I.; Mizrahi, D.M.; Suckeveriene, R.Y. Hybrid PANI-halamine design, synthesis and antibacterial activity. J. Water Process Eng. 2023, 56, 104539. [Google Scholar] [CrossRef]
- Brvar, M.; Perdih, A.; Oblak, M.; Mašič, L.P.; Solmajer, T. In silico discovery of 2-amino-4-(2, 4-dihydroxyphenyl) thiazoles as novel inhibitors of DNA gyrase B. Bioorg. Med. Chem. Lett. 2010, 20, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. New targets and screening approaches in antimicrobial drug discovery. Chem. Rev. 2005, 105, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Does the cell wall of bacteria remain a viable source of targets for novel antibiotics? Biochem. Pharmacol. 2006, 71, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Perdih, A.; Kovač, A.; Wolber, G.; Blanot, D.; Gobec, S.; Solmajer, T. Discovery of novel benzene 1, 3-dicarboxylic acid inhibitors of bacterial MurD and MurE ligases by structure-based virtual screening approach. Bioorg. Med. Chem. Lett. 2009, 19, 2668–2673. [Google Scholar] [CrossRef]
- Bahekar, S.S.; Shinde, D.B. Samarium (III) catalyzed one-pot construction of coumarins. Tetrahedron Lett. 2004, 45, 7999–8001. [Google Scholar] [CrossRef]
- Mesleh, M.F.; Cross, J.B.; Zhang, J.; Kahmann, J.; Andersen, O.A.; Barker, J.; Cheng, R.K.; Felicetti, B.; Wood, M.; Hadfield, A.T. Fragment-based discovery of DNA gyrase inhibitors targeting the ATPase subunit of GyrB. Bioorg. Med. Chem. Lett. 2016, 26, 1314–1318. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Emmerson, A.; Jones, A. The quinolones: Decades of development and use. J. Antimicrob. Chemoth. 2003, 51, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Tanitame, A.; Oyamada, Y.; Ofuji, K.; Fujimoto, M.; Iwai, N.; Hiyama, Y.; Suzuki, K.; Ito, H.; Terauchi, H.; Kawasaki, M. Synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitors. Pyrazole derivatives. J. Med. Chem. 2004, 47, 3693–3696. [Google Scholar] [CrossRef]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA topoisomerases. EcoSal Plus 2015, 6, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.A.; Ammar, Y.A.; Elhagali, G.A.; Eyada, H.A.; Aboul-Magd, D.S.; Ragab, A. Discovery a novel of thiazolo [3, 2-a] pyridine and pyrazolo [3, 4-d] thiazole derivatives as DNA gyrase inhibitors; design, synthesis, antimicrobial activity, and some in-silico ADMET with molecular docking study. J. Mol. Struct. 2023, 1287, 135671. [Google Scholar] [CrossRef]
- Kumar, S.; Arora, A.; Kumar, R.; Senapati, N.N.; Singh, B.K. Recent advances in synthesis of sugar and nucleoside coumarin conjugates and their biological impact. Carbohyd. Res. 2023, 530, 108857. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef]
- Matos, M.J.; Pérez-Cruz, F.; Vazquez-Rodriguez, S.; Uriarte, E.; Santana, L.; Borges, F.; Olea-Azar, C. Remarkable antioxidant properties of a series of hydroxy-3-arylcoumarins. Bioorg. Med. Chem. 2013, 21, 3900–3906. [Google Scholar] [CrossRef]
- Patra, S.; Patra, P. A Brief Review on the Design, Synthesis and Biological Evaluation of Pyrazolo [c] coumarin Derivatives. Polycycl. Aromat. Comp. 2024, 44, 818–859. [Google Scholar] [CrossRef]
- Jin, Y.; He, S.; Wu, F.; Luo, C.; Ma, J.; Hu, Y. Novel Coumarin-furo [2, 3-d] pyrimidinone hybrid derivatives as anticancer agents: Synthesis, biological evaluation and molecular docking. Eur. J. Pharm. Sci. 2023, 188, 106520. [Google Scholar] [CrossRef]
- Wang, S.-F.; Yin, Y.; Wu, X.; Qiao, F.; Sha, S.; Lv, P.-C.; Zhao, J.; Zhu, H.-L. Synthesis, molecular docking and biological evaluation of coumarin derivatives containing piperazine skeleton as potential antibacterial agents. Bioorg. Med. Chem. 2014, 22, 5727–5737. [Google Scholar] [CrossRef] [PubMed]
- Barot, K.P.; Jain, S.V.; Kremer, L.; Singh, S.; Ghate, M.D. Recent advances and therapeutic journey of coumarins: Current status and perspectives. Med. Chem. Res. 2015, 24, 2771–2798. [Google Scholar] [CrossRef]
- Kasumbwe, K.; Venugopala, K.N.; Mohanlall, V.; Odhav, B. Antimicrobial and antioxidant activities of substituted halogenated coumarins. J. Med. Plant Res. 2014, 8, 274–281. [Google Scholar] [CrossRef]
- Chen, S.H.; Chan, N.-L.; Hsieh, T.-s. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef]
- Schoeffler, A.J.; Berger, J.M. DNA topoisomerases: Harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008, 41, 41–101. [Google Scholar] [CrossRef]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef]
- Barančoková, M.; Kikelj, D.; Ilaš, J. Recent progress in the discovery and development of DNA gyrase B inhibitors. Future Med. Chem. 2018, 10, 1207–1227. [Google Scholar] [CrossRef]
- Bates, A.D.; Maxwell, A. DNA Topology; Oxford University Press: New York, NY, USA, 2005. [Google Scholar]
- Almutawa, F.; Alnomair, N.; Wang, Y.; Hamzavi, I.; Lim, H.W. Systematic review of UV-based therapy for psoriasis. Am. J. Clin. Dermatol. 2013, 14, 87–109. [Google Scholar] [CrossRef]
- Panno, M.L.; Giordano, F. Effects of psoralens as anti-tumoral agents in breast cancer cells. World J. Clin. Oncol. 2014, 5, 348. [Google Scholar] [CrossRef]
- Songca, S.P.; Adjei, Y. Applications of antimicrobial photodynamic therapy against bacterial biofilms. Int. J. Mol. Sci. 2022, 23, 3209. [Google Scholar] [CrossRef] [PubMed]
- Soukos, N.S.; Ximenez-Fyvie, L.A.; Hamblin, M.R.; Socransky, S.S.; Hasan, T. Targeted antimicrobial photochemotherapy. Antimicrob. Agents Chemother. 1998, 42, 2595–2601. [Google Scholar] [CrossRef] [PubMed]
- Vaccarin, C.; Gabbia, D.; Franceschinis, E.; De Martin, S.; Roverso, M.; Bogialli, S.; Sacchetti, G.; Tupini, C.; Lampronti, I.; Gambari, R. Improved Trimethylangelicin Analogs for Cystic Fibrosis: Design, Synthesis and Preliminary Screening. Int. J. Mol. Sci. 2022, 23, 11528. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Barret, R. Lipinski’s Rule of Five. In Medicinal Chemistry: Fundamentals; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Abo-Salem, H.M.; Ali, E.A.; El-Mowafi, S.A.; Abdel-Aziz, M.S.; El-Sawy, E.R.; Abd El Salam, H.A. New sulfonamide-tethered coumarin derivatives as potential DNA gyrase inhibitors: Design, synthesis, antimicrobial evaluation, and in silico study. J. Mol. Struct. 2024, 1296, 136860. [Google Scholar] [CrossRef]
- Parvez, A.; Meshram, J.; Tiwari, V.; Sheik, J.; Dongre, R.; Youssoufi, M.H.; Hadda, T.B. Pharmacophores modeling in terms of prediction of theoretical physico-chemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti’s protocol. Eur. J. Med. Chem. 2010, 45, 4370–4378. [Google Scholar] [CrossRef]
- Hahn, M. Receptor surface models. 1. Definition and construction. J. Med. Chem. 1995, 38, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Maxwell, A.; Burton, N.P.; O’Hagan, N. High-throughput assays for DNA gyrase and other topoisomerases. Nucleic Acids Res. 2006, 34, 104. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
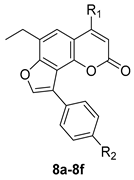 | ||
|---|---|---|
| Compound | R1 | R2 |
| 8a | Me | H |
| 8b | Me | Me |
| 8c | Me | OMe |
| 8d | Ph | H |
| 8e | Ph | Me |
| 8f | Ph | OMe |
 | ||||
|---|---|---|---|---|
| Compound | Inhibition Zone a (mm) | |||
| B. subtilis | S. aureus | E. coli | C. albicans | |
| 2a | 15 ± 1.41421 | 14 ± 1.41421 | - | 11 ± 0.81650 |
| 2b | 14 ± 0.81650 | 22 ± 1.41421 | - | 15 ± 1.41421 |
| 3a | 20 ± 0.81650 | 30 ± 1.41421 | 15 ± 1.41421 | 16 ± 0.81650 |
| 3b | 10 ± 1.41421 | 22 ± 0.81650 | 16 ± 1.41421 | 15 ± 1.41421 |
| 4a | 15 ± 0.81650 | 24 ± 1.41421 | - | 18 ± 1.41421 |
| 4b | 17 ± 0.81650 | 29 ± 0.81650 | - | 14 ± 1.41421 |
| 5a | 22 ± 0.81650 | 30 ± 0.81650 | 16 ± 1.41421 | 18 ± 0.81650 |
| 5b | 22 ± 1.41421 | 26 ± 0.81650 | - | - |
| 6a | 20 ± 1.41421 | 30 ± 1.41421 | 23 ± 1.41421 | 21 ± 0.81650 |
| 6b | 22 ± 1.41421 | 30 ± 1.41421 | 17 ± 1.41421 | 15 ± 1.41421 |
| 7a | 12 ± 1.41421 | 28 ± 1.41421 | 20 ± 0.81650 | - |
| 7b | 13 ± 1.41421 | 24 ± 1.41421 | 20 ± 1.41421 | - |
| 7c | 10 ± 1.41421 | - | 15 ± 1.41421 | - |
| 7d | 9 ± 1.41421 | - | 16 ± 1.41421 | - |
| 7e | - | - | 20 ± 0.81650 | - |
| 7f | - | - | 23 ± 0.81650 | 16 ± 0.81650 |
| 8a | 20 ± 1.41421 | 25 ± 1.41421 | 30 ± 1.41421 | - |
| 8b | 10 ± 1.41421 | 30 ± 1.41421 | 30 ± 1.41421 | - |
| 8c | 12 ± 1.41421 | 20 ± 0.81650 | 24 ± 1.41421 | - |
| 8d | 12 ± 0.81650 | 23 ± 1.41421 | 30 ± 0.81650 | - |
| 8e | 20 ± 1.41421 | 22 ± 1.41421 | 25 ± 0.81650 | - |
| 8f | 14 ± 1.41421 | 20 ± 1.41421 | 18 ± 0.81650 | - |
| DMF | - b | - | - | - |
| Xanthotoxin | 13 ± 0.81650 | - | 25 ± 0.81650 | 15 ± 0.81650 |
| Tobramycin | NT c | 21 ± 0.81650 | 20 ± 1.41421 | NT |
| Amphotercin B | NT | NT | NT | 14 ± 1.41421 |
| Compound | MIC (mg mL−1) a | MIC (mM) | ||
|---|---|---|---|---|
| Control b | Test c | Control | Test | |
| 2a | 6.25 | 6.25 | 22.78 | 22.78 |
| 2b | 6.25 | 6.25 | 18.58 | 18.58 |
| 3a | 0.78 | 0.78 | 3.04 | 3.04 |
| 3b | 3.125 | 3.125 | 9.82 | 9.82 |
| 4a | 3.125 | 0.78 | 10.40 | 2.60 |
| 4b | 3.125 | 3.125 | 8.62 | 8.62 |
| 5a | 3.125 | 0.78 | 11.07 | 2.76 |
| 5b | 3.125 | 0.78 | 9.07 | 2.26 |
| 6a | 3.125 | 0.39 | 11.23 | 1.40 |
| 6b | 3.125 | 1.5 | 9.18 | 4.41 |
| 7a | 6.25 | 1.5 | 19.39 | 4.65 |
| 7b | 1.5 | 0.19 | 4.46 | 0.56 |
| 7c | 12.5 | 3.125 | 35.47 | 8.87 |
| 7d | 6.25 | 6.25 | 16.26 | 16.26 |
| 7e | - d | - | - | - |
| 7f | - | - | - | - |
| 8a | 0.31 | 0.097 | 1.02 | 0.32 |
| 8b | 0.78 | 0.097 | 2.45 | 0.30 |
| 8c | 0.78 | 0.097 | 2.33 | 0.29 |
| 8d | 0.78 | 0.097 | 2.13 | 0.26 |
| 8e | 0.39 | 0.097 | 1.03 | 0.25 |
| 8f | 6.25 | 3.125 | 15.77 | 7.88 |
| DMF | - | - | - | - |
| Xanthotoxin | 12.5 | 1.5 | 57.82 | 6.94 |
| Compound | Docking Score (kcal/mol) | Predicted Binding Interactions | ||
|---|---|---|---|---|
| Binding Group | Amino Acid Residue | Interaction * | ||
| Reference ligand (II) | −8.45 | CO Amide | Arg136 | HB (2.0 Å) |
| Benzothiazole | Glu50, Arg76 | vdW | ||
| Pyrrole | Val43, Asn46, Val120, Val167 | vdW | ||
| 6a | −8.75 | Benzopyrone | Gly77 | HB (2.1 Å) |
| Benzopyrone | Glu50, Ile78 | vdW | ||
| Furan | Asn46, Ile78 | vdW | ||
| Fused phenyl | Val167 | vdW | ||
| 6b | −8.77 | Benzopyrone | Gly77 | HB (2.2 Å) |
| Benzopyrone | Glu50, Ile78 | vdW | ||
| Furan | Asn46 | vdW | ||
| Fused phenyl | Asn46 | vdW | ||
| Phenyl | Glu50, Arg76 | vdW | ||
| 7a | −8.72 | Benzopyrone | Gly77 | HB (2.0 Å) |
| Benzopyrone | Asn46, Glu50, Gly77, Ile78, Thr165 | vdW | ||
| Distal phenyl | Val43, Val71, Thr165, Val167 | vdW | ||
| Ethyl | Ile94 | vdW | ||
| 7b | −9.42 | Benzopyrone | Gly77 | HB (2.0 Å) |
| Benzopyrone | Ile78, Asn46, Glu50, Ile78, Thr165 | vdW | ||
| Distal phenyl | Val43, Val71, Val167 | vdW | ||
| Ethyl | Ile94 | vdW | ||
| 7c | −8.66 | Benzopyrone | Gly77 | HB (2.0 Å) |
| Benzopyrone | Asn46, Glu50, Ile78, Thr165 | vdW | ||
| Distal phenyl | Val43, Val120, Val167 | vdW | ||
| Distal methoxy | Val71, Asp73 | vdW | ||
| 7d | −8.42 | Benzopyrone | Asn46, Gly77, Ile94, Ile782 | vdW |
| Phenyl | Thr165, Val167 | vdW | ||
| Distal phenyl | Arg76, Pro79 | vdW | ||
| Spacer | Arg76, Arg136 | HB (2.8, 1.9 Å) | ||
| 7e | −8.58 | Benzopyrone | Gly77 | HB (2.0 Å) |
| Benzopyrone | Glu50, Thr165 | vdW | ||
| Phenyl | Glu50, Pro79 | vdW | ||
| Distal phenyl | Val43, Val120, Val167 | vdW | ||
| Distal methyl | Val71 | vdW | ||
| 7f | −9.95 | Benzopyrone | Gly77 | HB (2.1 Å) |
| Benzopyrone | Asn46, Glu50, Ile78, | vdW | ||
| Phenyl | Arg76, Pro79 | vdW | ||
| Distal phenyl | Val43, Val71, Val167 | vdW | ||
| Spacer | Met95 | vdW | ||
| Distal methoxy | Val71, Ile94 | vdW | ||
| 8a | −8.69 | Benzopyrone | Gly77 | HB (2.5 Å) |
| Benzopyrone | Glu50, Ile78 | vdW | ||
| Furan | Asn56, Ile78, Ile94 | vdW | ||
| Methyl | Arg76, Pro79 | vdW | ||
| Distal phenyl | Thr165, Val167 | vdW | ||
| 8b | −9.32 | Benzopyrone | Thr165 | HB (2.0 Å) |
| Benzopyrone | Glu50, Gly77, Ile78 | vdW | ||
| Furan | Asn46, Ile94 | vdW | ||
| Distal phenyl | Val71, Thr165 | vdW | ||
| Distal methyl | Val43, Val71, Val167 | vdW | ||
| 8c | −9.17 | Benzopyrone | Thr165 | HB (2.1 Å) |
| Benzopyrone | Glu50, Gly77, Arg76, Pro78 | vdW | ||
| Furan | Asn46, Ile78, Ile94 | vdW | ||
| Distal phenyl | Thr165 | vdW | ||
| Distal methoxy | Val71, Asp73 | vdW | ||
| 8d | −9.94 | Benzopyrone | Thr165 | HB (2.0 Å) |
| Benzopyrone | Gly77, Ile78, Thr165 | vdW | ||
| Furan | Asn46, Ile78, Val120 | vdW | ||
| Phenyl | Glu50, Arg76 | vdW | ||
| Distal phenyl | Val43, Val71, Val167 | vdW | ||
| 8e | −10.05 | Benzopyrone | Thr165 | HB (2.1 Å) |
| Benzopyrone | Glu50, Gly77, Ile78 | vdW | ||
| Furan | Ile78, Ile94 | vdW | ||
| Phenyl | Glu50, Arg76, Pro79 | vdW | ||
| Distal phenyl | Val43, Val167 | vdW | ||
| Distal methyl | Val71 | vdW | ||
| 8f | −10.10 | Benzopyrone | Gly77 | HB (2.3 Å) |
| Benzopyrone | Asn46, Ile78, Ile94 | vdW | ||
| Furan | Ile94 | vdW | ||
| Phenyl | Val43, Val167 | vdW | ||
| Distal phenyl | Glu50, Arg76, Pro79 | vdW | ||
| Compound | IC50 (μg mL−1) a | IC50 (μM) a |
|---|---|---|
| 8a | 2.40 ± 0.03559 | 7.89 ± 0.11694 |
| 8b | 12.30 ± 0.163299 | 38.63 ± 0.51292 |
| 8d | 0.28 ± 0.008165 | 0.76 ± 0.02228 |
| Novobiocin | 0.25 ± 0.024944 | 0.41 ± 0.04072 |
| Ciprofloxacin | 0.90 ± 0.073636 | 2.72 ± 0.22223 |
| Properties a | Tested Compounds | |
|---|---|---|
| Omeprazole | 8d | |
| Physicochemical properties | ||
| MW (g/mol) | 345.42 | 366.41 |
| Number of rotatable bonds | 5 | 3 |
| HBA | 5 | 3 |
| HBD | 1 | 0 |
| MR | 93.70 | 112.90 |
| TPSA (Å2) | 96.31 | 43.35 |
| Lipophilicity | ||
| Log Po/w (iLOGP) | 2.14 | 3.83 |
| Log Po/w (XLOGP) | 2.23 | 5.91 |
| Log Po/w (WLOGP) | 3.61 | 6.44 |
| Log Po/w (MLOGP) | 0.91 | 4.39 |
| Log Po/w (SILICOS-IT) | 3.16 | 6.83 |
| Consensus Log Po/w | 2.41 | 5.48 |
| Water solubility | ||
| Log S (ESOL) | −3.52 | −6.30 |
| Pharmacokinetics | ||
| GI absorption | High | High |
| BBB permeant | No | No |
| P-gp substrate | Yes | Yes |
| CYP1A2 inhibitor | Yes | Yes |
| CYP2C19 inhibitor | Yes | Yes |
| CYP2C9 inhibitor | Yes | No |
| CYP2D6 inhibitor | Yes | No |
| CYP3A4 inhibitor | Yes | No |
| Log Kp (skin permeation) cm/s | −6.82 | −4.34 |
| Drug likeness | ||
| Lipinski | Yes | Yes |
| Ghose | Yes | No |
| Veber | Yes | Yes |
| Egan | Yes | No |
| Muegge | Yes | No |
| Bioavailability score | 0.55 | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd El-Haleem, A.; Ammar, U.; Masci, D.; El-Ansary, S.; Abdel Rahman, D.; Abou-Elazm, F.; El-Dydamony, N. Discovery of Benzopyrone-Based Candidates as Potential Antimicrobial and Photochemotherapeutic Agents through Inhibition of DNA Gyrase Enzyme B: Design, Synthesis, In Vitro and In Silico Evaluation. Pharmaceuticals 2024, 17, 1197. https://doi.org/10.3390/ph17091197
Abd El-Haleem A, Ammar U, Masci D, El-Ansary S, Abdel Rahman D, Abou-Elazm F, El-Dydamony N. Discovery of Benzopyrone-Based Candidates as Potential Antimicrobial and Photochemotherapeutic Agents through Inhibition of DNA Gyrase Enzyme B: Design, Synthesis, In Vitro and In Silico Evaluation. Pharmaceuticals. 2024; 17(9):1197. https://doi.org/10.3390/ph17091197
Chicago/Turabian StyleAbd El-Haleem, Akram, Usama Ammar, Domiziana Masci, Sohair El-Ansary, Doaa Abdel Rahman, Fatma Abou-Elazm, and Nehad El-Dydamony. 2024. "Discovery of Benzopyrone-Based Candidates as Potential Antimicrobial and Photochemotherapeutic Agents through Inhibition of DNA Gyrase Enzyme B: Design, Synthesis, In Vitro and In Silico Evaluation" Pharmaceuticals 17, no. 9: 1197. https://doi.org/10.3390/ph17091197
APA StyleAbd El-Haleem, A., Ammar, U., Masci, D., El-Ansary, S., Abdel Rahman, D., Abou-Elazm, F., & El-Dydamony, N. (2024). Discovery of Benzopyrone-Based Candidates as Potential Antimicrobial and Photochemotherapeutic Agents through Inhibition of DNA Gyrase Enzyme B: Design, Synthesis, In Vitro and In Silico Evaluation. Pharmaceuticals, 17(9), 1197. https://doi.org/10.3390/ph17091197