
Discovery of Dual TRPA1 and TRPV1 Antagonists as Novel Therapeutic Agents for Pain

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Biological Activity
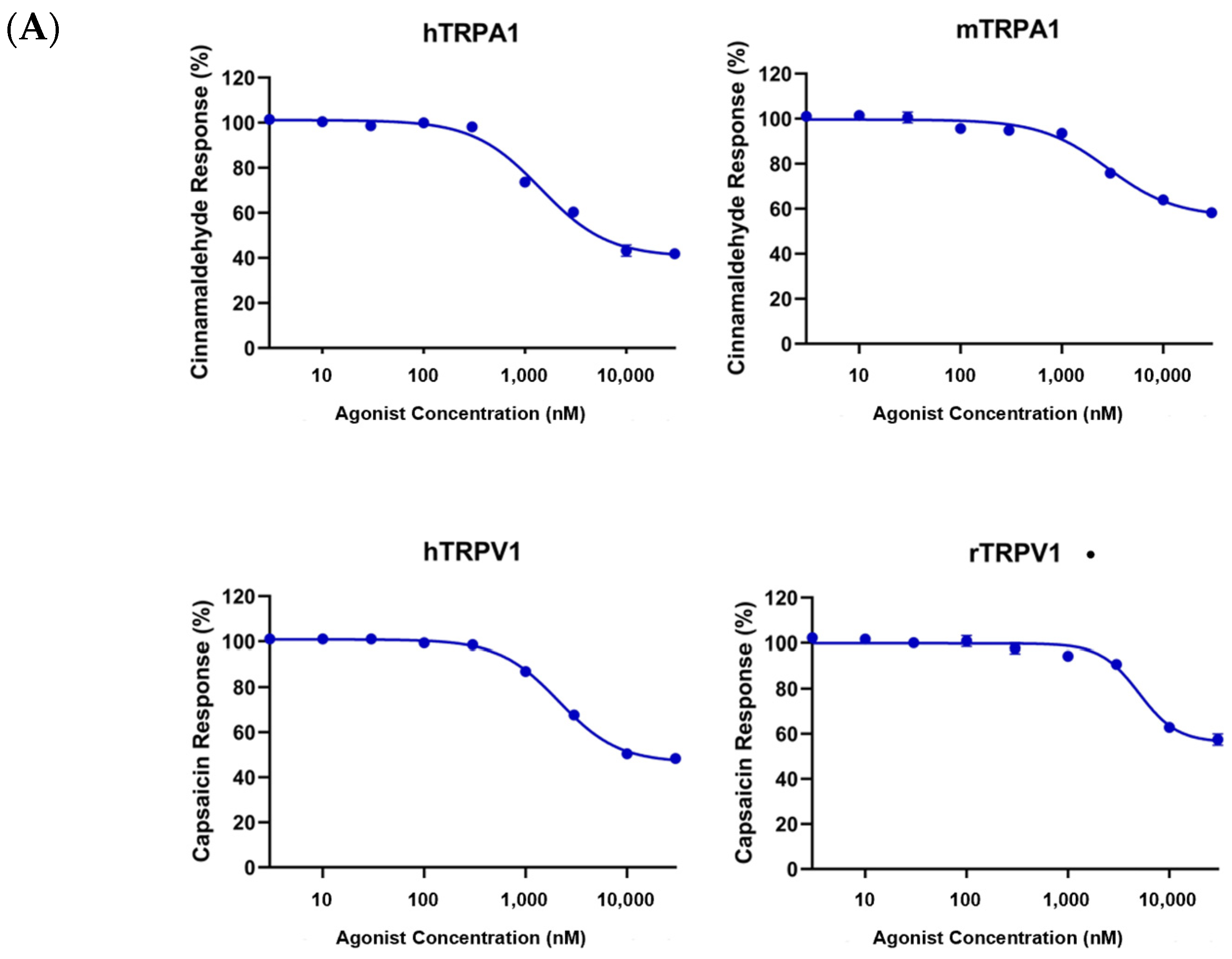
2.2.1. In Vitro Activity
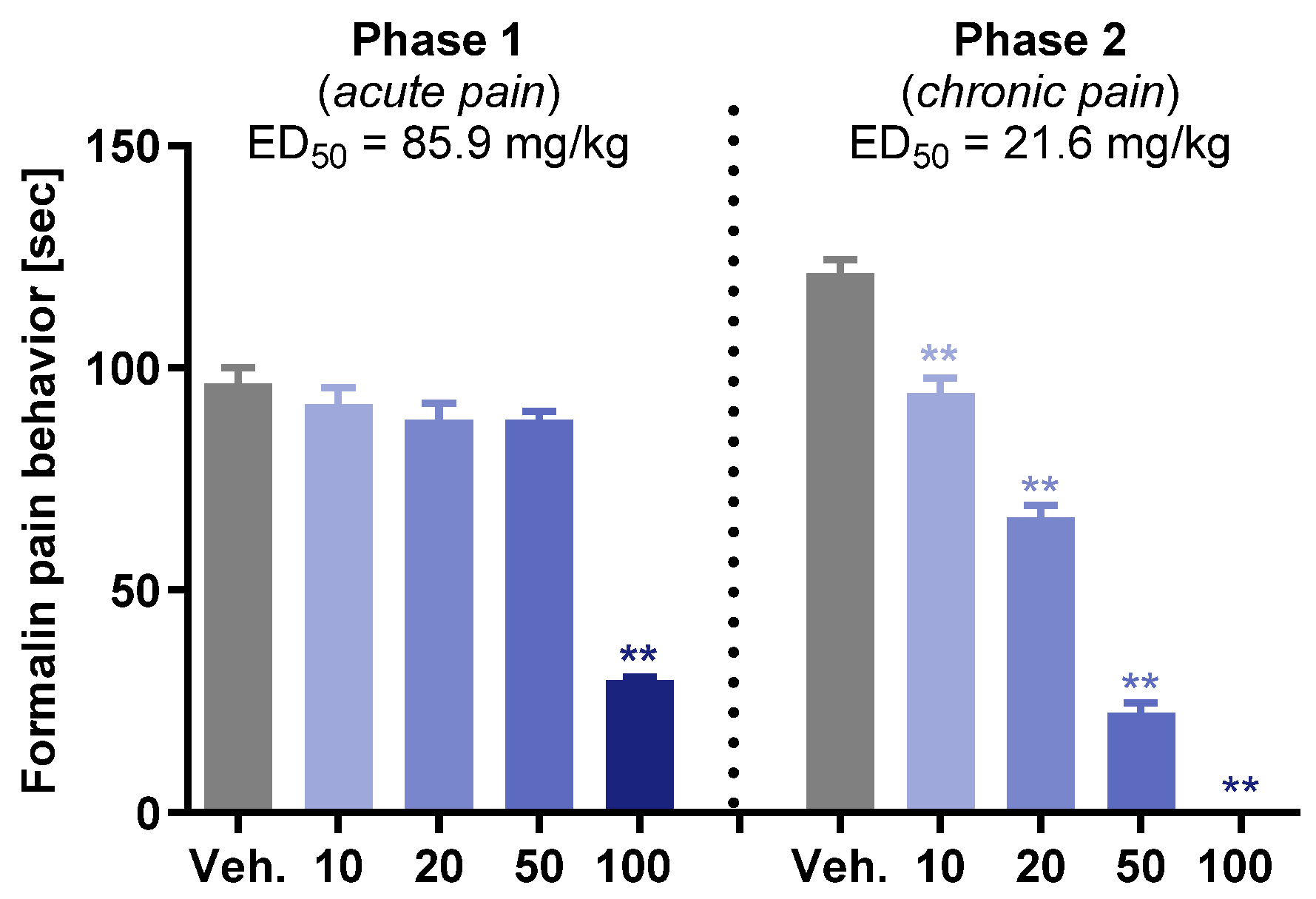
2.2.2. In Vivo Activity
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Reduction (Procedure A)
3.1.2. General Procedure for Cyanation (Procedure B)
3.1.3. General Procedure for Preparation of N’-Hydroxy-Imidamide (Procedure C)
3.1.4. General Procedure for Formation of Cyano Ester (Procedure D)
3.1.5. General Procedure for Krapcho Decarboxylation (Procedure E)
3.1.6. General Procedure for Hydrolysis (Procedure F)
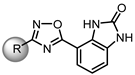
3.1.7. General Procedure for Condensation of 1,2,4-Oxadiazole (Procedure G)
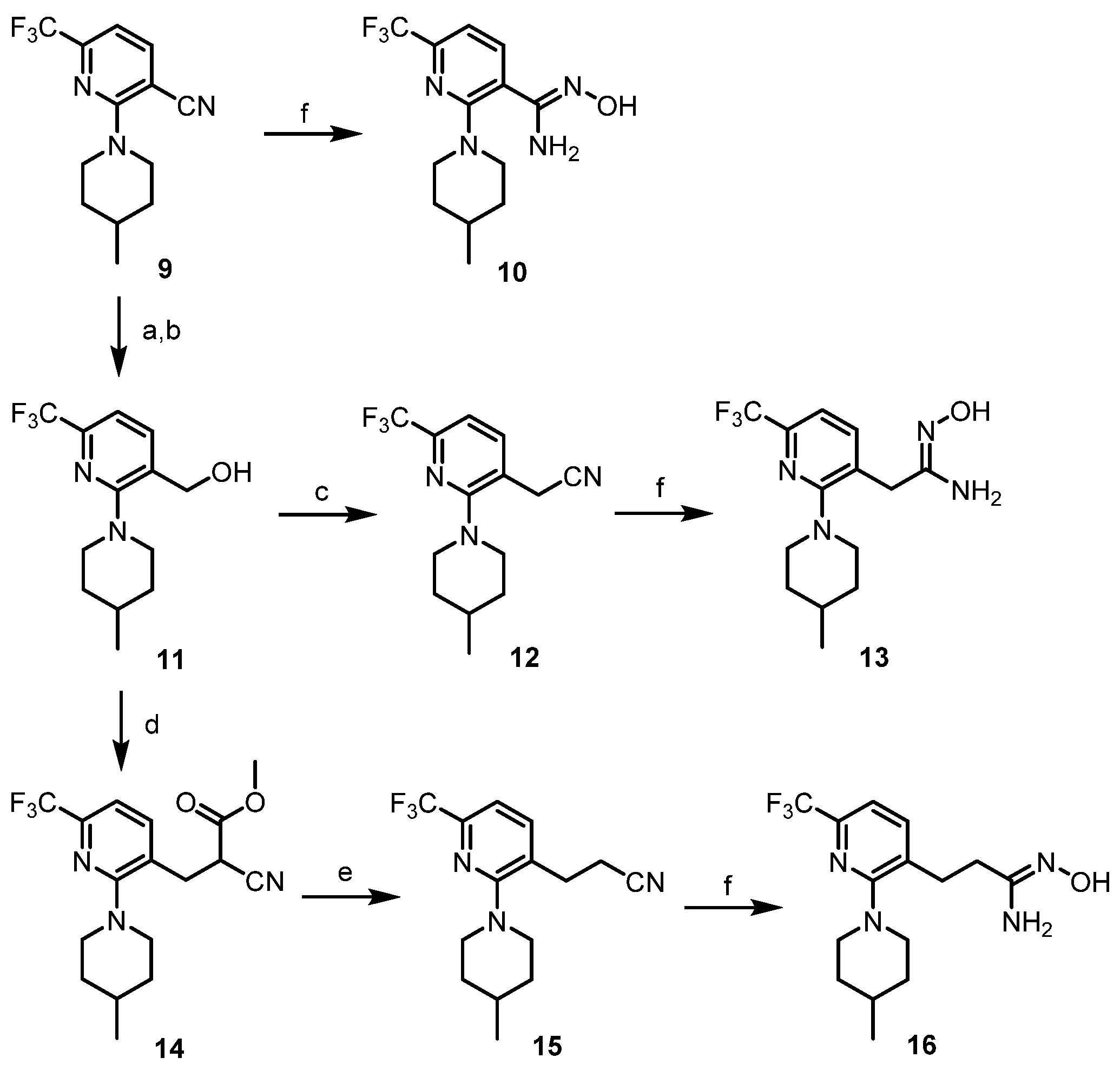
- N’-Hydroxy-2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)nicotinimidamide (10). Compound 10 was synthesized from 9 according to general procedure C. 62% yield, pale yellow oil; 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 5.83 (s, 2H), 3.88 (d, J = 12.8 Hz, 2H), 2.74 (t, J = 11.4 Hz, 2H), 1.59 (d, J = 12.8 Hz, 2H), 1.47–1.54 (m, 1H), 1.13–1.23 (m, 2H), 0.88 (d, J = 6.4 Hz, 3H).
- (2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)methanol (11). Potassium hydroxide (8.0 equiv.) was added to a solution of 9 (1.0 equiv.) in ethylene glycol and the mixture was refluxed for 24 h. After completion of the reaction, the mixture was cooled to 0 °C and diluted with water. The pH of the resulting mixture was adjusted to pH 2 using 1 N HCl and extracted with CH2Cl2. The organic layer was dried over MgSO4, and concentrated in vacuo. Compound 11 was synthesized from the obtained residue according to general procedure A. 66% yield, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 7.6 Hz, 1H), 7.30 (d, J = 7.2 Hz, 1H), 4.75 (s, 2H), 3.37 (d, J = 12.8 Hz, 2H), 2.89 (td, J = 12.4, 2.3 Hz, 2H), 1.77 (d, J = 10.8 Hz, 2H), 1.54–1.62 (m, 1H), 1.35 (qd, J = 12.4, 3.6 Hz, 2H), 0.99 (d, J = 6.4 Hz, 3H).
- 2-(2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)acetonitrile (12). Compound 12 was synthesized from 11 according to general procedure B. 96% yield, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 3.76 (s, 2H), 3.28 (d, J = 13.3 Hz, 2H), 2.88 (td, J = 12.6, 2.1 Hz, 2H), 1.76 (d, J = 12.8 Hz, 2H), 1.53–1.62 (m, 1H), 1.34 (qd, J = 12.2, 3.7 Hz, 2H), 1.00 (d, J = 6.4 Hz, 3H).
- N’-Hydroxy-2-(2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)acetimidamide (13). Compound 13 was synthesized from 12 according to general procedure C. 94% yield, white solid; 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.36 (d, J = 7.8 Hz, 1H), 5.53 (s, 2H), 3.38 (d, J = 12.8 Hz, 2H), 3.32 (s, 2H), 2.68 (t, J = 11.4 Hz, 2H), 1.66 (d, J = 12.8 Hz, 2H), 1.45–1.54 (m, 1H), 1.25 (qd, J = 12.0, 3.4 Hz, 2H), 0.92 (d, J = 6.4 Hz, 3H).
- Methyl 2-cyano-3-(2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)propanoate (14). Compound 14 was synthesized from 11 according to general procedure D. 76% yield, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 4.33 (dd, J = 9.1, 5.9 Hz, 1H), 4.25 (qd, J = 7.2, 1.4 Hz, 2H), 3.45 (dd, J = 14.6, 5.9 Hz, 1H), 3.32 (t, J = 11.0 Hz, 2H), 3.13 (dd, J = 14.4, 9.4 Hz, 1H), 2.95 (td, J = 12.5, 2.4 Hz, 1H), 2.83 (td, J = 12.3, 2.3 Hz, 1H), 1.77 (t, J = 12.2 Hz, 2H), 1.54–1.61 (m, 1H), 1.31–1.40 (m, 2H), 0.99 (d, J = 6.4 Hz, 3H).
- 3-(2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)propanenitrile (15). Compound 15 was synthesized from 14 according to general procedure E. 38% yield, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 7.8 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 3.32 (d, J = 12.9 Hz, 2H), 3.02 (t, J = 7.6 Hz, 2H), 2.87 (td, J = 12.6, 2.1 Hz, 2H), 2.74 (t, J = 7.4 Hz, 2H), 1.75 (d, J = 12.9 Hz, 2H), 1.55–1.61 (m, 1H), 1.32 (qd, J = 12.2, 3.9 Hz, 2H), 0.99 (d, J = 6.9 Hz, 3H).
- N’-Hydroxy-3-(2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)propanimidamide (16). Compound 16 was synthesized from 15 according to general procedure C. 85% yield, colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 8.80 (s, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 5.41 (s, 2H), 3.34 (d, J = 12.9 Hz, 2H), 2.82 (t, J = 7.8 Hz, 2H), 2.70 (t, J = 11.3 Hz, 2H), 2.30 (t, J = 8.0 Hz, 2H), 1.67 (d, J = 10.6 Hz, 2H), 1.45–1.53 (m, 1H), 1.24 (qd, J = 12.2, 3.4 Hz, 2H), 0.92 (d, J = 6.4 Hz, 3H).
- 4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazole-5-carboxamide (19). 17 (1.0 equiv.) was dissolved in ammonia solution 7 N in methanol (40 equiv.) and the mixture was heated at 60 °C for 15 h. After completion of the reaction, the solvent was concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired compound. 94% yield, white solid; 1H NMR (400 MHz, CDCl3) δ 7.70 (t, J = 1.8 Hz, 1H), 7.57 (dt, J = 7.3, 1.6 Hz, 1H), 7.44–7.51 (m, 2H), 5.75 (s, 2H).
- 4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazole-5-carbonitrile (20). Pyridine (30 equiv.) was added to a solution of 19 (1.0 equiv.) in THF at room temperature. After 30 min, trifluoroacetic anhydride (1.2 equiv.) was then added slowly at 0 °C, and the mixture was allowed to stir for an additional 1 h at room temperature. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired compound. 91% yield, white solid; 1H NMR (400 MHz, CDCl3) δ 8.14 (t, J = 1.8 Hz, 1H), 8.06 (dt, J = 7.3, 1.6 Hz, 1H), 7.46–7.52 (m, 2H).
- 4-(3-Chlorophenyl)-N’-hydroxy-2-(trifluoromethyl)thiazole-5-carboximidamide (21). Compound 21 was synthesized from 20 according to general procedure C. 57% yield, white solid; 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 7.74–7.75 (m, 1H), 7.68–7.71 (m, 1H), 7.48–7.49 (m, 2H), 6.17 (s, 2H).
- (4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)methanol (22). Compound 22 was synthesized from 17 according to general procedure A. 80% yield, white solid; 1H NMR (400 MHz, CDCl3) δ 7.66–7.67 (m, 1H), 7.51–7.54 (m, 1H), 7.39–7.40 (m, 2H), 5.02 (s, 2H).
- (2-(tert-Butyl)-4-(3-chlorophenyl)thiazol-5-yl)methanol (23). Compound 23 was synthesized from 18 according to general procedure A. 95% yield, yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.71–7.72 (m, 1H), 7.56 (dt, J = 7.2, 1.6 Hz, 1H), 7.32–7.39 (m, 2H), 4.88 (s, 2H), 1.47 (s, 9H).
- 2-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)acetonitrile (24). Compound 24 was synthesized from 22 according to general procedure B. 38% yield, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.59–7.61 (m, 1H), 7.44–7.46 (m, 3H), 4.04 (s, 2H).
- 2-(2-(tert-Butyl)-4-(3-chlorophenyl)thiazol-5-yl)acetonitrile (25). Compound 25 was synthesized from 23 according to general procedure B. 77% yield, yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.57–7.58 (m, 1H), 7.35–7.43 (m, 3H), 3.91 (s, 2H), 1.45 (s, 9H).
- 2-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)-N’-hydroxyacetimidamide (26). Compound 26 was synthesized from 24 according to general procedure C. 64% yield, pale yellow oil; 1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 7.68–7.69 (m, 1H), 7.61–7.64 (m, 1H), 7.50–7.52 (m, 2H), 5.74 (s, 2H), 3.76 (s, 2H).
- 2-(2-(tert-Butyl)-4-(3-chlorophenyl)thiazol-5-yl)-N’-hydroxyacetimidamide (27). Compound 27 was synthesized from 25 according to general procedure C. 91% yield, colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 7.67 (t, J = 1.8 Hz, 1H), 7.61 (dt, J = 7.4, 1.4 Hz, 1H), 7.38–7.47 (m, 2H), 5.59 (s, 2H), 3.56 (s, 2H), 1.35 (s, 9H).
- Methyl 3-(4-(3-chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)-2-cyanopropanoate (28). Compound 28 was synthesized from 22 according to general procedure D. 95% yield, colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.58–7.59 (m, 1H), 7.42–7.48 (m, 3H), 3.83 (s, 3H), 3.76 (dd, J = 7.5, 5.7 Hz, 1H), 3.69 (dd, J = 15.3, 5.9 Hz, 1H), 3.60 (q, J = 7.6 Hz, 1H).
- 3-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)propanenitrile (29). Compound 29 was synthesized from 28 according to general procedure E. 54% yield, yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.57–7.58 (m, 1H), 7.41–7.47 (m, 3H), 3.37 (t, J = 7.1 Hz, 2H), 2.70 (t, J = 7.4 Hz, 2H).
- 3-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)-N’-hydroxypropanimidamide (30). Compound 30 was synthesized from 29 according to general procedure C. 77% yield, colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.62–7.63 (m, 1H), 7.56–7.58 (m, 1H), 7.50–7.51 (m, 2H), 5.48 (s, 2H), 3.21 (t, J = 7.1 Hz, 2H), 2.36 (t, J = 7.4 Hz, 2H).
- Ethyl 3-(tert-butyl)-1-(3-chlorophenyl)-1H-pyrazole-5-carboxylate (32). To a solution of 31 (1.0 equiv.) in CH2Cl2 were added (3-chlorophenyl)boronic acid (2.0 equiv.), copper(II) acetate (1.5 equiv.), and pyridine (2.0 equiv.). The mixture was stirred for 22 h at room temperature, filtered through Celite, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired compound. 84% yield, yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.44–7.45 (m, 1H), 7.30–7.37 (m, 3H), 6.88 (s, 1H), 4.23 (q, J = 7.2 Hz, 2H), 1.34 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H).
- 2-(3-(tert-Butyl)-1-(3-chlorophenyl)-1H-pyrazol-5-yl)acetonitrile (33). Compound 33 was synthesized from 32 according to general procedures A and B. 84% yield, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.38–7.45 (m, 3H), 7.29 (dt, J = 7.3, 1.8 Hz, 1H), 6.41 (s, 1H), 3.73 (s, 2H), and 1.33 (s, 9H).
- 2-(3-(tert-Butyl)-1-(3-chlorophenyl)-1H-pyrazol-5-yl)-N’-hydroxyacetimidamide (34). Compound 34 was synthesized from 33 according to general procedure C. 95% yield, white solid; 1H NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 7.59–7.60 (m, 1H), 7.40–7.53 (m, 3H), 6.24 (s, 1H), 5.51 (s, 2H), 3.36 (s, 2H), 1.22 (s, 9H).
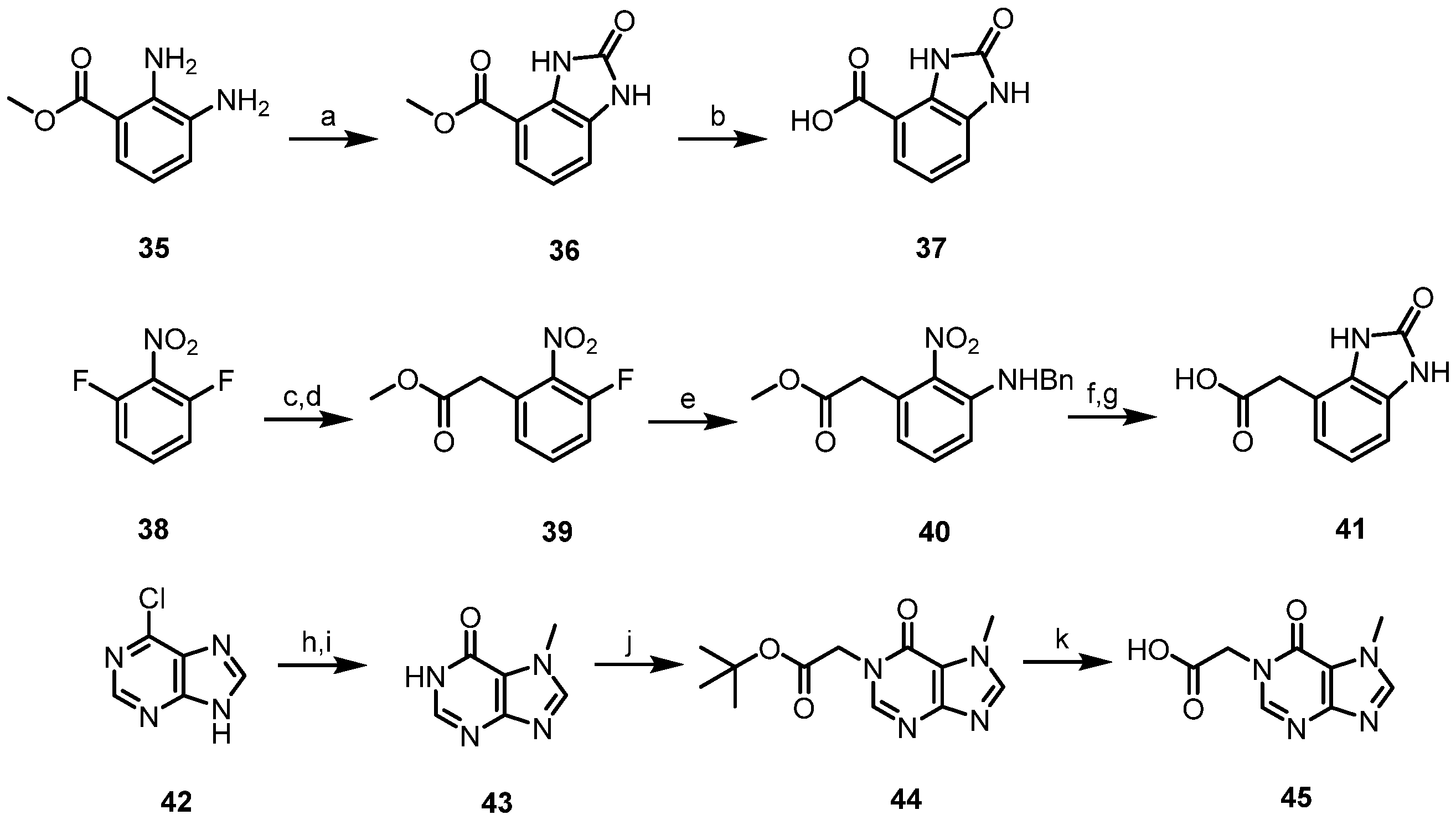
- Methyl 2-oxo-2,3-dihydro-1H-benzo[d]imidazole-4-carboxylate (36). To a solution of methyl 2,3-diaminobenzoate (1.0 equiv.) in THF were added 1,1′-carbonyldiimidazole (1.05 equiv.) and triethylamine (1.0 equiv.) and the mixture was refluxed for 21 h. After completion of the reaction, the solvent was concentrated in vacuo. After recrystallization with hexane, the desired compound was isolated. 95% yield, light brown solid; 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 10.68 (s, 1H), 7.44 (dd, J = 8.0, 1.1 Hz, 1H), 7.13 (d, J = 7.4 Hz, 1H), 7.01 (t, J = 7.8 Hz, 1H), 3.82 (s, 3H).
- 2-Oxo-2,3-dihydro-1H-benzo[d]imidazole-4-carboxylic acid (37). Compound 37 was synthesized from 36 according to general procedure F. 75% yield, brown solid; 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 10.36 (s, 1H), 7.40 (dd, J = 8.0, 1.1 Hz, 1H), 7.09 (d, J = 7.4 Hz, 1H), and 6.97 (t, J = 7.8 Hz, 1H).
- Methyl 2-(3-fluoro-2-nitrophenyl)acetate (39). Dimethylmalonate (1.5 equiv.) was added slowly to a suspension of sodium hydride with 60% dispersion in paraffin liquid (1.5 equiv.) in DMF at 0 °C, and the mixture was stirred for 30 min at room temperature. 1,3-difluoro-2-nitrobenzene (1.0 equiv.) was then added slowly at 0 °C, and the mixture was allowed to stir at 50 °C for 15 h. After dilution with water at 0 °C, the reaction mixture was quenched with 1 N HCl and extracted with EtOAc. The organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired compound. 90% yield, pale yellow solid; 1H NMR (400 MHz, CDCl3) δ 7.47 (td, J = 8.3, 5.5 Hz, 1H), 7.21 (t, J = 8.9 Hz, 1H), 7.15 (d, J = 7.3 Hz, 1H), 3.82 (s, 2H), and 3.70 (s, 3H).
- Methyl 2-(3-(benzylamino)-2-nitrophenyl)acetate (40). Methyl 2-(3-fluoro-2-nitrophenyl)acetate (1.0 equiv.), synthesized from 39 according to general procedure, was added to a solution of benzylamine (1.5 equiv.) in DMF. E. Triethylamine (1.5 equiv.) was then added, and the mixture was stirred at 60 °C for 13 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired compound. 89% yield, orange oil; 1H NMR (400 MHz, CDCl3) δ 7.57 (s, 1H), 7.23–7.37 (m, 6H), 6.76 (d, J = 8.6 Hz, 1H), 6.53 (d, J = 7.3 Hz, 1H), 4.47 (d, J = 5.5 Hz, 2H), 3.88 (s, 2H), and 3.71 (s, 3H).
- 2-(2-Oxo-2,3-dihydro-1H-benzo[d]imidazol-4-yl)acetic acid (41). Palladium 10% on activated carbon was added to a solution of 40 (1.0 equiv.) in methanol and the mixture was stirred under hydrogen for 1 h at room temperature. The mixture was filtered through Celite and the filterate was concentrated in vacuo. 1,1′-carbonyldiimidazole (1.1 equiv.) was added to a solution of obtained residue in THF and the mixture was stirred for 1 h at room temperature. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel. Compound 41 was synthesized from the obtained residue according to general procedure F. 86% yield, light brown solid; 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 10.54 (s, 1H), and 6.73–6.84 (m, 3H), 3.58 (s, 2H).
- 7-Methyl-1,7-dihydro-6H-purin-6-one (43). Under a nitrogen atmosphere, methylmagnesium chloride 3.0 M solution in THF (1.1 equiv.) was slowly added to a solution of 6-chloro-9H-purine (1.0 equiv.) in anhydrous THF, and the mixture was stirred for 1 h at room temperature. Iodomethane (3.0 equiv.) was then added, and the mixture was allowed to stir at 50 °C for 15 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel. The obtained residue was dissolved in formic acid and heated to 75 °C. After concentration in vacuo, the residue was re-dissolved in ethanol, and the mixture was refluxed for an additional 30 min. The solvent was concentrated in vacuo to afford the desired compound, which was used in the next step without further purification. 86% yield, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 8.27 (s, 1H), 7.95 (s, 1H), and 3.93 (s, 3H).
- tert-Butyl 2-(7-methyl-6-oxo-6,7-dihydro-1H-purin-1-yl)acetate (44). To a stirred solution of 43 (1.0 equiv.), tert-butyl bromoacetate (1.0 equiv.) and potassium carbonate (1.2 equiv.) in DMF was added, and the mixture was stirred at 50 °C for 6 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired compound. 36% yield, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.21 (s, 1H), 8.16 (s, 1H), 4.67 (s, 2H), 3.92 (s, 3H), and 1.39 (s, 9H).
- 2-(7-Methyl-6-oxo-6,7-dihydro-1H-purin-1-yl)acetic acid (45). 44 was dissolved in hydrochloric acid and the mixture was stirred for 30 min at room temperature. After completion of the reaction, the solvent was concentrated in vacuo to afford the desired compound, which was used in the next step without further purification. 99% yield, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.27 (s, 1H), 8.25 (s, 1H), 4.70 (s, 2H), 3.93 (s, 3H).
- 4-(3-(4-Chlorophenethyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (46). Compound 46 was synthesized from 3-(4-chlorophenyl)-N’-hydroxypropanimidamide and 37 according to general procedure G. 72% yield, 99.01% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.74 (s, 1H), 7.55 (dd, J = 8.0, 1.1 Hz, 1H), 7.26–7.31 (m, 4H), 7.18 (dd, J = 7.8, 0.9 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 3.11–3.14 (m, 2H), 3.02–3.06 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.39, 170.36, 155.56, 140.06, 131.41, 131.31, 130.88, 129.24, 128.79, 121.63, 119.73, 113.06, 104.91, 31.68, 27.42; HRMS (FAB) calc. for C17H13ClN4O2 [M + H]+ 341.0805, found: 341.0795.
- 4-(3-((4′-(Trifluoromethyl)-[1,1′-biphenyl]-3-yl)methyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (47). Compound 47 was synthesized from N’-hydroxy-2-(4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)acetimidamide and 37 according to general procedure G. 55% yield, 99.47% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.74 (s, 1H), 7.86 (d, J = 8.2 Hz, 2H), 7.77–7.79 (m, 3H), 7.60 (td, J = 4.3, 2.3 Hz, 1H), 7.55 (dd, J = 8.0, 1.1 Hz, 1H), 7.43–7.48 (m, 2H), 7.16 (d, J = 7.3 Hz, 1H), 7.09 (t, J = 7.8 Hz, 1H), 4.24 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.87, 170.07, 155.55, 144.47, 139.39, 137.33, 131.42, 129.92, 129.71, 129.26, 128.43 (d, J = 31.5 Hz), 128.42, 128.08, 126.35, 126.31, 126.27, 126.22, 121.64, 119.89, 113.11, 104.89, 31.95; HRMS (FAB) calc. for C23H15F3N4O2 [M + H]+ 437.1225, found: 437.1241.
- 4-(3-((4′-(Trifluoromethyl)-[1,1′-biphenyl]-4-yl)methyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (48). Compound 48 was synthesized from N’-hydroxy-2-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)acetimidamide and 37 according to general procedure G. 71% yield, 98.85% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 10.75 (s, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 8.2 Hz, 2H), 7.52–7.57 (m, 3H), 7.17 (d, J = 6.9 Hz, 1H), 7.10 (t, J = 7.8 Hz, 1H), 4.22 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.92, 170.03, 155.57, 144.36, 137.76, 136.79, 131.41, 130.38, 129.23, 128.29 (d, J = 31.6 Hz), 127.92, 127.79, 126.32, 126.30, 126.26, 121.66, 119.93, 113.13, 104.86, 31.60; HRMS (FAB) calc. for C23H15F3N4O2 [M + H]+ 437.1225, found: 437.1208.
- 4-(3-(2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (49). Compound 49 was synthesized from 10 and 37 according to general procedure G. 59% yield, 99.35% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 10.96 (s, 1H), 8.79 (d, J = 7.3 Hz, 1H), 7.65 (dd, J = 8.0, 1.1 Hz, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 7.3 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 3.66 (d, J = 13.3 Hz, 2H), 2.81 (t, J = 11.7 Hz, 2H), 1.59 (d, J = 12.3 Hz, 2H), 1.48–1.53 (m, 1H), 1.23 (qd, J = 12.1, 3.4 Hz, 2H), 0.88 (d, J = 5.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 173.32, 167.48, 159.33, 155.65, 144.14, 131.48, 129.39, 121.82 (d, J = 275.9 Hz), 121.68, 119.89, 114.22, 113.32, 111.34, 104.61, 49.66, 33.86, 30.63, 22.32; HRMS (FAB) calc. for C21H19F3N6O2 [M + H]+ 445.1600, found: 445.1592.
- 4-(3-((2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)methyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (50). Compound 50 was synthesized from 13 and 37 according to general procedure G. 30% yield, 99.42% purity, brown solid; 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 10.75 (s, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.55 (dd, J = 8.2, 0.9 Hz, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 4.23 (s, 2H), 3.39 (d, J = 12.8 Hz, 2H), 2.74 (t, J = 11.4 Hz, 2H), 1.66 (d, J = 12.8 Hz, 2H), 1.46–1.51 (m, 1H), 1.20–1.29 (m, 2H), 0.90 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 173.97, 169.32, 162.34, 155.58, 143.80, 141.25, 131.42, 129.23, 128.00, 122.12 (d, J = 272.2 Hz), 121.67, 119.98, 114.77, 113.18, 104.82, 50.75, 34.31, 30.69, 27.88, 22.38; HRMS (FAB) calc. for C22H21F3N6O2 [M + H]+ 459.1756, found: 459.1762.
- 4-(3-(2-(2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)ethyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (51). Compound 51 was synthesized from 16 and 37 according to general procedure G. 57% yield, 99.45% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.66 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.56 (dd, J = 8.0, 1.1 Hz, 1H), 7.37 (d, J = 7.4 Hz, 1H), 7.18 (dd, J = 7.8, 0.9 Hz, 1H), 7.11 (t, J = 7.8 Hz, 1H), 3.34–3.36 (m, 2H), 3.13–3.19 (m, 4H), 2.71 (t, J = 11.4 Hz, 2H), 1.64 (d, J = 11.0 Hz, 2H), 1.43–1.48 (m, 1H), 1.19–1.26 (m, 2H), 0.86 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 173.50, 170.42, 162.50, 155.50, 142.99 (q, J = 33.4 Hz), 140.01, 132.27, 131.41, 129.25, 122.22 (d, J = 271.1 Hz), 121.65, 119.74, 114.80, 113.08, 104.92, 50.86, 34.37, 30.76, 28.36, 25.49, 22.25; HRMS (FAB) calc. for C23H23F3N6O2 [M + H]+ 473.1913, found: 473.1906.
- 4-(3-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (52). Compound 52 was synthesized from 21 and 37 according to general procedure G. 64% yield, 99.28% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 10.75 (s, 1H), 7.89 (t, J = 1.8 Hz, 1H), 7.76 (dt, J = 7.3, 1.4 Hz, 1H), 7.55–7.58 (m, 2H), 7.51 (t, J = 8.0 Hz, 1H), 7.21 (dd, J = 7.8, 0.9 Hz, 1H), 7.12 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 174.21, 162.07, 155.77 (d, J = 40.1 Hz), 155.54, 154.85, 134.78, 133.44, 131.52, 130.73, 130.31, 129.84, 129.48, 128.88, 124.19, 121.76, 120.10, 119.90 (d, J = 270.2 Hz), 113.67, 103.99; HRMS (FAB) calc. for C19H9ClF3N5O2S [M + H]+ 464.0196, found: 464.0211.
- 4-(3-(2-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)ethyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (53). Compound 53 was synthesized from 30 and 37 according to general procedure G. 37% yield, 98.60% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.66 (s, 1H), 7.57–7.59 (m, 2H), 7.50 (d, J = 6.9 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.37–7.40 (m, 1H), 7.18 (dd, J = 7.5, 1.1 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 3.63 (t, J = 7.1 Hz, 2H), 3.17 (t, J = 7.1 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.66, 169.42, 155.54, 151.57, 150.96 (d, J = 40.1 Hz), 139.59, 135.49, 133.93, 131.38, 131.09, 129.31, 129.10, 128.76, 127.81, 121.59, 119.72, 118.89, 113.14, 104.71, 27.50, 24.14; HRMS (FAB) calc. for C21H13ClF3N5O2S [M + H]+ 492.0509, found: 492.0519.
- 4-(3-((2-(tert-Butyl)-4-(3-chlorophenyl)thiazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (54). Compound 54 was synthesized from 27 and 37 according to general procedure G. 43% yield, 98.83% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 10.70 (s, 1H), 7.74 (s, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.57 (dd, J = 8.0, 1.1 Hz, 1H), 7.43–7.51 (m, 2H), 7.18 (d, J = 7.8 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H), 4.47 (s, 2H), 1.35 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 179.07, 174.29, 169.32, 155.52, 149.84, 136.84, 133.82, 131.47, 131.03, 129.34, 128.75, 128.43, 127.63, 127.18, 121.71, 120.05, 113.29, 104.67, 37.92, 30.98, 24.41; HRMS (FAB) calc. for C23H20ClN5O2S [M + H]+ 466.1104, found: 466.1100.
- 4-(3-((3-(tert-Butyl)-1-(3-chlorophenyl)-1H-pyrazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (55). Compound 55 was synthesized from 34 and 37 according to general procedure G. 33% yield, 99.30% purity, light brown solid; 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 10.62 (s, 1H), 7.64 (t, J = 2.1 Hz, 1H), 7.46–7.54 (m, 3H), 7.43 (dt, J = 7.8, 1.8 Hz, 1H), 7.17 (dd, J = 7.8, 0.9 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 6.32 (s, 1H), 4.33 (s, 2H), 1.22 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 174.00, 168.20, 162.29, 155.49, 141.18, 138.05, 133.97, 131.42, 131.36, 129.27, 127.94, 125.02, 123.75, 121.66, 119.93, 113.19, 105.51, 104.74, 32.41, 30.77, 24.04; HRMS (FAB) calc. for C23H21ClN6O2 [M + H]+ 449.1493, found: 449.1487.
- 4-((3-(4-Chlorophenethyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (56). Compound 56 was synthesized from 3-(4-chlorophenyl)-N’-hydroxypropanimidamide and 41 according to general procedure G. 38% yield, 97.77% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.68 (s, 1H), 7.24–7.27 (m, 2H), 7.17–7.20 (m, 2H), 6.82–6.89 (m, 2H), 6.77 (dd, J = 7.3, 1.4 Hz, 1H), 4.31 (s, 2H), 2.89–2.93 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 178.07, 169.91, 155.79, 139.73, 131.31, 130.84, 130.38, 129.32, 128.72, 121.97, 121.19, 115.08, 108.23, 31.84, 27.86, 27.30; HRMS (FAB) calc. for C18H15ClN4O2 [M + H]+ 355.0962, found: 355.0951.
- 4-((3-((4′-(Trifluoromethyl)-[1,1′-biphenyl]-3-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (57). Compound 57 was synthesized from N’-hydroxy-2-(4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)acetimidamide and 41 according to general procedure G. 38% yield, 99.26% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.67 (s, 1H), 7.77–7.81 (m, 4H), 7.57–7.61 (m, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 6.78–6.87 (m, 3H), 4.31 (s, 2H), 4.11 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 178.46, 169.71, 155.77, 144.45, 139.39, 137.26, 130.37, 129.89, 129.56, 129.39, 128.44 (d, J = 32.5 Hz), 128.21, 128.03, 126.37, 126.33, 126.25, 126.19, 122.07, 121.16, 114.95, 108.25, 31.74, 27.98; HRMS (FAB) calc. for C24H17F3N4O2 [M + H]+ 451.1382, found: 451.1393.
- 4-((3-((4′-(Trifluoromethyl)-[1,1′-biphenyl]-4-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (58). Compound 58 was synthesized from N’-hydroxy-2-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)acetimidamide and 41 according to general procedure G. 56% yield, 98.98% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.67 (s, 1H), 7.83 (d, J = 8.2 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 6.78–6.88 (m, 3H), 4.32 (s, 2H), 4.08 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 178.43, 169.68, 155.77, 144.34, 137.73, 136.69, 130.37, 130.25, 129.38, 128.31 (d, J = 31.5 Hz), 127.92, 127.76, 126.32, 126.28, 126.25, 122.07, 121.19, 114.91, 108.27, 31.44, 27.99; HRMS (FAB) calc. for C24H17F3N4O2 [M + H]+ 451.1382, found: 451.1393.
- 4-((3-(2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (59). Compound 59 was synthesized from 10 and 41 according to general procedure G. 36% yield, 98.31% purity, yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.69 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 6.83–6.91 (m, 3H), 4.43 (s, 2H), 3.51 (d, J = 13.3 Hz, 2H), 2.68 (t, J = 11.9 Hz, 2H), 1.38–1.45 (m, 3H), 0.92–1.01 (m, 2H), 0.81 (d, J = 5.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 178.80, 167.77, 158.87, 155.80, 146.65, 143.16, 130.43, 129.44, 122.21, 121.72 (d, J = 272.1 Hz), 121.18, 114.79, 113.80, 111.05, 108.34, 49.15, 33.61, 30.58, 28.05, 22.15; HRMS (FAB) calc. for C22H21F3N6O2 [M + H]+ 459.1756, found: 459.1749.
- 4-((3-((2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (60). Compound 60 was synthesized from 13 and 41 according to general procedure G. 73% yield, 98.85% purity, light brown solid; 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 10.67 (s, 1H), 7.69 (d, J = 7.3 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 6.76–6.87 (m, 3H), 4.31 (s, 2H), 4.10 (s, 2H), 3.27–3.30 (m, 2H), 2.65 (t, J = 11.4 Hz, 1H), 1.56 (d, J = 12.8 Hz, 2H), 1.39–1.47 (m, 1H), 1.08 (qd, J = 12.0, 3.3 Hz, 2H), 0.84 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 178.39, 168.92, 162.31, 155.76, 143.64 (d, J = 33.6 Hz), 141.14, 130.35, 129.30, 128.00, 122.08 (d, J = 272.1 Hz), 121.96, 121.15, 114.90, 114.69, 108.26, 50.55, 34.15, 30.58, 27.93, 22.31; HRMS (FAB) calc. for C23H23F3N6O2 [M + H]+ 473.1913, found: 473.1916.
- 4-((3-(2-(2-(4-Methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)ethyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (61). Compound 61 was synthesized from 16 and 41 according to general procedure G. 68% yield, 99.58% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.67 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 6.81–6.87 (m, 2H), 6.76 (dd, J = 7.1, 1.6 Hz, 1H), 4.30 (s, 2H), 3.29 (d, J = 12.8 Hz, 2H), 2.96–3.06 (m, 4H), 2.67 (t, J = 11.4 Hz, 2H), 1.62 (d, J = 10.5 Hz, 2H), 1.43–1.48 (m, 1H), 1.12–1.22 (m, 2H), 0.89 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 178.13, 170.07, 162.38, 155.78, 142.93 (d, J = 33.4 Hz), 139.77, 131.94, 130.38, 129.35, 122.20 (d, J = 271.1 Hz), 121.96, 121.12, 115.01, 114.65, 108.20, 50.78, 34.38, 30.76, 28.14, 27.86, 25.12, 22.34; HRMS (FAB) calc. for C24H25F3N6O2 [M + H]+ 487.2069, found: 487.2074.
- 4-((3-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (62). Compound 62 was synthesized from 21 and 41 according to general procedure G. 31% yield, 99.61% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.69 (s, 1H), 7.77 (t, J = 1.8 Hz, 1H), 7.65 (dt, J = 7.8, 1.4 Hz, 1H), 7.51–7.53 (m, 1H), 7.45 (t, J = 8.0 Hz, 1H), 6.83–6.90 (m, 3H), 4.43 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 179.39, 161.76, 155.79, 155.41 (d, J = 39.3 Hz), 154.52, 134.49, 133.42, 130.68, 130.38, 130.20, 129.63, 129.38, 128.75, 123.72, 122.20, 121.23, 119.47 (d, J = 219.5 Hz), 114.25, 108.45, 28.00; HRMS (FAB) calc. for C20H11ClF3N5O2S [M + H]+ 478.0352, found: 478.0356.
- 4-((3-((4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (63). Compound 63 was synthesized from 26 and 41 according to general procedure G. 31% yield, 99.42% purity, yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.67 (s, 1H), 7.68 (s, 1H), 7.59 (dt, J = 6.7, 1.7 Hz, 1H), 7.46–7.52 (m, 2H), 6.82–6.88 (m, 2H), 6.78 (dd, J = 7.4, 1.4 Hz, 1H), 4.56 (s, 2H), 4.34 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 179.09, 168.23, 155.75, 152.65 (d, J = 40.1 Hz), 152.36, 134.97, 134.03, 133.75, 131.23, 130.38, 129.47, 129.39, 128.84, 127.81, 122.03, 121.48, 121.14, 114.68, 108.31, 28.03, 24.22; HRMS (FAB) calc. for C21H13ClF3N5O2S [M + H]+ 492.0509, found: 492.0497.
- 4-((3-(2-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)ethyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (64). Compound 64 was synthesized from 30 and 41 according to general procedure G. 69% yield, 96.24% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.67 (s, 1H), 7.59–7.60 (m, 1H), 7.45–7.53 (m, 3H), 6.81–6.87 (m, 2H), 6.73 (dd, J = 6.6, 2.1 Hz, 1H), 4.29 (s, 2H), 3.39 (t, J = 7.3 Hz, 2H), 3.08 (t, J = 7.3 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 178.43, 169.13, 155.77, 151.54, 151.35 (d, J = 39.1 Hz), 139.46, 135.51, 133.94, 131.16, 130.38, 129.31, 129.20, 128.86, 127.84, 121.92, 121.13, 120.23 (d, J = 269.2 Hz), 114.87, 108.23, 27.82, 27.33, 24.28; HRMS (FAB) calc. for C22H15ClF3N5O2S [M + H]+ 506.0665, found: 506.0667.
- 4-((3-((2-(tert-Butyl)-4-(3-chlorophenyl)thiazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (65). Compound 65 was synthesized from 27 and 41 according to general procedure G. 56% yield, 99.36% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 10.67 (s, 1H), 7.65–7.66 (m, 1H), 7.58 (td, J = 4.2, 2.3 Hz, 1H), 7.40–7.46 (m, 2H), 6.78–6.88 (m, 3H), 4.34 (s, 2H), 4.32 (s, 2H), 1.34 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 179.05, 178.84, 168.94, 155.76, 149.81, 136.74, 133.78, 130.92, 130.38, 129.41, 128.67, 128.40, 127.54, 127.10, 122.01, 121.13, 114.77, 108.29, 37.90, 30.97, 28.04, 24.17; HRMS (FAB) calc. for C24H22ClN5O2S [M + H]+ 480.1261, found: 480.1257.
- 4-((3-((3-(tert-Butyl)-1-(3-chlorophenyl)-1H-pyrazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (66). Compound 66 was synthesized from 34 and 41 according to general procedure G. 68% yield, 99.57% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.66 (s, 1H), 7.55–7.56 (m, 1H), 7.38–7.46 (m, 3H), 6.81–6.87 (m, 2H), 6.72 (dd, J = 7.1, 1.6 Hz, 1H), 6.18 (s, 1H), 4.30 (s, 2H), 4.19 (s, 2H), 1.21 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 178.58, 167.86, 162.19, 155.76, 141.07, 137.98, 133.89, 131.22, 130.37, 129.34, 127.87, 124.87, 123.62, 121.90, 121.12, 114.87, 108.23, 105.47, 32.35, 30.75, 27.90, 23.81; HRMS (FAB) calc. for C24H23ClN6O2 [M + H]+ 463.1649, found: 463.1641.
- 7-Methyl-1-((3-((4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,7-dihydro-6H-purin-6-one (67). Compound 67 was synthesized from N’-hydroxy-2-(4’-(trifluoromethyl)-[1,1’-biphenyl]-3-yl)acetimidamide and 45 according to general procedure G. 55% yield, 99.91% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.18 (s, 1H), 7.76–7.81 (m, 4H), 7.57–7.61 (m, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 5.50 (s, 2H), 4.14 (s, 2H), 3.87 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.97, 169.87, 157.06, 154.05, 147.85, 145.83, 144.40, 139.37, 136.98, 129.91, 129.57, 128.45 (d, J = 31.5 Hz), 128.24, 128.01, 126.35, 126.31, 126.27, 126.24, 115.12, 42.08, 33.92, 31.60; HRMS (FAB) calc. for C23H17F3N6O2 [M + H]+ 467.1443, found: 467.1437.
- 7-Methyl-1-((3-((4’-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,7-dihydro-6H-purin-6-one (68). Compound 68 was synthesized from N’-hydroxy-2-(4’-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)acetimidamide and 45 according to general procedure G. 49% yield, 99.53% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.19 (s, 1H), 7.84 (d, J = 8.2 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 5.50 (s, 2H), 4.11 (s, 2H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.95, 169.87, 157.06, 154.05, 147.83, 145.85, 144.29, 137.82, 136.39, 130.25, 128.32 (d, J = 31.5 Hz), 127.93, 127.79, 126.31, 126.27, 126.25, 115.13, 42.09, 33.96, 31.34; HRMS (FAB) calc. for C23H17F3N6O2 [M + H]+ 467.1443, found: 467.1444.
- 7-Methyl-1-((3-(2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)-1,2,4-oxadiazol-5-yl)methyl)-1,7-dihydro-6H-purin-6-one (69). Compound 69 was synthesized from 10 and 45 according to general procedure G. 59% yield, 98.20% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.43 (d, J = 7.6 Hz, 1H), 8.39 (s, 1H), 8.20 (s, 1H), 7.46 (d, J = 7.9 Hz, 1H), 5.51 (s, 2H), 3.89 (s, 3H), 3.59 (d, J = 13.3 Hz, 2H), 2.75 (t, J = 11.8 Hz, 2H), 1.52 (d, J = 11.7 Hz, 2H), 1.43–1.48 (m, 1H), 1.05–1.14 (m, 2H), 0.85 (d, J = 5.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.82, 169.68, 159.10, 157.06, 154.05, 147.79, 146.65, 143.65, 145.80, 129.41, 121.94 (d, J = 271.2 Hz), 115.12, 114.59, 50.04, 41.95, 34.00, 33.93, 30.63, 22.25; HRMS (FAB) calc. for C21H21F3N8O2 [M + H]+ 475.1818, found: 475.1809.
- 7-Methyl-1-((3-((2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-1,7-dihydro-6H-purin-6-one (70). Compound 70 was synthesized from 13 and 45 according to general procedure G. 61% yield, 99.53% purity, light brown solid; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.19 (s, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.38 (d, J = 7.3 Hz, 1H), 5.50 (s, 2H), 4.13 (s, 2H), 3.90 (s, 3H), 3.28–3.30 (m, 2H), 2.66 (t, J = 12.3 Hz, 2H), 1.56 (d, J = 12.8 Hz, 2H), 1.39–1.47 (m, 1H), 1.08–1.19 (m, 2H), 0.85 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.92, 169.15, 162.33, 157.03, 154.00, 147.77, 145.81, 143.93, 141.17, 127.74, 122.03 (d, J = 265.4 Hz), 115.11, 114.70, 50.59, 41.94, 34.16, 33.93, 30.60, 27.92, 22.27; HRMS (FAB) calc. for C22H23F3N8O2 [M + H]+ 489.1974, found: 489.1985.
- 7-Methyl-1-((3-(2-(2-(4-methylpiperidin-1-yl)-6-(trifluoromethyl)pyridin-3-yl)ethyl)-1,2,4-oxadiazol-5-yl)methyl)-1,7-dihydro-6H-purin-6-one (71). Compound 71 was synthesized from 16 and 45 according to general procedure G. 60% yield, 99.69% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.20 (s, 1H), 7.79 (d, J = 7.3 Hz, 1H), 7.32 (d, J = 7.3 Hz, 1H), 5.50 (s, 2H), 3.91 (s, 3H), 3.26–3.30 (m, 2H), 3.04–3.08 (m, 2H), 2.95–2.99 (m, 2H), 2.66 (td, J = 12.1, 1.7 Hz, 2H), 1.59 (d, J = 12.3 Hz, 2H), 1.40–1.49 (m, 1H), 1.10–1.19 (m, 2H), 0.86 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.73, 170.22, 162.38, 157.08, 154.02, 147.80, 145.79, 142.95 (d, J = 33.4 Hz), 139.73, 131.85, 122.19 (d, J = 271.2 Hz), 115.12, 114.66, 50.77, 41.96, 34.37, 33.93, 30.71, 27.97, 24.96, 22.28; HRMS (FAB) calc. for C23H25F3N8O2 [M + H]+ 503.2131, found: 503.2123.
- 1-((3-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)-1,2,4-oxadiazol-5-yl)methyl)-7-methyl-1,7-dihydro-6H-purin-6-one (72). Compound 72 was synthesized from 21 and 45 according to general procedure G. 76% yield, 99.89% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.21 (s, 1H), 7.74 (t, J = 1.8 Hz, 1H), 7.60 (dt, J = 7.8, 1.3 Hz, 1H), 7.45–7.48 (m, 1H), 7.36 (t, J = 8.0 Hz, 1H), 5.60 (s, 2H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 176.91, 161.91, 157.08, 155.58 (d, J = 40.1 Hz), 154.99, 154.01, 147.70, 145.85, 134.41, 133.32, 130.51, 130.14, 129.69, 128.76, 123.17, 119.68 (d, J = 264.4 Hz), 115.09, 41.95, 33.95; HRMS (FAB) calc. for C19H11ClF3N7O2S [M + H]+ 494.0414, found: 494.0426.
- 1-((3-((4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-7-methyl-1,7-dihydro-6H-purin-6-one (73). Compound 73 was synthesized from 26 and 45 according to general procedure G. 48% yield, 99.23% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.20 (s, 1H), 7.67 (t, J = 1.6 Hz, 1H), 7.56 (dt, J = 7.0, 1.7 Hz, 1H), 7.44–7.50 (m, 2H), 5.52 (s, 2H), 4.59 (s, 2H), 3.89 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 177.59, 173.55, 165.98, 159.79, 157.09, 154.00, 152.46, 147.73, 145.94, 135.37, 134.01, 130.72, 129.67, 128.77, 127.80, 121.47 (d, J = 63.0 Hz), 115.11, 41.95, 33.96, 24.12; HRMS (FAB) calc. for C20H13ClF3N7O2S [M + H]+ 508.0570, found: 508.0564.
- 1-((3-(2-(4-(3-Chlorophenyl)-2-(trifluoromethyl)thiazol-5-yl)ethyl)-1,2,4-oxadiazol-5-yl)methyl)-7-methyl-1,7-dihydro-6H-purin-6-one (74). Compound 74 was synthesized from 30 and 45 according to general procedure G. 83% yield, 99.39% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.19 (s, 1H), 7.60–7.61 (m, 1H), 7.42–7.53 (m, 3H), 5.48 (s, 2H), 3.88 (s, 3H), 3.38 (t, J = 7.3 Hz, 2H), 3.11 (t, J = 7.3 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 176.00, 169.29, 157.06, 153.98, 151.54, 151.18, 147.74, 145.79, 139.31, 135.49, 133.93, 131.10, 129.19, 128.86, 127.81, 120.23 (d, J = 269.2 Hz), 115.11, 42.01, 33.87, 27.13, 24.16; HRMS (FAB) calc. for C21H15ClF3N7O2S [M + H]+ 522.0727, found: 522.0730.
- 1-((3-((2-(tert-Butyl)-4-(3-chlorophenyl)thiazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-7-methyl-1,7-dihydro-6H-purin-6-one (75). Compound 75 was synthesized from 27 and 45 according to general procedure G. 72% yield, 99.75% purity, pale yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.20 (s, 1H), 7.65–7.66 (m, 1H), 7.54–7.57 (m, 1H), 7.40–7.41 (m, 2H), 5.53 (s, 2H), 4.35 (s, 2H), 3.90 (s, 3H), 1.34 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 179.12, 176.40, 169.11, 157.09, 154.05, 149.94, 147.79, 145.82, 136.67, 133.75, 130.90, 128.64, 128.42, 127.53, 126.76, 115.13, 42.13, 37.90, 33.95, 30.95, 24.05; HRMS (FAB) calc. for C23H22ClN7O2S [M + H]+ 496.1322, found: 496.1307.
- 1-((3-((3-(tert-Butyl)-1-(3-chlorophenyl)-1H-pyrazol-5-yl)methyl)-1,2,4-oxadiazol-5-yl)methyl)-7-methyl-1,7-dihydro-6H-purin-6-one (76). Compound 76 was synthesized from 34 and 45 according to general procedure G. 76% yield, 99.29% purity, white solid; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.20 (s, 1H), 7.55–7.56 (m, 1H), 7.36–7.43 (m, 3H), 6.15 (s, 1H), 5.49 (s, 2H), 4.21 (s, 2H), 3.90 (s, 3H), 1.19 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 176.17, 168.00, 162.16, 157.06, 154.02, 147.77, 145.81, 141.04, 137.79, 133.88, 131.23, 127.90, 124.87, 123.67, 115.12, 105.41, 42.01, 33.93, 32.33, 30.70, 23.70; HRMS (FAB) calc. for C23H23ClN8O2 [M + H]+ 479.1711, found: 479.1702.
3.2. In Vitro Assay
Fluorescence Imaging Plate Reader Assay
3.3. In Vivo Assay
3.3.1. Animals
3.3.2. Formalin Test
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chronic Pain: Medication Decisions. Available online: https://www.mayoclinic.org/chronic-pain-medication-decisions/art-20360371 (accessed on 8 February 2023).
- Yao, K.; Dou, B.; Zhang, Y.; Chen, Z.; Li, Y.; Fan, Z.; Ma, Y.; Du, S.; Wang, J.; Xu, Z.; et al. Inflammation—The role of TRPA1 channel. Front. Physiol. 2023, 14, 1093925. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, S.; Nikolaeva-Koleva, M.; Alarcón-Alarcón, D.; Butrón, L.; González-Rodríguez, S. Is TRPA1 burning down TRPV1 as druggable target for the treatment of chronic pain? Int. J. Mol. Sci. 2019, 20, 2906. [Google Scholar] [CrossRef] [PubMed]
- Skerratt, S. Recent progress in the discovery and development of TRPA1 modulators. Prog. Med. Chem. 2017, 56, 81–115. [Google Scholar] [PubMed]
- Iftinca, M.; Defaye, M.; Altier, C. TRPV1-targeted drugs in development for human pain conditions. Drugs 2021, 81, 7–27. [Google Scholar] [CrossRef] [PubMed]
- Gladkikh, I.N.; Sintsova, O.V.; Leychenko, E.V.; Kozlov, S.A. TRPV1 ion channel: Structure features, activity modulators, and therapeutic potential. Biochemistry 2021, 86, S50–S70. [Google Scholar] [CrossRef]
- Bamps, D.; Vriens, J.; de Hoon, J.; Voets, T. TRP channel cooperation for nociception: Therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 655–677. [Google Scholar] [CrossRef]
- Fallah, H.P.; Ahuja, E.; Lin, H.; Qi, J.; He, Q.; Gao, S.; An, H.; Zhang, J.; Xie, Y.; Liang, D. A review on the role of TRP channels and their potential as drug targets and insight into the TRP channel drug discovery methodologies. Front. Pharmacol. 2022, 13, 914499. [Google Scholar] [CrossRef]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef]
- Koivisto, A.P.; Voets, T.; Ladarola, M.J.; Szallasi, A. Targeting TRP channels for pain relief: A review of current evidence from bench to bedside. Curr. Opin. Pharmacol. 2024, 75, 102447. [Google Scholar] [CrossRef] [PubMed]
- Horváth, Á.; Biró-Süto, T.; Kántás, B.; Payrits, M.; Skoda-Földes, R.; Szánti-Pintér, E.; Helyes, Z.; Szöke, É. Antinociceptive effects of lipid raft disruptors, a novel carboxamido-steroid and methyl β-cyclodextrin, in mice by inhibiting transient receptor potential vanilloid 1 and ankyrin 1 channel activation. Front. Physiol. 2020, 11, 559109. [Google Scholar] [CrossRef]
- Pyo, H.J.; An, X.; Cho, H. The role of free fatty acid receptor pathways in a selective regulation of TRPA1 and TRPV1 by resolvins in primary sensory neurons. J. Cell Physiol. 2022, 237, 3651–3660. [Google Scholar] [CrossRef]
- Horváth, Á.; Payrits, M.; Steib, A.; Kántás, B.; Biró-Sütó, T.; Erostyák, J.; Makkai, G.; Sághy, É.; Helyes, Z.; Szöke, É. Analgesic effects of lipid raft disruption by sphingomyelinase and myriocin via transient receptor potential vanilloid 1 and transient receptor potential ankyrin 1 ion channel modulation. Front. Pharmacol. 2021, 11, 593319. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, S.R.; Chen, H.; Pan, H.L. Endogenous transient receptor potential ankyrin 1 and vanilloid 1 activity potentiates glutamatergic input to spinal lamina I neurons in inflammatory pain. J. Neurochem. 2019, 149, 381–398. [Google Scholar] [CrossRef]
- Duitama, M.; Moreno, Y.; Santander, S.P.; Casas, Z.; Sutachan, J.J.; Torres, Y.P.; Albarracín, S.L. TRP channels as molecular targets to relieve cancer pain. Biomolecules 2022, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Su, C.J.; Xu, J.H.; Liu, X.; Zhao, F.L.; Pan, J.; Shi, A.M.; Hu, D.M.; Yu, Y.L.; Liu, T.; Zhang, Y.S. X-ray induces mechanical and heat allodynia in mouse via TRPA1 and TRPV1 activation. Mol. Pain 2019, 15, 1–13. [Google Scholar]
- Zhu, H.; Wang, Y.; He, Y.; Yu, W. Inflammation-mediated macrophage polarization induces TRPV1/TRPA1 heteromers in endometriosis. Am. J. Transl. Res. 2022, 14, 3066–3078. [Google Scholar]
- Thammanichanon, P.; Kaewpitak, A.; Binlateh, T.; Pavasant, P.; Leethanakul, C. Varied temporal expression patterns of trigeminal TRPA1 and TRPV1 and the neuropeptide CGRP during orthodontic force-induced pain. Arch. Oral Biol. 2021, 128, 105170. [Google Scholar] [CrossRef]
- Wang, S.; Brigoli, B.; Lim, J.; Karley, A.; Chung, M.K. Roles of TRPV1 and TRPA1 in spontaneous pain from inflamed masseter muscle. Neuroscience 2018, 384, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Roy, T.K.; Uniyal, A.; Kotiyal, A.; Tiwari, V. Multifactorial pathways in burn injury-induced chronic pain: Novel targets and their pharmacological modulation. Mol. Biol. Rep. 2022, 49, 12121–12132. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, Y.; Wang, M.; Zhang, H.; Chen, Y.; Adcock, I.M.; Chung, K.F.; Mo, J.; Zhang, Y.; Li, F. TRPV1 and TRPA1 in lung inflammation and airway hyperresponsiveness induced by fine particulate matter (PM2.5). Oxid. Med. Cell Long. 2019, 2019, 7450151. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Y.; Xu, M.; Zhang, H.; Chen, Y.; Chung, K.F.; Adcock, I.M.; Li, F. Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic. Biol. Med. 2019, 134, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ying, S.; Zhao, X.; Liu, J.; Wang, Y. Increased expression of lung TRPV1/TRPA1 in a cough model of bleomycin-induced pulmonary fibrosis in guinea pigs. BMC Pulm. Med. 2019, 19, 27. [Google Scholar] [CrossRef]
- Lee, L.Y.; Hsu, C.C.; Lin, Y.J.; Lin, R.L.; Khosravi, M. Interaction between TRPA1 and TRPV1: Synergy on pulmonary sensory nerves. Pulm. Pharmacol. Ther. 2015, 35, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Csekő, K.; Beckers, B.; Keszthelyi, D.; Helyes, Z. Role of TRPV1 and TRPA1 ion channels in inflammatory bowel diseases: Potential therapeutic targets? Pharmaceuticals 2019, 12, 48. [Google Scholar] [CrossRef]
- Gouin, O.; L’Herondelle, K.; Lebonvallet, N.; Le Gall-Ianotto, C.; Sakka, M.; Buhé, V.; Plée-Gautier, E.; Carré, L.; Lefeuvre, L.; Misery, L.; et al. TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: Pro-inflammatory response induced by their activation and their sensitization. Protein Cell 2017, 8, 644–661. [Google Scholar] [CrossRef]
- Schwartz, E.S.; Christianson, J.A.; Chen, X.; La, J.H.; Davis, B.M.; Albers, K.M.; Gebhart, G.F. Synergistic role of TRPV1 and TRPA1 in pancreatic pain and inflammation. Gastroenterology 2011, 140, 1283–1291. [Google Scholar] [CrossRef]
- Fernandes, E.S.; Fernandes, M.A.; Keeble, J.E. The functions of TRPA1 and TRPV1: Moving away from sensory nerves. Br. J. Pharmacol. 2012, 166, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Wilzopolski, J.; Kietzmann, M.; Mishra, S.K.; Stark, H.; Bäumer, W.; Rossbach, K. TRPV1 and TRPA1 channels are both involved downstream of histamine-induced itch. Biomolecules 2021, 11, 1166. [Google Scholar] [CrossRef]
- Tsagareli, M.G.; Nozadze, I.; Tsiklauri, N.; Carstens, M.I.; Gurtskaia, G.; Carstens, E. Thermal hyperalgesia and mechanical allodynia elicited by histamine and non-histaminergic itch mediators: Respective involvement of TRPV1 and TRPA1. Neuroscience 2020, 449, 35–45. [Google Scholar] [CrossRef]
- Kalangara, J.P.; Vanijcharoenkarn, K.; Chisolm, S.; Kuruvilla, M.E. Neuropathic pain and itch: Mechanisms in allergic conjunctivitis. Curr. Opin. Allergy Clin. Immunol. 2022, 20, 298–303. [Google Scholar] [CrossRef]
- Garami, A.; Shimansky, Y.P.; Rumbus, Z.; Vizin, R.C.L.; Farkas, N.; Hegyi, J.; Szakacs, Z.; Solymar, M.; Csenkey, A.; Chiche, D.A.; et al. Hyperthermia induced by transient receptor potential vanilloid-1 (TRPV1) antagonists in human clinical trials: Insights from mathematical modeling and meta-analysis. Pharmacol. Ther. 2020, 208, 107474. [Google Scholar] [CrossRef] [PubMed]
- Payrits, M.; Saghy, E.; Matyus, P.; Czompa, A.; Ludmerczki, R.; Deme, R.; Sandor, Z.; Helyes, Z.S.; Szoke, E. A novel 3-(4,5-diphenyl-1,3-oxazol-2-yl)propanal oxime compound is a potent transient receptor potential ankyrin 1 and vanilloid 1 (TRPA1 and V1) receptor antagonist. Neuroscience 2016, 324, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Horváth, Á.; Tékus, V.; Bencze, N.; Szentes, N.; Scheich, B.; Bölcskei, K.; Szőke, É.; Mócsai, A.; Tóth-Sarudy, É.; Mátyus, P.; et al. Analgesic effects of the novel semicarbazide-sensitive amine oxidaseinhibitor SZV 1287 in mouse pain models with neuropathicmechanisms: Involvement of transient receptor potential vanilloid 1 and ankyrin 1 receptors. Pharmacol. Res. 2018, 131, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Horváth, Á.; Menghis, A.; Botz, B.; Borbély, É.; Kemény, Á.; Tékus, V.; Csepregi, J.Z.; Mócsai, A.; Juhász, T.; Zákány, R.; et al. Analgesic and anti-inflammatory effects of the novel semicarbazide-sensitive amine-oxidase inhibitor SZV-1287 in chronic arthritis models of the mouse. Sci. Rep. 2017, 7, 39863. [Google Scholar] [CrossRef]
- Horváth, Á.I.; Szentes, N.; Tékus, V.; Payrits, M.; Szőke, É.; Oláh, E.; Garami, A.; Fliszár-Nyúl, E.; Poór, M.; Sár, S.; et al. Proof-of-concept for the analgesic effect and thermoregulatory safety of orally administered multi-target compound SZV 1287 in mice: A novel drug candidate for neuropathic pain. Biomedicines 2021, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, P.; Lu, S.; Guo, R.; Gao, W.; Tong, H.; Yin, Y.; Han, X.; Liu, T.; Chen, X.; et al. Liquiritin, a novel inhibitor of TRPV1 and TRPA1, protects against LPS induced acute lung injury. Cell Calcium 2020, 88, 102198. [Google Scholar] [CrossRef]
- Schenkel, L.B.; Olivieri, P.R.; Boezio, A.A.; Deak, H.L.; Emkey, R.; Graceffa, R.F.; Gunaydin, H.; Guzman-Perez, A.; Lee, J.H.; Teffera, Y.; et al. Optimization of a novel quinazolinone-based series of transient receptor potential A1 (TRPA1) antagonists demonstrating potent in vivo activity. J. Med. Chem. 2016, 59, 2794–2809. [Google Scholar] [CrossRef]
- Napoletano, M.; Trevisani, M.; Pavani, M.G.; Fruttarolo, F. TRPV1 Vanilloid Receptor Antagonists with a Bicyclic Portion. Patent WO2011120604A1, 13 February 2017. [Google Scholar]
- Kim, M.S.; Ryu, H.; Kang, D.W.; Cho, S.H.; Seo, S.; Park, Y.S.; Kim, M.Y.; Kwak, E.J.; Kim, Y.S.; Bhondwe, R.S.; et al. 2-(3-Fluoro-4- methylsulfonylaminophenyl)propanamides as potent transient receptor potential vanilloid 1 (TRPV1) antagonists: Structure-activity relationships of 2-amino derivatives in the N-(6-trifluoromethylpyridin-3-ylmethyl) C-region. J. Med. Chem. 2012, 55, 8392–8408. [Google Scholar] [CrossRef]
- Krapcho, A.P.; Weimaster, J.F.; Eldridge, J.M.; Jahngen, E.G.E., Jr.; Lovey, A.J.; Stephens, W.P. Synthetic applications and mechanism studies of the decarbalkoxylations of geminal diesters and related systems effected in Me2SO by water and/or by water with added salts. J. Org. Chem. 1978, 43, 138–147. [Google Scholar] [CrossRef]
- Ann, J.; Kim, H.S.; Thorat, S.A.; Kim, H.; Ha, H.J.; Choi, K.; Kim, Y.H.; Kim, M.; Hwang, S.W.; Pearce, L.V.; et al. Discovery of nonpungent transient receptor potential vanilloid 1 (TRPV1) agonist as strong topical analgesic. J. Med. Chem. 2020, 63, 418–424. [Google Scholar]
- Zuo, D.; Hong, M.; Jung, A.; Lee, S.; Do, N.; Jung, S.; Jeon, Y.; Jeong, J.W.; Huang, G.; Li, L.X.; et al. Discovery of N-(1-(2-hydroxyethyl) quinolin-2-one)-N’-(1-phenyl-1H-pyrazol-5-yl)methyl) urea as mode-selective TRPV1 antagonist. Bioorg. Med. Chem. Lett. 2024, 101, 129656. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.Y.S.; Clark, C.G.; Saubern, S.; Adams, J.; Winters, M.P.; Chan, D.M.T.; Combs, A. New aryl/heteroaryl C-N bond cross-coupling reactions via arylboronic acid/cupric acetate arylation. Tetrahedron Lett. 1998, 39, 2941–2944. [Google Scholar] [CrossRef]
- Dubuisson, D.; Dennis, S.G. The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 1977, 4, 161–174. [Google Scholar] [CrossRef]
- Tjølsen, A.; Berge, O.G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The formalin test: An evaluation of the method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| % Inhibition 1 | |||||
| Compound | R | hTRPA1 | mTRPA1 | hTRPV1 | rTRPV1 |
| AM-0902 | 100% | 125% | |||
| BCTC | 100% | 100% | |||
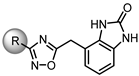
| 46 | 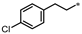 | 33% | 27% | 7% | 10% |
| 47 | 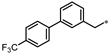 | 38% | 26% | 15% | 17% |
| 48 | 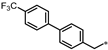 | NE | 21% | 45% | 33% |
| 49 |  | 40% | 14% | 12% | 15% |
| 50 | 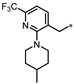 | 60% | 44% | 52% | 48% |
| 51 |  | NE | 21% | 20% | 11% |
| 52 |  | NE | 40% | 8% | NE |
| 53 | 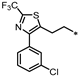 | NE | 48% | 13% | NE |
| 54 |  | NE | 4% | 43% | 25% |
| 55 | 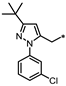 | 29% | 60% | 28% | NE |
 | |||||
| % Inhibition 1 | |||||
| Compound | R | hTRPA1 | mTRPA1 | hTRPV1 | rTRPV1 |
| 56 | 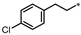 | 32% | 29% | 34% | NE |
| 57 | 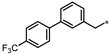 | 23% | 24% | 45% | 29% |
| 58 | 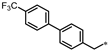 | 33% | 15% | 31% | 20% |
| 59 | 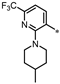 | 40% | 24% | 13% | 1% |
| 60 | 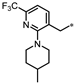 | 22% | 32% | 24% | 29% |
| 61 | 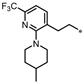 | 29% | 25% | 64% | 62% |
| 62 | 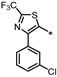 | NE | 16% | 23% | 28% |
| 63 | 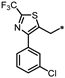 | 17% | 17% | 51% | 30% |
| 64 | 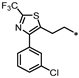 | NE | 20% | 21% | 24% |
| 65 |  | 24% | 23% | 69% | 40% |
| 66 | 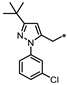 | 34% | 55% | 77% | 46% |
 | |||||
| % Inhibition 1 | |||||
| Compound | R | hTRPA1 | mTRPA1 | hTRPV1 | rTRPV1 |
| 67 | 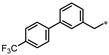 | NE | 16% | NE | NE |
| 68 |  | 35% | 35% | 7% | 18% |
| 69 | 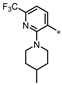 | NE | 16% | 16% | NE |
| 70 | 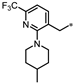 | 9% | 34% | 22% | NE |
| 71 |  | 11% | 28% | NE | NE |
| 72 | 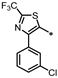 | 35% | 46% | 31% | 4% |
| 73 | 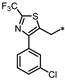 | NE | 22% | 10% | NE |
| 74 | 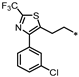 | 5% | 14% | 3% | NE |
| 75 | 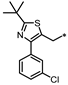 | 11% | 43% | 14% | NE |
| 76 | 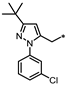 | 7% | 50% | 7% | NE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, N.; Zuo, D.; Kim, M.; Kim, M.; Ha, H.-J.; Blumberg, P.M.; Ann, J.; Hwang, S.W.; Lee, J. Discovery of Dual TRPA1 and TRPV1 Antagonists as Novel Therapeutic Agents for Pain. Pharmaceuticals 2024, 17, 1209. https://doi.org/10.3390/ph17091209
Do N, Zuo D, Kim M, Kim M, Ha H-J, Blumberg PM, Ann J, Hwang SW, Lee J. Discovery of Dual TRPA1 and TRPV1 Antagonists as Novel Therapeutic Agents for Pain. Pharmaceuticals. 2024; 17(9):1209. https://doi.org/10.3390/ph17091209
Chicago/Turabian StyleDo, Nayeon, Dongxu Zuo, Miri Kim, Minseok Kim, Hee-Jin Ha, Peter M. Blumberg, Jihyae Ann, Sun Wook Hwang, and Jeewoo Lee. 2024. "Discovery of Dual TRPA1 and TRPV1 Antagonists as Novel Therapeutic Agents for Pain" Pharmaceuticals 17, no. 9: 1209. https://doi.org/10.3390/ph17091209
APA StyleDo, N., Zuo, D., Kim, M., Kim, M., Ha, H.-J., Blumberg, P. M., Ann, J., Hwang, S. W., & Lee, J. (2024). Discovery of Dual TRPA1 and TRPV1 Antagonists as Novel Therapeutic Agents for Pain. Pharmaceuticals, 17(9), 1209. https://doi.org/10.3390/ph17091209