

RP-CAD for Lipid Quantification: Systematic Method Development and Intensified LNP Process Characterization


Abstract
:
1. Introduction
2. Results
2.1. Lipid Quantification—Method Development, Calibration, and Validation
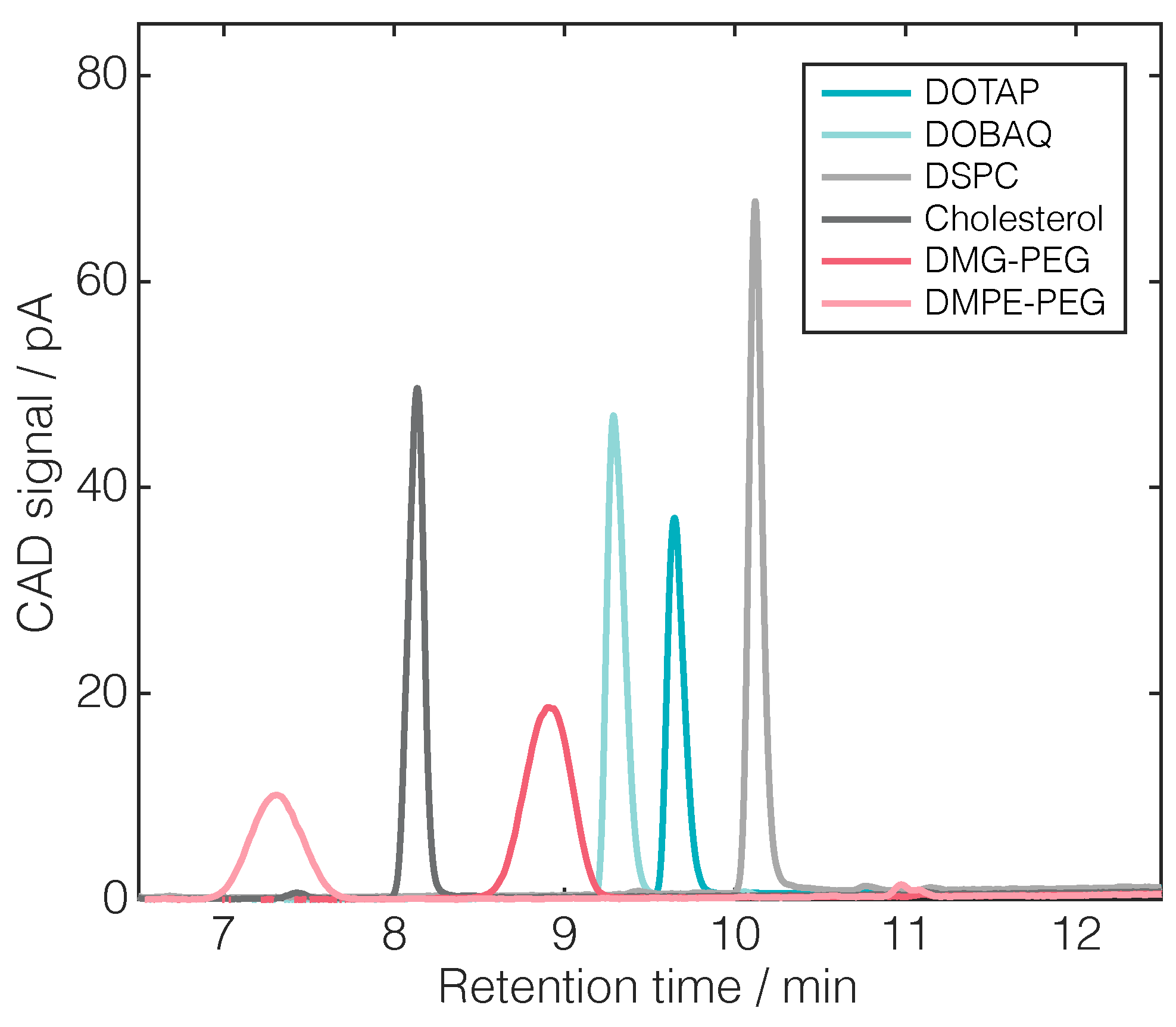
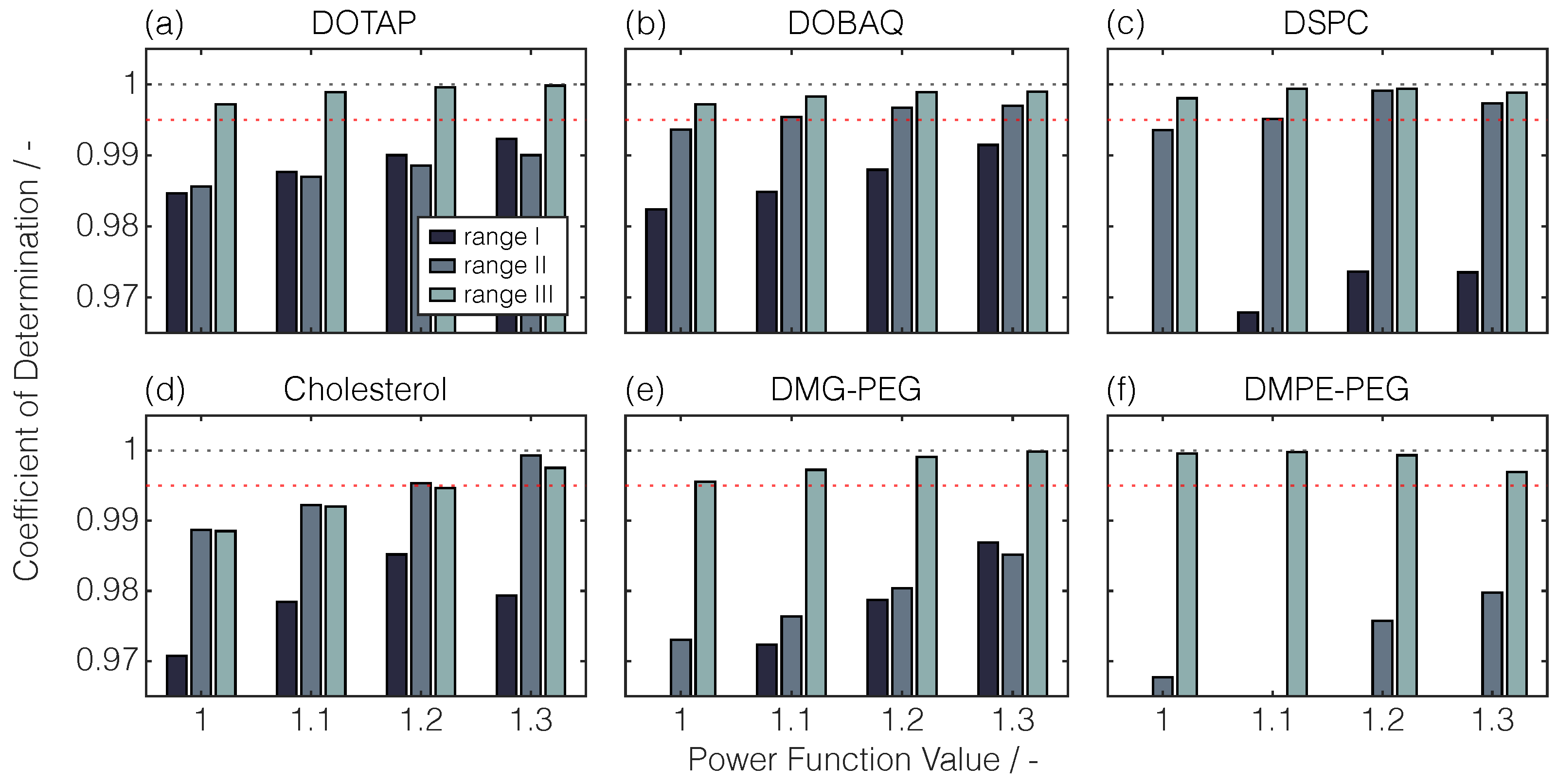
2.1.1. Method Development
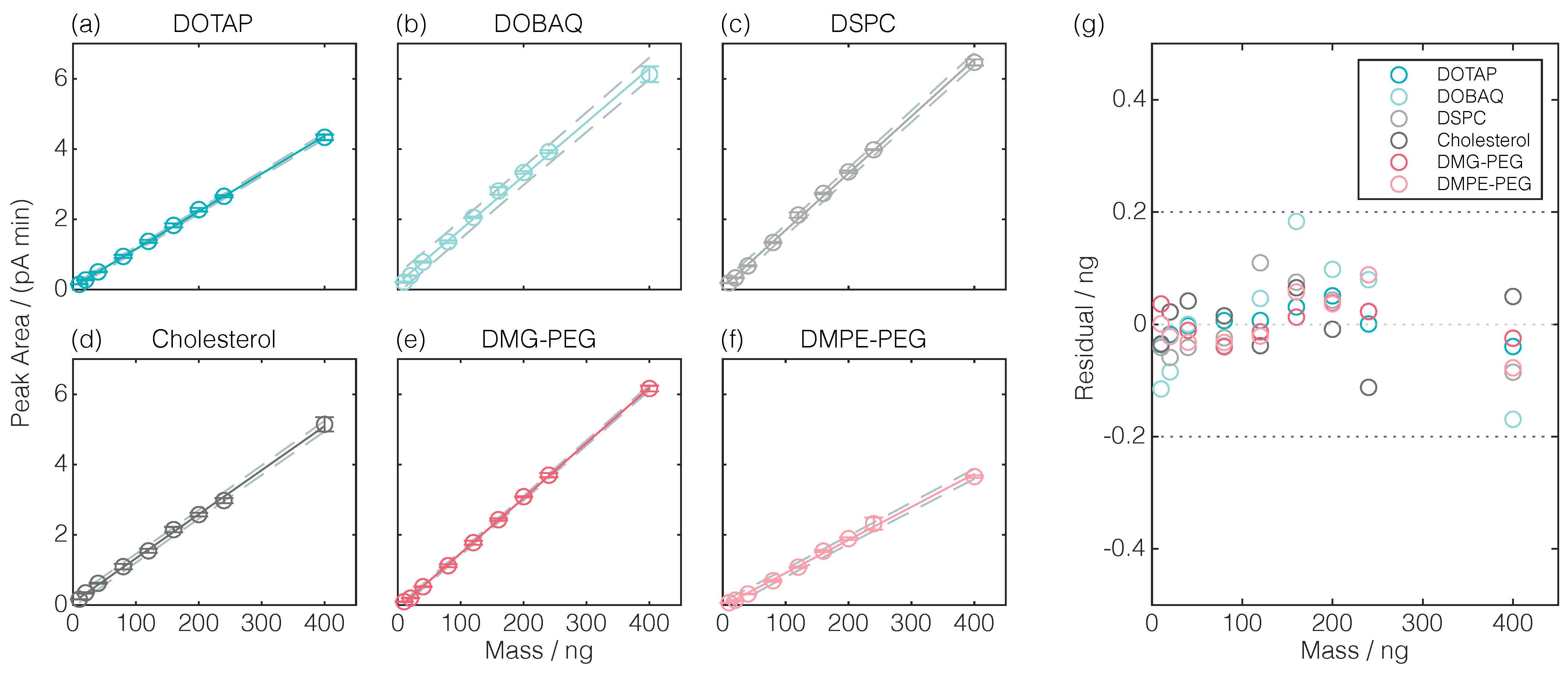
2.1.2. Method Calibration and Validation
2.2. Applicability for Lipid Nanoparticle Process Characterization
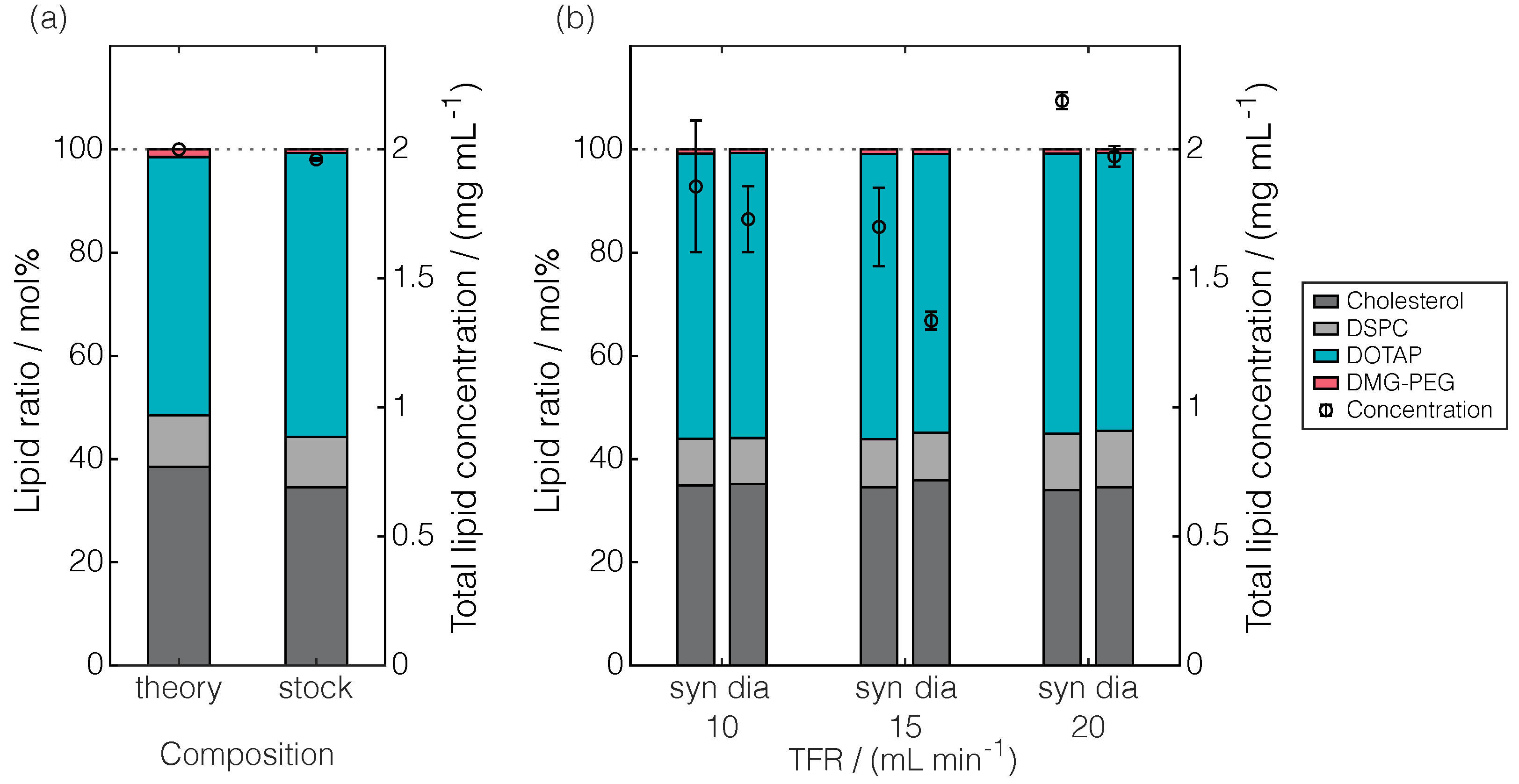
2.2.1. Standard Analytical Panel
2.2.2. Charged Aerosol Detection for Lipid Quantification and Process Performance
3. Discussion
3.1. RP-CAD for Lipid Quantification
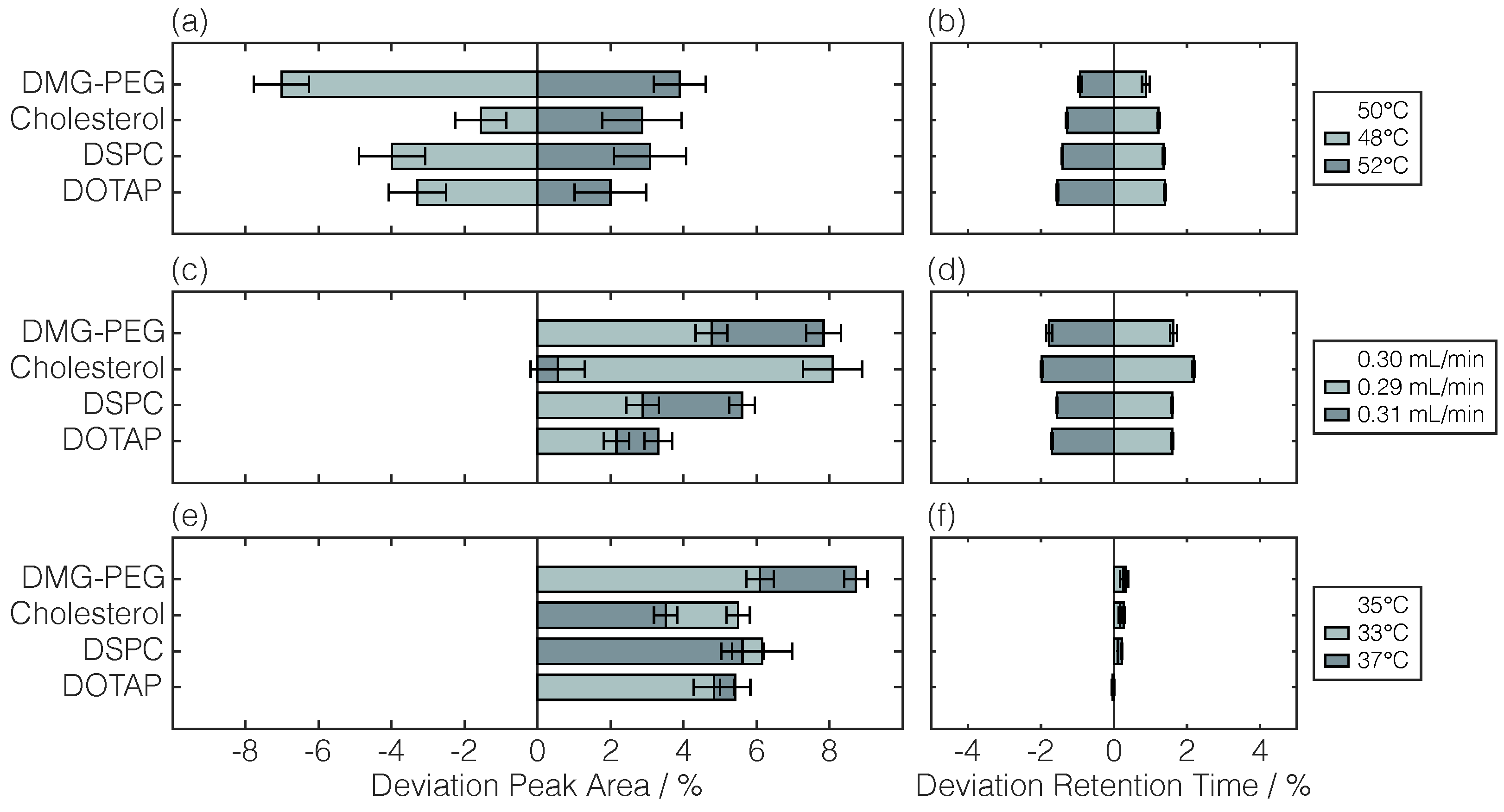
3.2. RP-CAD Complements LNP Attributes and Reveals Process Performance
4. Materials and Methods
4.1. Materials and Buffers
4.2. Development of the RP-HPLC-CAD Method
4.2.1. Preparation of Lipid Stock Solutions
4.2.2. Instrumentation
4.2.3. Variation of PFV and the Linearity Range
4.2.4. RP-CAD Method Calibration and Validation
4.3. Lipid Nanoparticle Process Characterization
4.3.1. Preparation of Aqueous Nucleic Acid Stock Solution
4.3.2. Microfluidics and Purification
4.3.3. Lipid Concentration, Molar Ratio, and Recovery
4.3.4. Particle Size and Surface Charge
4.3.5. Nucleic Acid Encapsulation
4.4. Statistical Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| ACN | Acetonitril |
| CL | Cationic lipid |
| CAD | Charged aerosol detection |
| DLS | Dynamic light scattering |
| DMG-PEG | 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 |
| DMPE-PEG | 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene |
| glycol)-2000] (ammonium salt) | |
| DOBAQ | N-(4-carboxybenzyl)-N,N-dimethyl-2,3-bis(oleoyloxy) propan-1-aminium |
| DOTAP | 1,2-Dioleoyl-3-trimethylammonium-propane (chloride salt) |
| DSPC | 1,2-Dioctadecanoyl-sn-glycero-3-phosphocholine |
| ELS | Electrophoretic light scattering |
| FRR | Flow rate ratio |
| HPLC | High performance liquid chromatography |
| ICH | International Council for Harmonisation |
| LNP | Lipid nanoparticle |
| mRNA | Messenger RNA |
| PDI | Polydispersity index |
| PEG | Polyethylene glycol |
| PEGL | Polyethylene glycol lipid |
| PFV | Power function value |
| RI | Refractive index |
| RP | Reversed-phase |
| siRNA | Small-interfering RNA |
| TE | Tris-EDTA |
| TEM | Transmission electron microscopy |
| TFA | Trifluoroacetic acid |
| TFR | Total flow rate |
| Tris | Tris(hydroxymethyl)aminomethane |
Appendix A



References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C 2012, 116, 18440–18450. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Albertsen, C.H.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Leung, A.K.; Tam, Y.Y.C.; Chen, S.; Hafez, I.M.; Cullis, P.R. Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J. Phys. Chem. B 2015, 119, 8698–8706. [Google Scholar] [CrossRef]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing considerations for the development of lipid nanoparticles using microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; Tam, Y.Y.C.; Cullis, P.R. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale 2019, 11, 21733–21739. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; van der Meel, R.; Zaifman, J.; Darjuan, M.M.; Grisch-Chan, H.M.; Thöny, B.; Tam, Y.Y.C.; Cullis, P.R. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 2019, 11, 9023–9031. [Google Scholar] [CrossRef]
- Fan, Y.; Yen, C.W.; Lin, H.C.; Hou, W.; Estevez, A.; Sarode, A.; Goyon, A.; Bian, J.; Lin, J.; Koenig, S.G.; et al. Automated high-throughput preparation and characterization of oligonucleotide-loaded lipid nanoparticles. Int. J. Pharm. 2021, 599. [Google Scholar] [CrossRef]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.C.; Lin, P.J.C.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther.-Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem.-Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Love, K.T.; Dorkin, J.R.; Sirirungruang, S.; Zhang, Y.; Chen, D.; Bogorad, R.L.; Yin, H.; Chen, Y.; Vegas, A.J.; et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl. Acad. Sci. USA 2014, 111, 3955–3960. [Google Scholar] [CrossRef]
- Ball, R.; Bajaj, P.; Whitehead, K. Achieving long-term stability of lipid nanoparticles: Examining the effect of pH, temperature, and lyophilization. Int. J. Nanomed. 2016, 12, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.L.; Hajj, K.A.; Vizelman, J.; Bajaj, P.; Whitehead, K.A. Lipid Nanoparticle Formulations for Enhanced Co-delivery of siRNA and mRNA. Nano Lett. 2018, 18, 3814–3822. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef]
- Mildner, R.; Hak, S.; Parot, J.; Hyldbakk, A.; Borgos, S.; Some, D.; Johann, C.; Caputo, F. Improved multidetector asymmetrical-flow field-flow fractionation method for particle sizing and concentration measurements of lipid-based nanocarriers for RNA delivery. Eur. J. Pharm. Biopharm. 2021, 163, 252–265. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, J.E.; Lee, J.C.; Maharjan, R.; Oh, H.; Lee, K.; Kim, N.A.; Jeong, S.H. Optimization of HPLC–CAD method for simultaneous analysis of different lipids in lipid nanoparticles with analytical QbD. J. Chromatogr. A 2023, 1709, 464375. [Google Scholar] [CrossRef]
- Bender, V.; Fuchs, L.; Süss, R. RP-HPLC-CAD method for the rapid analysis of lipids used in lipid nanoparticles derived from dual centrifugation. Int. J. Pharm. X 2024, 7, 100255. [Google Scholar] [CrossRef]
- Okuda, K.; Sato, Y.; Iwakawa, K.; Sasaki, K.; Okabe, N.; Maeki, M.; Tokeshi, M.; Harashima, H. On the size-regulation of RNA-loaded lipid nanoparticles synthesized by microfluidic device. J. Control. Release 2022, 348, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Kulkarni, J.A.; Huynh, A.; Chen, S.; van der Meel, R.; Tam, Y.Y.C.; Cullis, P.R. Characterization of Lipid Nanoparticles Containing Ionizable Cationic Lipids Using Design-of-Experiments Approach. Langmuir 2021, 37, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC-Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef]
- Gamache, P.H.; McCarthy, R.S.; Freeto, S.M.; Asa, D.J.; Woodcock, M.J.; Laws, K.; Cole, R.O. HPLC analysis of nonvolatile analytes using charged aerosol detection. LC-GC Europe 2005, 18, 345–354. [Google Scholar]
- Gamache, P.H.; Kaufman, S.L. Principles of Charged Aerosol Detection; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–65. [Google Scholar] [CrossRef]
- Moreau, R.A. The analysis of lipids via HPLC with a charged aerosol detector. Lipids 2006, 41, 727–734. [Google Scholar] [CrossRef]
- Soliven, A.; Ahmad, I.A.H.; Tam, J.; Kadrichu, N.; Challoner, P.; Markovich, R.; Blasko, A. A simplified guide for charged aerosol detection of non-chromophoric compounds—Analytical method development and validation for the HPLC assay of aerosol particle size distribution for amikacin. J. Pharm. Biomed. Anal. 2017, 143, 68–76. [Google Scholar] [CrossRef]
- Tam, J.; Ahmad, I.A.; Blasko, A. A four parameter optimization and troubleshooting of a RPLC—Charged aerosol detection stability indicating method for determination of S-lysophosphatidylcholines in a phospholipid formulation. J. Pharm. Biomed. Anal. 2018, 155, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.A.H.; Blasko, A.; Tam, J.; Variankaval, N.; Halsey, H.M.; Hartman, R.; Regalado, E.L. Revealing the inner workings of the power function algorithm in Charged Aerosol Detection: A simple and effective approach to optimizing power function value for quantitative analysis. J. Chromatogr. A 2019, 1603, 1–7. [Google Scholar] [CrossRef]
- Pawellek, R.; Muellner, T.; Gamache, P.; Holzgrabe, U. Power function setting in charged aerosol detection for the linearization of detector response—Optimization strategies and their application. J. Chromatogr. A 2021, 1637. [Google Scholar] [CrossRef]
- Díaz-López, R.; Libong, D.; Tsapis, N.; Fattal, E.; Chaminade, P. Quantification of pegylated phospholipids decorating polymeric microcapsules of perfluorooctyl bromide by reverse phase HPLC with a charged aerosol detector. J. Pharm. Biomed. Anal. 2008, 48, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, M.; Suryavanshi, V.; Panduranga, N.S.; Jayaraman, K.; Bajpai, L.; Fish, W.; Hu, Y.; Bhutani, H. Development of HPLC-CAD stability indicating assay method for polyethylene glycol-conjugated phospholipid (DMPE-PEG 2000) and identification of its degradation products. J. Pharm. Biomed. Anal. 2021, 198. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Rahnfeld, L.; Luciani, P. Analytical profiling and stability evaluation of liposomal drug delivery systems: A rapid UHPLC-CAD-based approach for phospholipids in research and quality control. Talanta 2020, 220, 121320. [Google Scholar] [CrossRef]
- Li, L.; Foley, J.P.; Helmy, R. Simultaneous separation of small interfering RNA and lipids using ion-pair reversed-phase liquid chromatography. J. Chromatogr. A 2019, 1601, 145–154. [Google Scholar] [CrossRef]
- Kinsey, C.; Lu, T.; Deiss, A.; Vuolo, K.; Klein, L.; Rustandi, R.R.; Loughney, J.W. Determination of lipid content and stability in lipid nanoparticles using ultra high-performance liquid chromatography in combination with a Corona Charged Aerosol Detector. Electrophoresis 2022, 43, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, C.; Wu, X.; Cui, Y.; Liu, X.; Jin, Y.; Li, Y.; Wang, L. Validation of an HPLC-CAD Method for Determination of Lipid Content in LNP-Encapsulated COVID-19 mRNA Vaccines. Vaccines 2023, 11, 937. [Google Scholar] [CrossRef]
- ICH. Q2(R2) Guideline on Validation of Analytical Procedures. 2022. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q2r2-guideline-validation-analytical-procedures-step-5-revision-1_en.pdf (accessed on 8 July 2024).
- Murugaiah, V.; Zedalis, W.; Lavine, G.; Charisse, K.; Manoharan, M. Reversed-phase high-performance liquid chromatography method for simultaneous analysis of two liposome-formulated short interfering RNA duplexes. Anal. Biochem. 2010, 401, 61–67. [Google Scholar] [CrossRef]
- Studzińska, S.; Pietrzak, L.; Buszewski, B. The Effects of Stationary Phases on Retention and Selectivity of Oligonucleotides in IP-RP-HPLC. Chromatographia 2014, 77, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Quarry, M.A.; Glajch, J.L. Solvent-strength selectivity in reversed-phase HPLC. Chromatographia 1987, 24, 33–44. [Google Scholar] [CrossRef]
- Stroock, A.D.; Dertinger, S.K.W.; Ajdari, A.; Mezić, I.; Stone, H.A.; Whitesides, G.M. Chaotic Mixer for Microchannels. Science 2002, 295, 647–651. [Google Scholar] [CrossRef]
- EMA. Onpattro: Summary of Product Characteristics. 2018. Available online: https://www.ema.europa.eu/en/documents/product-information/onpattro-epar-product-information_en.pdf (accessed on 8 July 2024).
- Suzuki, Y.; Hyodo, K.; Tanaka, Y.; Ishihara, H. SiRNA-lipid nanoparticles with long-term storage stability facilitate potent gene-silencing in vivo. J. Control. Release 2015, 220, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Jakubek, Z.J.; Chen, S.; Zaifman, J.; Tam, Y.Y.C.; Zou, S. Lipid Nanoparticle and Liposome Reference Materials: Assessment of Size Homogeneity and Long-Term −70 °C and 4 °C Storage Stability. Langmuir 2022, 39, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol. Ther.-Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | Retention Time | Linear Regression | |||
|---|---|---|---|---|---|
| Slope | y-Intercept | ||||
| min | pA min (ng)−1 | pA min | - | - | |
| DOTAP | 9.69 | 0.0107 | 0.0774 | 0.9995 | 0.0010 |
| DOBAQ | 9.33 | 0.0153 | 0.1735 | 0.9966 | 0.0146 |
| DSPC | 10.13 | 0.0162 | 0.0671 | 0.9990 | 0.0050 |
| CHOL | 8.14 | 0.0125 | 0.0742 | 0.9988 | 0.0035 |
| DMG-PEG | 8.91 | 0.0157 | −0.0973 | 0.9998 | 0.0009 |
| DMPE-PEG | 7.31 | 0.0094 | −0.0291 | 0.9981 | 0.0031 |
| Total Flow Rate | Synthesized | Dialyzed |
|---|---|---|
| mL min−1 | % | % |
| 10 | 97.07 | 98.04 |
| 15 | 98.45 | 99.88 |
| 20 | 99.99 | 100.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckert, N.; Dietrich, A.; Hubbuch, J. RP-CAD for Lipid Quantification: Systematic Method Development and Intensified LNP Process Characterization. Pharmaceuticals 2024, 17, 1217. https://doi.org/10.3390/ph17091217
Beckert N, Dietrich A, Hubbuch J. RP-CAD for Lipid Quantification: Systematic Method Development and Intensified LNP Process Characterization. Pharmaceuticals. 2024; 17(9):1217. https://doi.org/10.3390/ph17091217
Chicago/Turabian StyleBeckert, Nicole, Annabelle Dietrich, and Jürgen Hubbuch. 2024. "RP-CAD for Lipid Quantification: Systematic Method Development and Intensified LNP Process Characterization" Pharmaceuticals 17, no. 9: 1217. https://doi.org/10.3390/ph17091217
APA StyleBeckert, N., Dietrich, A., & Hubbuch, J. (2024). RP-CAD for Lipid Quantification: Systematic Method Development and Intensified LNP Process Characterization. Pharmaceuticals, 17(9), 1217. https://doi.org/10.3390/ph17091217