
Dysregulation of Mitochondrial Homeostasis in Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress in Mitochondria
Cellular Effects of Oxidative Stress in Cardiovascular Disease
3. Mitochondrial Membrane Dynamics
3.1. Mitochondria Permeability Transition (MPT) in Ischemia Reperfusion Injury
3.2. Mitochondrial Permeability Transition and Post Cardiac Arrest Syndrome
3.3. Mitochondrial Permeability Transition and COVID-19 Vasculopathy
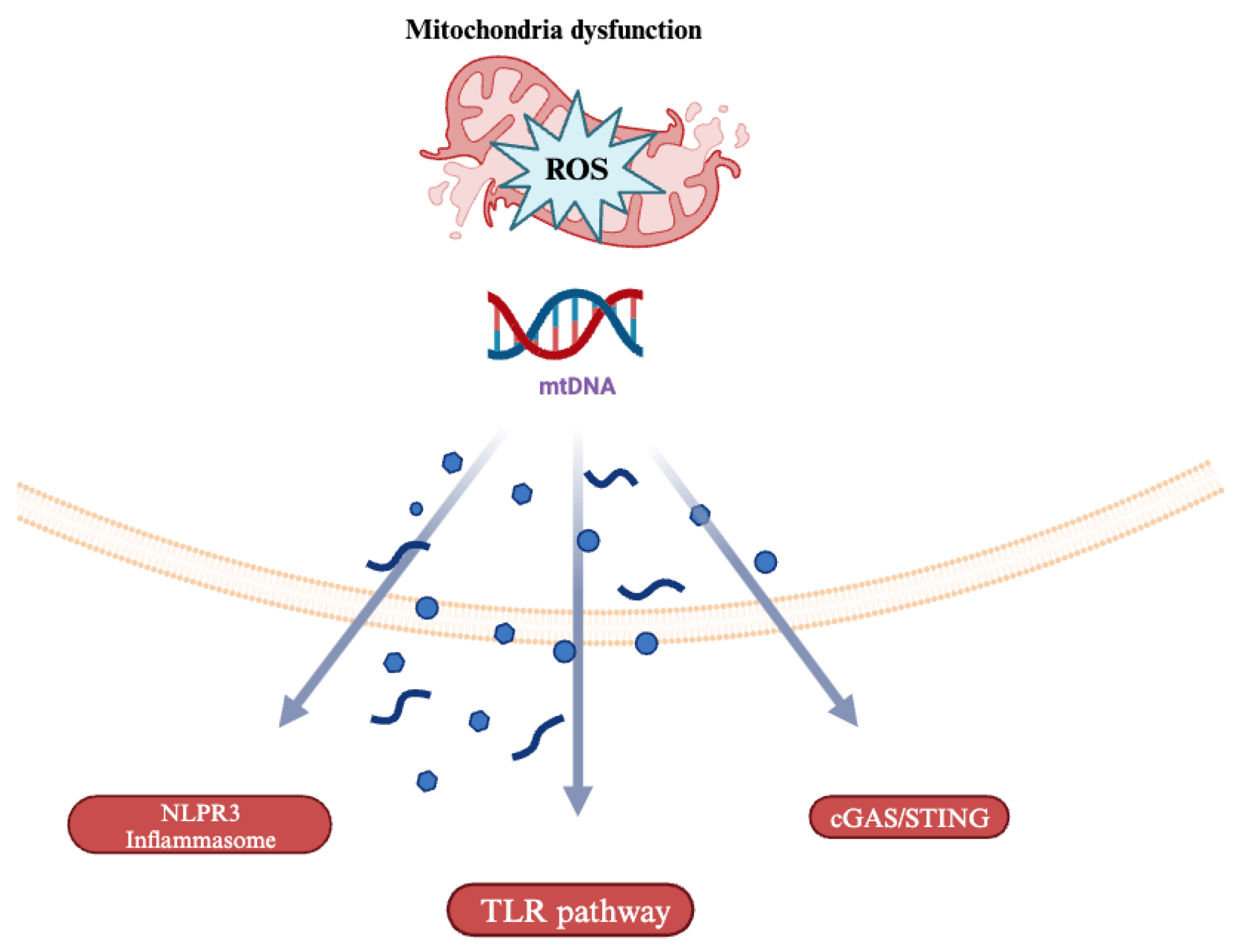
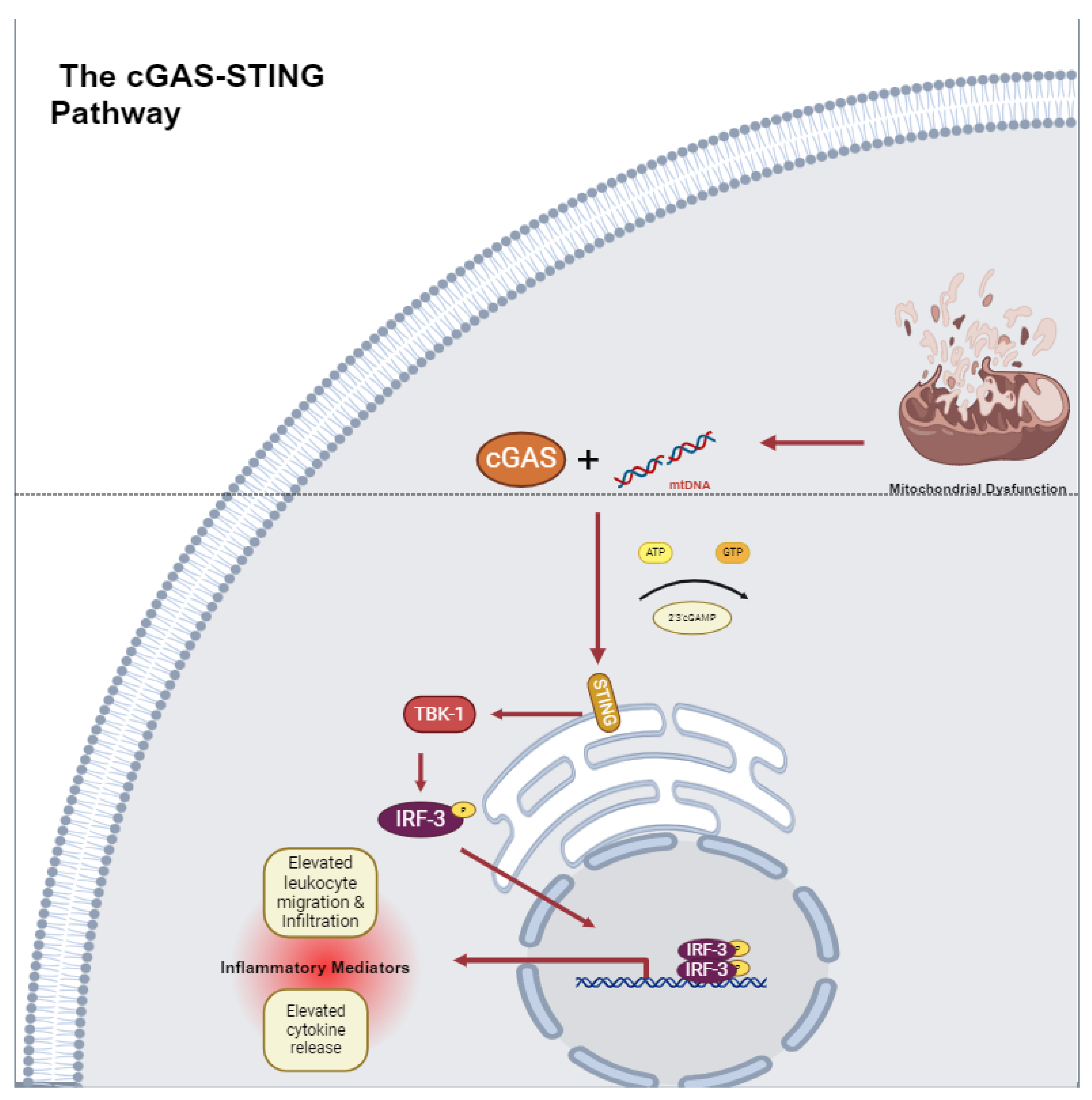
4. Mitochondrial DNA (mtDNA)-Driven Inflammation
The cGAS-STING Pathway in Cardiovascular Disease
5. Mitochondrial Dynamics and Cardiovascular Disease
5.1. Mitochondrial Dynamics and Vascular Smooth Muscle Cells
5.2. Mitochondrial Dynamics and Endothelial Cells
6. Mitophagy and Cardiovascular Disease
6.1. PINK1 and Parkin
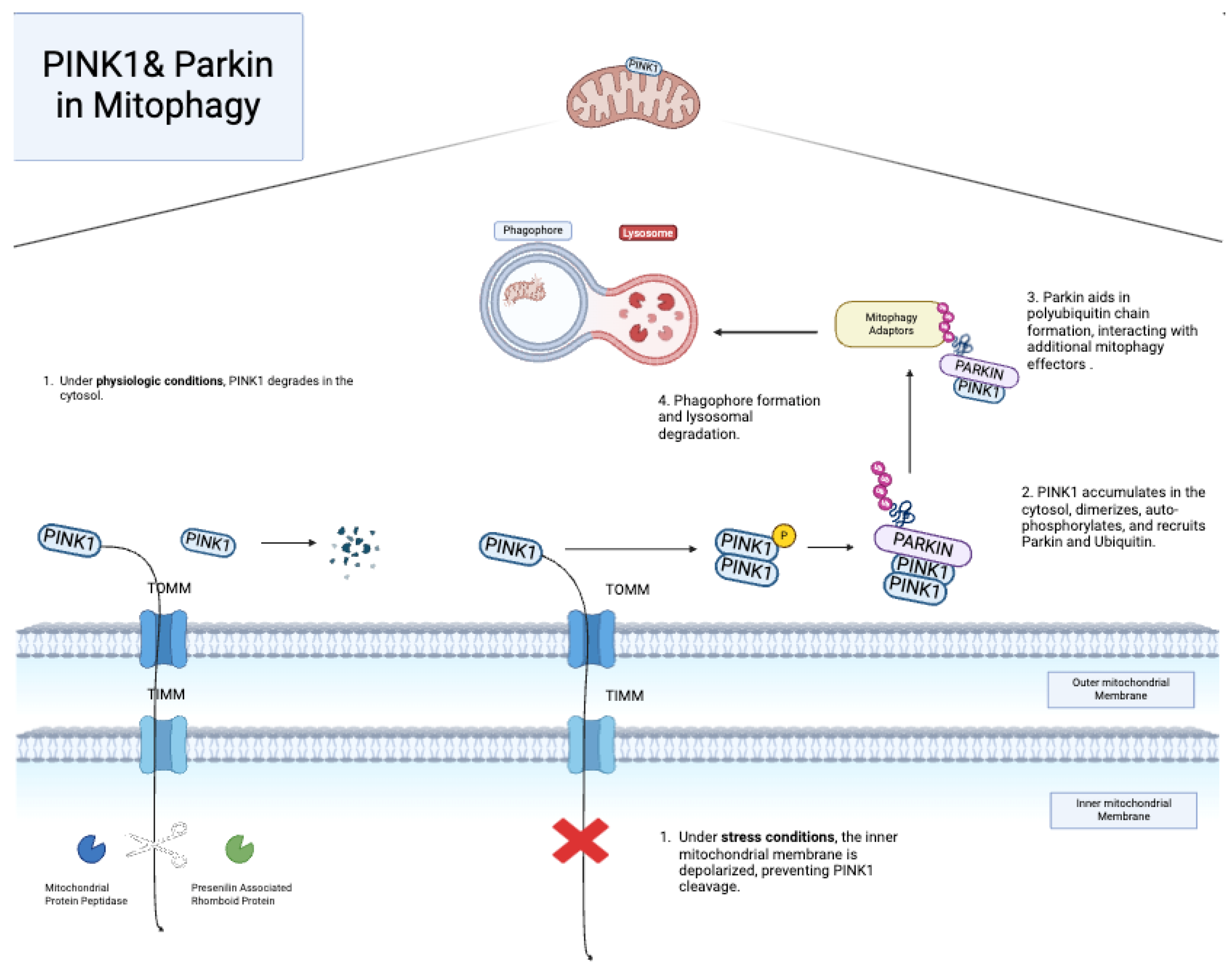
6.2. Other Mitophagy Pathways
6.3. Mitochondrial Therapeutics and Their Potential in Non-Cardiovascular Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CVD | cardiovascular disease |
| ATP | Adenosine Triphosphate |
| AD | Alzheimer’s disease |
| ADP | Adenosine Diphosphate |
| ApoE | Apo Lipoprotein E |
| Bcl-2 | B-cell lymphoma |
| CytC | cytochrome C |
| BNIP3 | BCL-2-interacting protein-3-like protein |
| Ca2+ | calcium |
| CANX | calnexin |
| cGAMP | 2′3′-cyclic guanosine monophosphate |
| cGAS | cyclic GMP-AMP synthase |
| CRISPR | Cluster Regulatory Interspaced Short Palindromic Repeats |
| CSA | cyclosporine A |
| DNA | Deoxyribonucleic Acid |
| DRP-1 | Dynamin-related protein |
| EC | endothelial cell |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| FIS-1 | fission protein 1 |
| Mff | Mitochondrial Fission Factor |
| MiD49 | Mitochondrial Dynamics Protein 49 |
| MiD51 | Mitochondrial Dynamics Protein 51 |
| FUNDC-1 | Fun14 Domain Containing 1 protein |
| HAEC | human aortic endothelial cell |
| HCAEC | human coronary artery endothelial cell |
| HFD | high-fat diet |
| HUVEC | human umbilical vein endothelial cell |
| I-CAM | Intercellular Adhesion Molecule |
| I-R | ischemia reperfusion |
| IMM | inner mitochondrial membrane |
| INF-2 | Inverted Formin 2 |
| IRF-3 | interferon gamma regulatory factor 3 |
| NF-KB | nuclear factor-kappa beta |
| LC3 | Light Chain 3 |
| LDL | Low-Density Lipoprotein |
| LPS | Lipopolysaccharide |
| LV | Left Ventricle |
| MCEC | mouse coronary endothelial cell |
| mDIVI | Mitochondrial Division Inhibitor |
| MELAS | mitochondrial encephalopathy, lactic acidosis, and stroke-like syndrome |
| MFN | Mitofusin |
| OPA-1 | Optic Atrophy 1 |
| MI | myocardial infarction |
| MMP | Mixed Metalloproteinase |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| MPT | mitochondria permeability transition |
| MST-1 | mammalian Ste2-like kinase |
| MtDNA | mitochondrial DNA |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NC | normal chow |
| NLRP3 | NOD-, LRR-, and pyrin-domain-containing protein 3 |
| O2 | oxygen |
| OMM | outer mitochondrial membrane |
| OSS | oscillating shear stress |
| PA | palmitic acid |
| PCAS | post cardiac arrest syndrome |
| PCI | Percutaneous Coronary Intervention |
| PDGF | Platelet-Derived Growth Factor |
| PINK1 | PTEN-induced putative kinase protein |
| PKM-2 | pyruvate kinase myozyme 2 |
| PMN | Polymorphonuclear Leukocyte |
| pO2 | partial pressure of blood oxygen |
| PTPC | permeability transition pore complex |
| RCD | regulated cell death |
| ROS | reactive oxidative species |
| ROSC | Return of Spontaneous Circulation |
| SAVI | STING-Associated Vasculopathy |
| Spire1C | Spire Homolog 1C |
| sPLA2-IIA | phospholipase A2 2A |
| STING | stimulator of interferon genes |
| TAC | temporary aortic constriction |
| TBHP | tert-butyl hydroperoxide |
| TBK-1 | TANK-binding kinase 1 |
| TFAM | Transcription Factor A |
| TLR | Toll-Like Receptors |
| V-CAM | Vascular Cell Adhesion Molecule |
| VEGF | Vascular Endothelial Growth Factor |
| VR-EPC | vascular-resident endothelial progenitor cell |
| VSMC | vascular smooth muscle cell |
| WT | wild-type |
| ∆ψm | mitochondrial membrane potential |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. GBD-NHLBI-JACC Global Burden of Cardiovascular Diseases Writing Group. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Kundu, T.K.; Velayutham, M.; Zweier, J.L. Aldehyde oxidase functions as a superoxide generating NADH oxidase: An important redox regulated pathway of cellular oxygen radical formation. Biochemistry 2012, 51, 2930–2939. [Google Scholar] [CrossRef]
- Schröder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef]
- Penna, C.; Mancardi, D.; Rastaldo, R.; Pagliaro, P. Cardioprotection: A radical view. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2009, 1787, 781–793. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. From Mitochondria to Atherosclerosis: The Inflammation Path. Biomedicines 2021, 9, 258. [Google Scholar] [CrossRef]
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.-F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef]
- Moriya, J. Critical roles of inflammation in atherosclerosis. J. Cardiol. 2019, 73, 22–27. [Google Scholar] [CrossRef]
- Koga, Y.; Akita, Y.; Junko, N.; Yatsuga, S.; Povalko, N.; Fukiyama, R.; Ishii, M.; Matsuishi, T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 2006, 66, 1766–1769. [Google Scholar] [CrossRef]
- Kantrow, S.P.; Piantadosi, C.A. Release of cytochrome c from liver mitochondria during permeability transition. Biochem. Biophys. Res. Commun. 1997, 232, 669–671. [Google Scholar] [CrossRef]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol.-Cell Physiol. 1990, 258, C755–C786. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Li, H.; Wang, Y.; Peng, X.; Shao, H.; Li, H.; Bai, S.; Xu, C. Exogenous spermine inhibits hypoxia/ischemia-induced myocardial apoptosis via regulation of mitochondrial permeability transition pore and associated pathways. Exp. Biol. Med. 2016, 241, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Zahrebelski, G.; Nieminen, A.L.; Al-Ghoul, K.; Qian, T.; Herman, B.; Lemasters, J.J. Progression of subcellular changes during chemical hypoxia to cultured rat hepatocytes: A laser scanning confocal microscopic study. Hepatology 1995, 21, 1361–1372. [Google Scholar]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Upadhaya, S.; Madala, S.; Baniya, R.; Subedi, S.K.; Saginala, K.; Bachuwa, G. Impact of cyclosporine A use in the prevention of reperfusion injury in acute myocardial infarction: A meta-analysis. Cardiol. J. 2017, 24, 43–50. [Google Scholar] [CrossRef]
- Pottecher, J.; Kindo, M.; Chamaraux-Tran, T.-N.; Charles, A.-L.; Lejay, A.; Kemmel, V.; Vogel, T.; Chakfe, N.; Zoll, J.; Diemunsch, P.; et al. Skeletal muscle ischemia-reperfusion injury and cyclosporine A in the aging rat. Fundam. Clin. Pharmacol. 2016, 30, 216–225. [Google Scholar] [CrossRef]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, L.; Xu, J.; Gao, J.; Ye, S.; Li, Z.; Chen, Y.; Zhang, X. Limb Ischemic Postconditioning Alleviates Postcardiac Arrest Syndrome through the Inhibition of Mitochondrial Permeability Transition Pore Opening in a Porcine Model. BioMed Res. Int. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Jankauskas, S.S.; Kansakar, U.; Varzideh, F.; Avvisato, R.; Santulli, G. COVID-19 causes the opening of the mitochondrial permeability transition pore in human endothelial cells. Vasc. Pharmacol. 2024, 155, 107349. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef]
- Groot, G.S.; Kroon, A.M. Mitochondrial DNA from various organisms does not contain internally methylated cytosine in -CCGG- sequences. Biochim. Biophys. Acta 1979, 564, 355–357. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Sun, S.; Sursal, T.; Adibnia, Y.; Zhao, C.; Zheng, Y.; Li, H.; Otterbein, L.E.; Hauser, C.J.; Itagaki, K. Mitochondrial DAMPs Increase Endothelial Permeability through Neutrophil Dependent and Independent Pathways. PLoS ONE 2013, 8, e59989. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef]
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The cGAS–STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.-C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Montealegre Sanchez, G.A.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Dong, M.; Chen, M.; Zhang, Y.; He, X.; Min, J.; Tan, Y.; Wei, H.; Li, X.; Chen, X.; Zheng, L.; et al. Oscillatory shear stress promotes endothelial senescence and atherosclerosis via STING activation. Biochem. Biophys. Res. Commun. 2024, 715, 149979. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Y.; Zhang, L.; Ren, P.; Zhang, C.; Li, Y.; Azares, A.R.; Zhang, M.; Guo, J.; Ghaghada, K.B.; et al. Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation 2020, 141, 42–66. [Google Scholar] [CrossRef]
- Mao, Y.; Luo, W.; Zhang, L.; Wu, W.; Yuan, L.; Xu, H.; Song, J.; Fujiwara, K.; Abe, J.; LeMaire, S.A.; et al. STING–IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Zhang, H.; Park, H.; Choi, K.-S.; Lee, H.-W.; Agrawal, V.; Kim, Y.-M.; Kwon, Y.-G. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat. Commun. 2015, 6, 6943. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Mao, Y.; Luo, W.; Wu, W.; Xu, H.; Wang, X.L.; Shen, Y.H. Palmitic acid dysregulates the Hippo–YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS–STING–IRF3 signaling mechanism. J. Biol. Chem. 2017, 292, 15002–15015. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Ban, T.; Heymann, J.A.W.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef]
- Ishida, M. Mitochondrial fusion and fission in vascular disease. Hypertens. Res. 2024, 47, 2935–2938. [Google Scholar] [CrossRef]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Wang, L.; Yu, T.; Lee, H.; O’Brien, D.K.; Sesaki, H.; Yoon, Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc. Res. 2015, 106, 272–283. [Google Scholar] [CrossRef]
- Salabei, J.K.; Hill, B.G. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 2013, 1, 542–551. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, K.; Gao, W.; Li, Q.; Chen, L.; Wang, G.; Tang, J. Overexpression of Mitofusin 2 inhibited oxidized low-density lipoprotein induced vascular smooth muscle cell proliferation and reduced atherosclerotic lesion formation in rabbit. Biochem. Biophys. Res. Commun. 2007, 363, 411–417. [Google Scholar] [CrossRef]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef]
- Li, M.; Zhu, Y.; Jaiswal, S.K.; Liu, N.-F. Mitochondria Homeostasis and Vascular Medial Calcification. Calcif. Tissue Int. 2021, 109, 113–120. [Google Scholar] [CrossRef]
- Rashdan, N.A.; Sim, A.M.; Cui, L.; Phadwal, K.; Roberts, F.L.; Carter, R.; Ozdemir, D.D.; Hohenstein, P.; Hung, J.; Kaczynski, J.; et al. Osteocalcin Regulates Arterial Calcification Via Altered Wnt Signaling and Glucose Metabolism. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2020, 35, 357–367. [Google Scholar] [CrossRef]
- Chen, W.R.; Zhou, Y.J.; Sha, Y.; Wu, X.P.; Yang, J.Q.; Liu, F. Melatonin attenuates vascular calcification by inhibiting mitochondria fission via an AMPK/Drp1 signalling pathway. J. Cell. Mol. Med. 2020, 24, 6043–6054. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vasc. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Z.; Chen, K.; Jin, H.; Wu, J.; Huang, Z.; Lu, X.; Zheng, X. Dynamin-related protein 1 mediates the therapeutic effect of isoliquiritigenin in diabetic intimal hyperplasia via regulation of mitochondrial fission. Hypertens. Res. 2024, 47, 1908–1924. [Google Scholar] [CrossRef]
- Zou, R.; Shi, W.; Qiu, J.; Zhou, N.; Du, N.; Zhou, H.; Chen, X.; Ma, L. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc. Diabetol. 2022, 21, 106. [Google Scholar] [CrossRef]
- Chen, T.; Deng, S.; Lin, R. The inhibitory effect of Isoliquiritigenin on the proliferation of human arterial smooth muscle cell. BMC Pharmacol. Toxicol. 2017, 18, 57. [Google Scholar] [CrossRef]
- Jin, H.; Jiang, Y.; Du, F.; Guo, L.; Wang, G.; Kim, S.C.; Lee, C.W.; Shen, L.; Zhao, R. Isoliquiritigenin Attenuates Monocrotaline-Induced Pulmonary Hypertension via Inhibition of the Inflammatory Response and PASMCs Proliferation. Evid.-Based Complement. Altern. Med. Ecam 2019, 2019, 4568198. [Google Scholar] [CrossRef] [PubMed]
- Torres, G.; Morales, P.E.; García-Miguel, M.; Norambuena-Soto, I.; Cartes-Saavedra, B.; Vidal-Peña, G.; Moncada-Ruff, D.; Sanhueza-Olivares, F.; San Martín, A.; Chiong, M. Glucagon-like peptide-1 inhibits vascular smooth muscle cell dedifferentiation through mitochondrial dynamics regulation. Biochem. Pharmacol. 2016, 104, 52–61. [Google Scholar] [CrossRef]
- Lugus, J.J.; Ngoh, G.A.; Bachschmid, M.M.; Walsh, K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J. Mol. Cell. Cardiol. 2011, 51, 885–893. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Rao, G.; Murphy, B.; Dey, A.; Dwivedi, S.K.D.; Zhang, Y.; Roy, R.V.; Chakraborty, P.; Bhattacharya, R.; Mukherjee, P. Cystathionine beta synthase regulates mitochondrial dynamics and function in endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 9372–9392. [Google Scholar] [CrossRef]
- Zhu, T.; Hu, Q.; Yuan, Y.; Yao, H.; Zhang, J.; Qi, J. Mitochondrial dynamics in vascular remodeling and target-organ damage. Front. Cardiovasc. Med. 2023, 10, 1067732. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Trudeau, K.; Molina, A.J.A.; Guo, W.; Roy, S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: Implications for diabetic retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Marx, N.; Lam, C.S.P.; Schnaidt, S.; Ofstad, A.P.; Brueckmann, M.; Jamal, W.; et al. On behalf of the EMPEROR-Reduced Trial Committees and Investigators. Effect of Empagliflozin on Cardiovascular and Renal Outcomes in Patients with Heart Failure by Baseline Diabetes Status: Results From the EMPEROR-Reduced Trial. Circulation 2021, 143, 337–349. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liu, J.; Piao, H.; Zhu, Z.; Wei, R.; Liu, K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 2022, 13, 1039241. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Veyron, S.; Soya, N.; Eldeeb, M.A.; Lukacs, G.L.; Fon, E.A.; Trempe, J.-F. Mechanism of PINK1 activation by autophosphorylation and insights into assembly on the TOM complex. Mol. Cell 2022, 82, 44–59.e6. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, B.; Wang, H.; He, H.; Wu, Q.; Qin, X.; Yang, X.; Chen, L.; Xu, G.; Yuan, Z.; et al. Disruption of the superoxide anions-mitophagy regulation axis mediates copper oxide nanoparticles-induced vascular endothelial cell death. Free Radic. Biol. Med. 2018, 129, 268–278. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, Z.; Mao, L.; Xu, G.; Li, N.; Zhang, M.; Weng, P.; Zheng, L.; Dong, X.; Hu, S.; et al. PINK1/TAX1BP1-directed mitophagy attenuates vascular endothelial injury induced by copper oxide nanoparticles. J. Nanobiotechnology 2022, 20, 149. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, H.; Yang, J.; Li, B.; Ding, J.; Cheng, S.; Bsoul, N.; Zhang, C.; Li, J.; Liu, H.; et al. Activating Parkin-dependent mitophagy alleviates oxidative stress, apoptosis, and promotes random-pattern skin flaps survival. Commun. Biol. 2022, 5, 616. [Google Scholar] [CrossRef]
- Zhu, W.; Yuan, Y.; Liao, G.; Li, L.; Liu, J.; Chen, Y.; Zhang, J.; Cheng, J.; Lu, Y. Mesenchymal stem cells ameliorate hyperglycemia-induced endothelial injury through modulation of mitophagy. Cell Death Dis. 2018, 9, 837. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, K.-A.; Kim, J.-H.; Kim, E.-H.; Bae, O.-N. Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species. Antioxidants 2020, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef]
- Zekri-Nechar, K.; Zamorano-León, J.J.; Cortina-Gredilla, M.; López-de-Andrés, A.; Jiménez-García, R.; Navarro-Cuellar, C.; López-Farré, A.; Martínez-Martínez, C.H. Mitochondrial mitophagy protection combining rivaroxaban and aspirin in high glucose-exposed human coronary artery endothelial cell. An in vitro study. Diabetes Vasc. Dis. Res. 2022, 19, 14791641221129877. [Google Scholar] [CrossRef]
- Xi, J.; Rong, Y.; Zhao, Z.; Huang, Y.; Wang, P.; Luan, H.; Xing, Y.; Li, S.; Liao, J.; Dai, Y.; et al. Scutellarin ameliorates high glucose-induced vascular endothelial cells injury by activating PINK1/Parkin-mediated mitophagy. J. Ethnopharmacol. 2021, 271, 113855. [Google Scholar] [CrossRef]
- Zhang, Y.; Weng, J.; Huan, L.; Sheng, S.; Xu, F. Mitophagy in atherosclerosis: From mechanism to therapy. Front. Immunol. 2023, 14, 1165507. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Peng, X.; Chen, H.; Li, Y.; Huang, D.; Huang, B.; Sun, D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol. Int. 2020, 44, 1481–1490. [Google Scholar] [CrossRef]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubašić, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, Q.; Sun, H.; Liu, L.; Wang, C.; Li, Z.; Xu, Y.; Wang, L.; Zhang, L.; Zhang, H.; et al. BNIP3 (BCL2 interacting protein 3) regulates pluripotency by modulating mitochondrial homeostasis via mitophagy. Cell Death Dis. 2022, 13, 334. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, W.; Chen, H.; Jiang, L.; Zhu, R.; Feng, D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 2016, 12, 1675–1676. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dai, X.; Wu, S.; Xu, W.; Song, P.; Huang, K.; Zou, M.-H. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat. Commun. 2021, 12, 2616. [Google Scholar] [CrossRef]
- Tassone, G.; Kola, A.; Valensin, D.; Pozzi, C. Dynamic Interplay between Copper Toxicity and Mitochondrial Dysfunction in Alzheimer’s Disease. Life 2021, 11, 386. [Google Scholar] [CrossRef]
- Ho, H.J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88. [Google Scholar] [CrossRef]
- Białas, A.J.; Sitarek, P.; Miłkowska-Dymanowska, J.; Piotrowski, W.J.; Górski, P. The Role of Mitochondria and Oxidative/Antioxidative Imbalance in Pathobiology of Chronic Obstructive Pulmonary Disease. Oxidative Med. Cell. Longev. 2016, 2016, 7808576. [Google Scholar] [CrossRef]
- Grattagliano, I.; Russmann, S.; Diogo, C.; Bonfrate, L.; Oliveira, P.J.; Wang, D.Q.; Portincasa, P. Mitochondria in chronic liver disease. Curr. Drug Targets 2011, 12, 879–893. [Google Scholar] [CrossRef]
- Lau, C.H.; Liang, Q.L.; Zhu, H. Next-generation CRISPR technology for genome, epigenome and mitochondrial editing. Transgenic Res. 2024, 33, 323–357. [Google Scholar] [CrossRef]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, R.; Wang, H.; Kazaleh, M.; Ailawadi, G.; Salmon, M. Dysregulation of Mitochondrial Homeostasis in Cardiovascular Diseases. Pharmaceuticals 2025, 18, 112. https://doi.org/10.3390/ph18010112
Patil R, Wang H, Kazaleh M, Ailawadi G, Salmon M. Dysregulation of Mitochondrial Homeostasis in Cardiovascular Diseases. Pharmaceuticals. 2025; 18(1):112. https://doi.org/10.3390/ph18010112
Chicago/Turabian StylePatil, Ricky, Hui Wang, Matthew Kazaleh, Gorav Ailawadi, and Morgan Salmon. 2025. "Dysregulation of Mitochondrial Homeostasis in Cardiovascular Diseases" Pharmaceuticals 18, no. 1: 112. https://doi.org/10.3390/ph18010112
APA StylePatil, R., Wang, H., Kazaleh, M., Ailawadi, G., & Salmon, M. (2025). Dysregulation of Mitochondrial Homeostasis in Cardiovascular Diseases. Pharmaceuticals, 18(1), 112. https://doi.org/10.3390/ph18010112