
Prodrug Approach as a Strategy to Enhance Drug Permeability

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
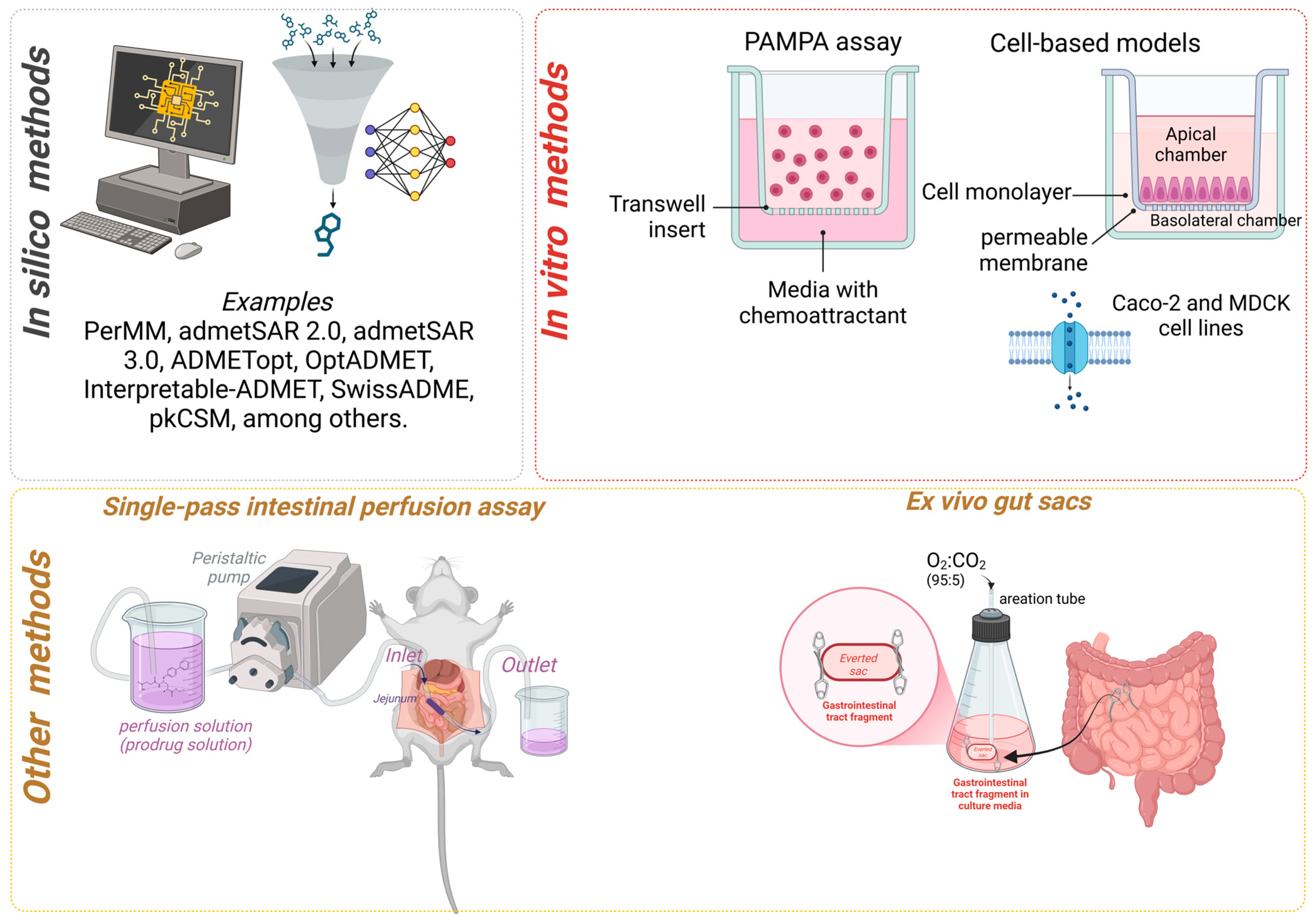
2. Permeability Determination Methods
2.1. In Silico Determination Methods
2.2. In Vitro Determination of Permeability
2.3. PAMPA Assay
2.4. Cell-Based Models
2.5. Other Methods to Determine the Permeability of Prodrugs
3. Examples of Market Prodrugs Designed to Enhance Drug Permeability
3.1. Prodrugs to Enhance Ocular, Skin, and CNS Permeability
3.2. The Use of Drug Transporters in the Pharmaceutical Market
4. New Technologies in Prodrugs to Enrich Permeability Properties
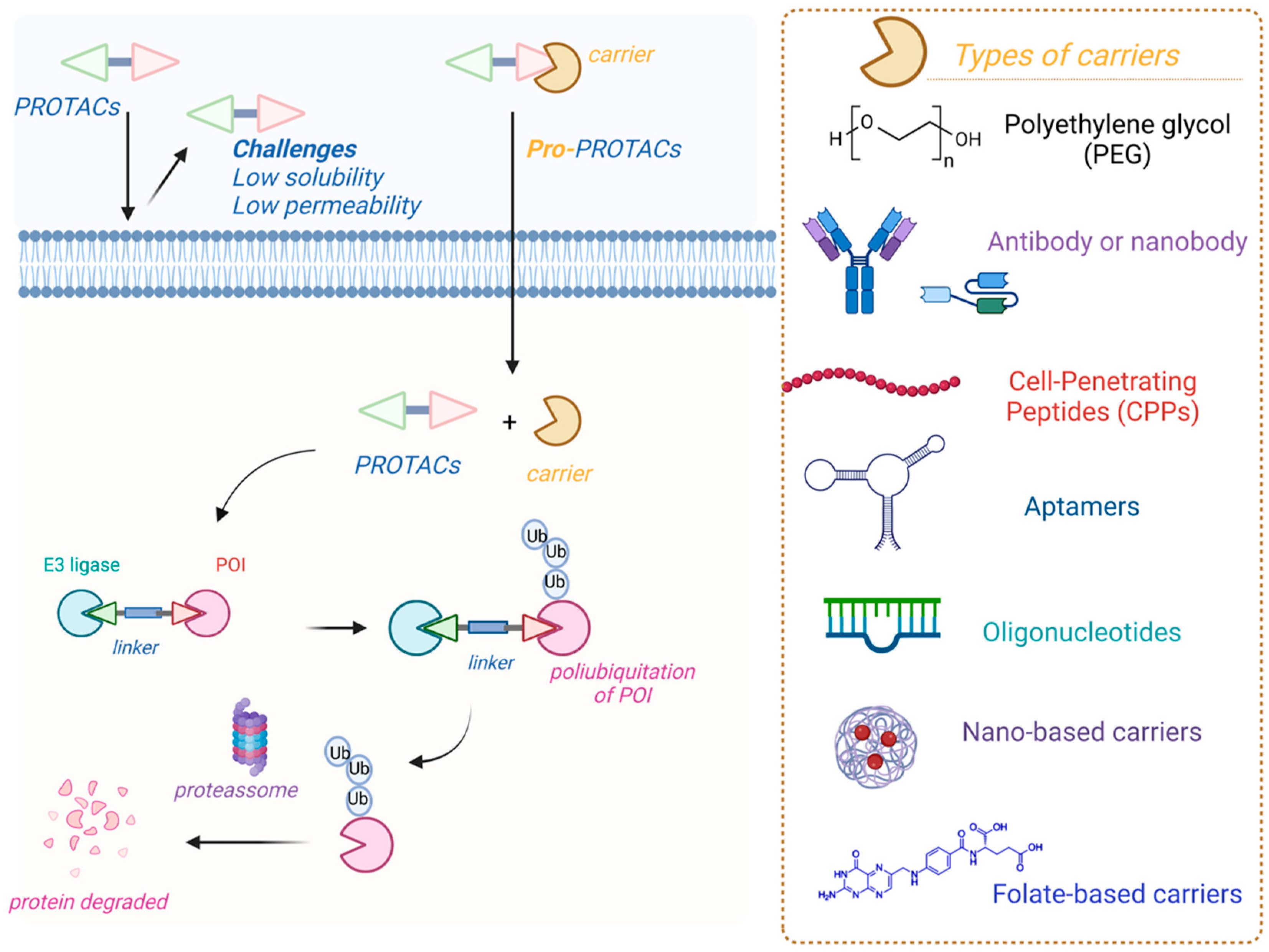
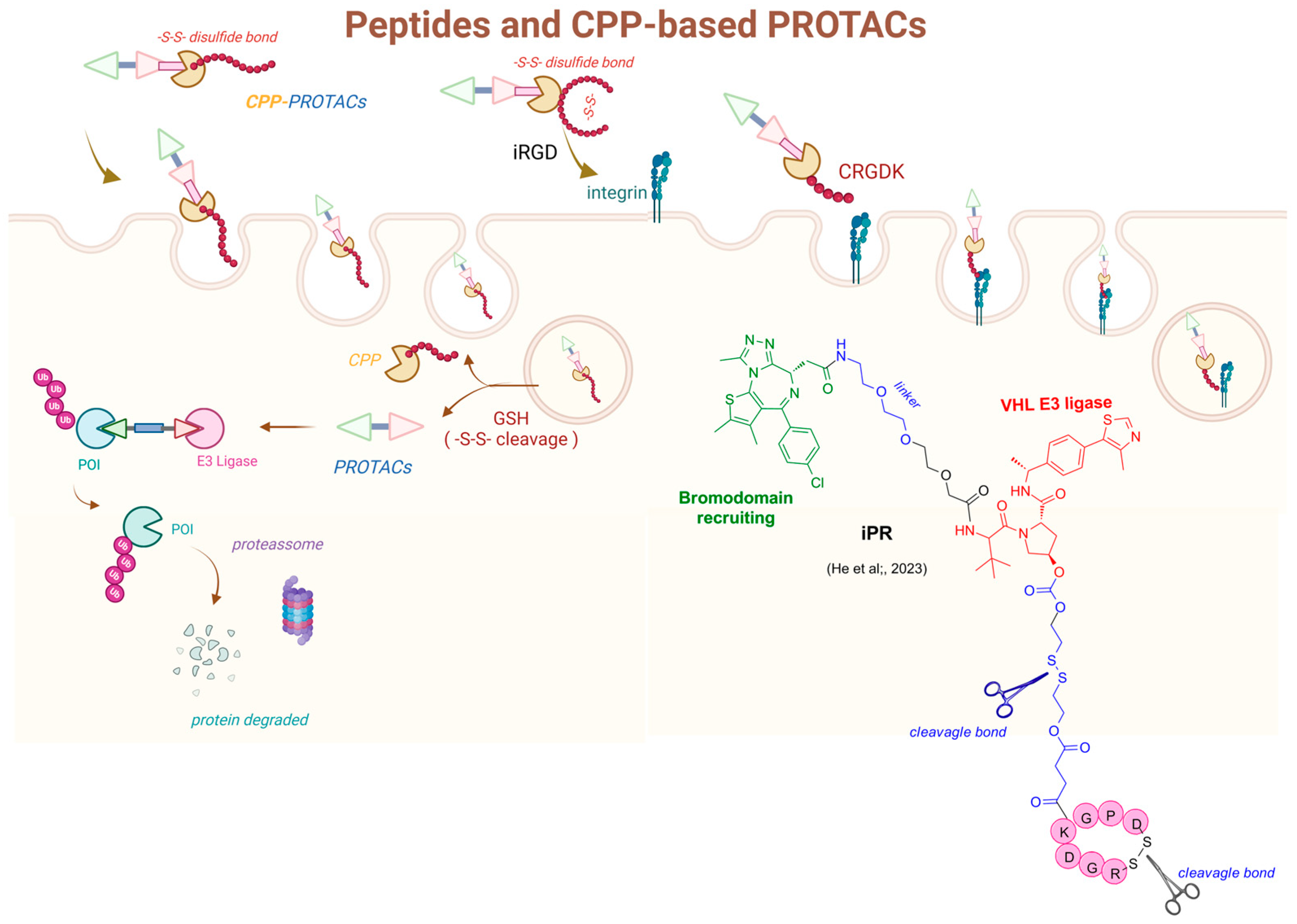
4.1. Prodrugs of PROteolysis TArgeting Chimeras (PROTACs)
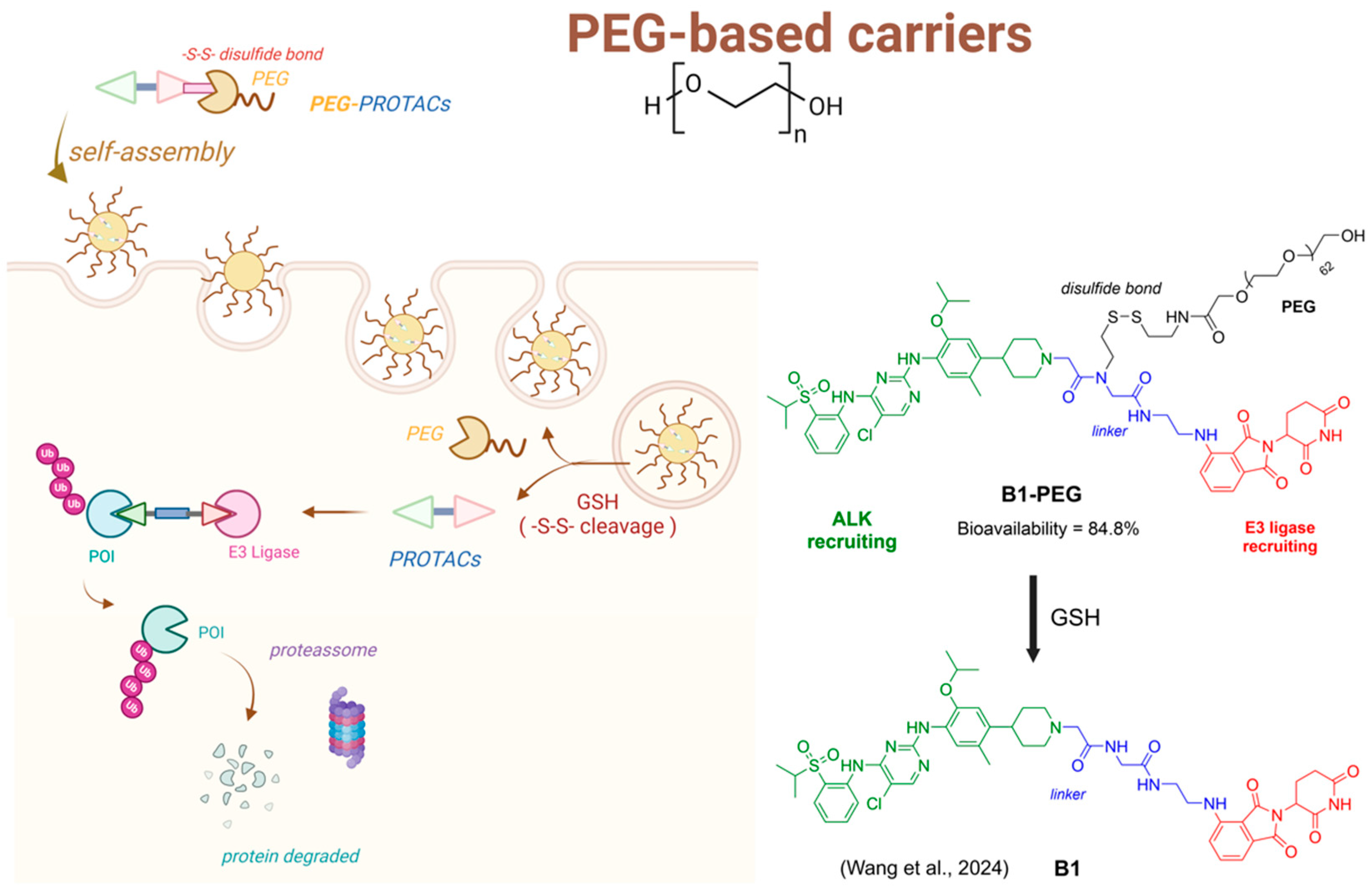
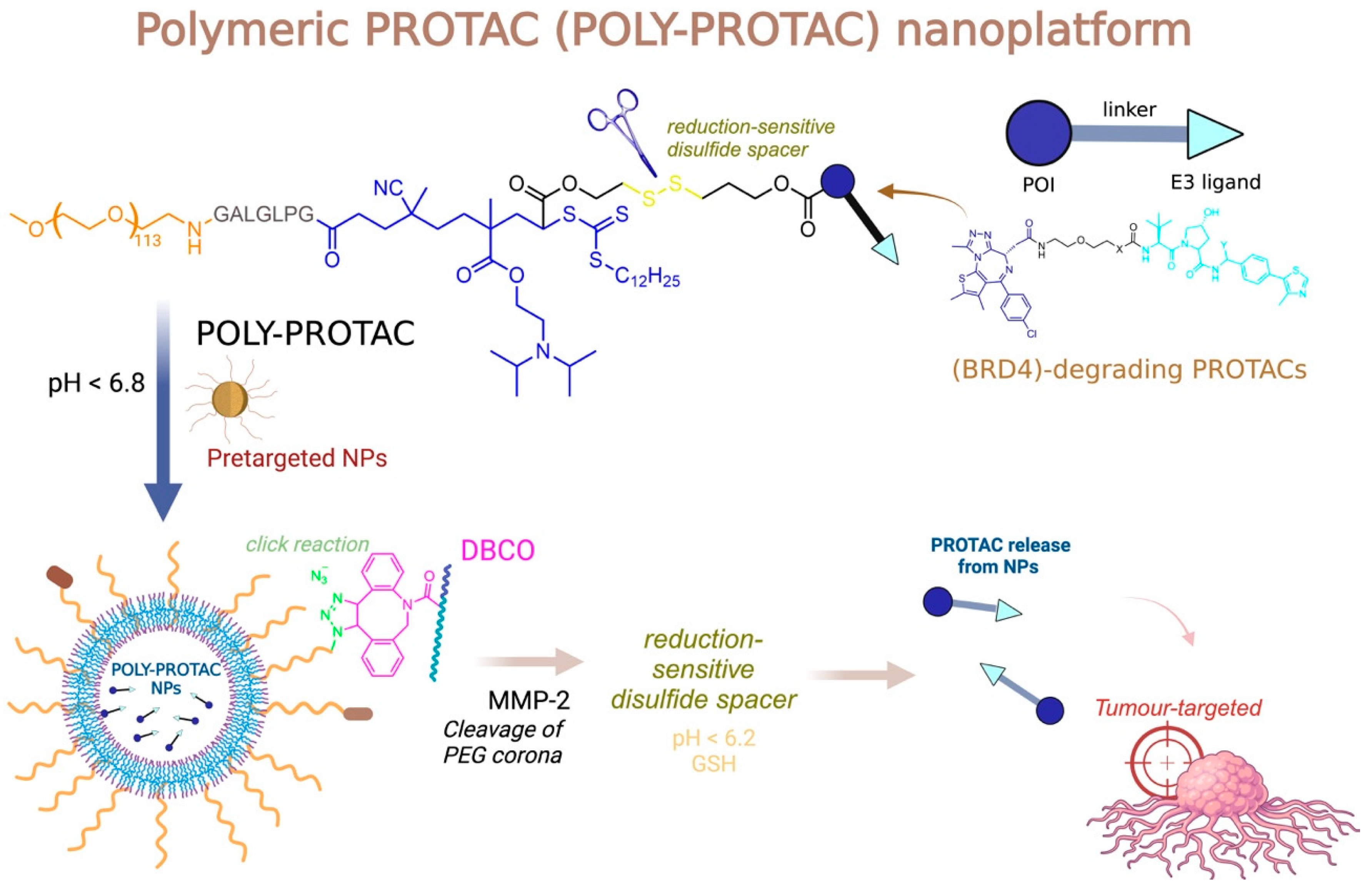
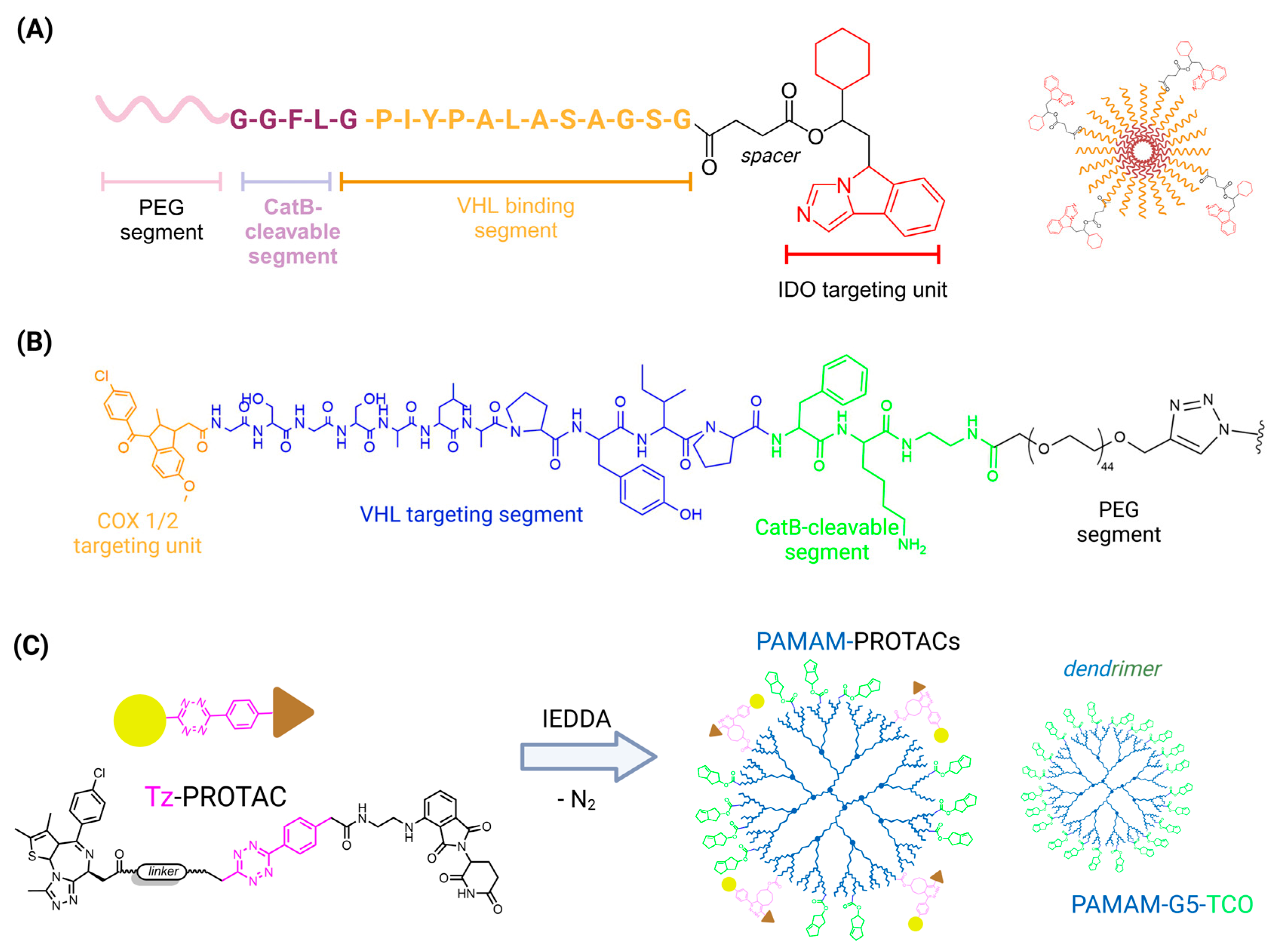
4.2. PROTACs and Polymers
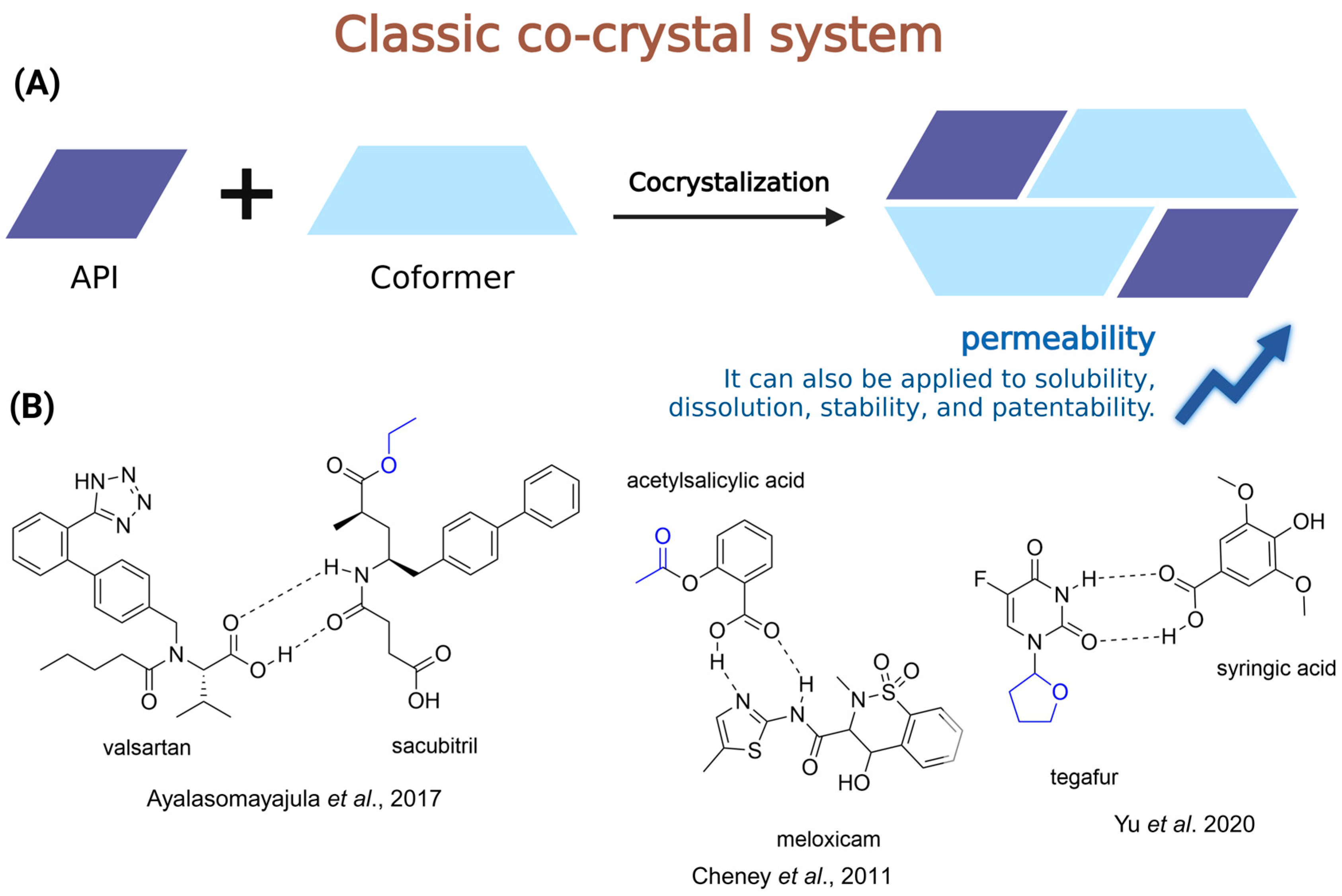
4.3. Co-Crystal and Prodrugs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5′-DFUR | 5′-deoxy-5-fluorouridine |
| 5-FU | 5-fluorouracil |
| AADC | L-amino acid decarboxylase |
| AAMD | All-atom MD |
| ABC | ATP-Binding Cassette |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| ACV | Acyclovir |
| ADEPT | Antibody-directed enzyme prodrug therapy |
| ADME | Absorption, distribution, metabolism, and excretion |
| ALK | Anaplastic lymphoma kinase |
| ANNs | Artificial neural networks |
| API | Adenosine triphosphate |
| AT1 | Active pharmaceutical ingredients |
| ATP | Angiotensin II type-1 |
| AUC | Area under the curve |
| BBB | Blood–brain barrier |
| BCS | Biopharmaceutical Classification System |
| BNP | Brain natriuretic peptide |
| BRD4 | Bromodomain-containing protein 4 |
| CDSs | Chemical delivery systems |
| CES1 | Carboxylesterase 1 |
| CGMD | Coarse-grained MD |
| CME | Cystoid macular edema |
| CNS | Central nervous system |
| COX-1 | Cyclooxygenase-1 |
| COX-2 | Cyclooxygenase-2 |
| CPPs | Cell-penetrating peptides |
| CRBN | Cereblon |
| DAR | Drug-to-antibody ratio |
| DBCO | Dibenzocyclooctyne |
| DC50 | Degradation Concentration 50% |
| deuHTBZ | Deuterated dihydrotetrabenazine isomers |
| DL | Deep learning |
| DMF | Dimethyl fumarate |
| dThdPase | Cytidine deaminase and thymidine phosphorylase |
| E. coli | Escherichia coli |
| EC50 | Effective concentration |
| EML4-ALK | Echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase |
| ESBLs | Extended-spectrum β-lactamases |
| FDA | Food and Drug Administration |
| FOLR1 | Folate receptor-1 |
| FRs | Folate receptors |
| GI50 | Growth Inhibition 50% |
| Gly | Glycine |
| GSH | Glutathione |
| HCAR2 | Hydroxycarboxylic acid receptor 2 |
| HCV | Chronic hepatitis C virus |
| His | Histidine |
| HIV | Human immunodeficiency virus |
| hPEPT-1 | Human peptide transporter 1 |
| IC50 | Inhibitory concentration |
| IMPDH | Inosine monophosphate dehydrogenase |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| LAT1 | l-type amino acid transporter |
| LC-MS | Liquid Chromatography–Mass Spectrometry |
| LDV | Ledipasvir |
| Leu | Leucine |
| LogD | Logarithm of the distribution coefficient |
| LogP | Logarithm of the octanol/water partition coefficient |
| MBLs | Metallo-β-lactamases |
| MD | Molecular dynamics |
| MDCK | Madin–Darby Canine Kidney |
| ML | Machine learning |
| MOFs | Metal–organic frameworks |
| NDDSs | Nanosized Drug Delivery Systems |
| NF-κB | Factor nuclear kappa B |
| Nrf2 | Erythroid-derived factor 2 |
| NSAID | Non-steroidal anti-inflammatory drugs |
| NSCLC | Non-small cell lung cancer |
| OATP | Organic Anion Transporting Polypeptide |
| PAMPA | Parallel Artificial Membrane Permeability Assay |
| Papp | Apparent permeability coefficient |
| PBPs | Penicillin-binding proteins |
| PCFT | Proton-coupled folate transporter |
| PD-1 | Cell death protein 1 |
| Pe | Permeability coefficient |
| Peff | Effective permeation |
| PEG | Polyethylene glycol |
| PGE2 | Prostaglandin E2 |
| POI | Protein of interest |
| POM | Pivaloyloxymethyl |
| PROTACs | PROteolysis TArgeting Chimeras |
| PS-1 | Presenilin-1 |
| RAFT | Reversible Addition Fragmentation chain Transfer |
| RFC | Reduced folate carrier |
| RIPK2 | Serine- and threonine-protein kinase 2 |
| Ro5 | Rule of five |
| SLC | Solute-Linked Carrier |
| SLCO | Solute Carrier Organic Anion |
| SOF | Sofosbuvir |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAF | Tenofovir alafenamide |
| TEER | Transepithelial electrical resistance |
| TFV | Tenofovir |
| TFV-DP | Tenofovir diphosphate |
| Tyr | Tyrosine |
| Val | Valine |
| VHL | von Hippel–Lindau protein |
| VMAT2 | Vesicular monoamine transporter 2 |
| WHO | World Health Organization |
References
- Sertkaya, A.; Beleche, T.; Jessup, A.; Sommers, B.D. Costs of Drug Development and Research and Development Intensity in the US, 2000–2018. JAMA Netw. Open 2024, 7, e2415445. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in Clinical Success Rates and Therapeutic Focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- Alavijeh, M.S.; Palmer, A.M. The Pivotal Role of Drug Metabolism and Pharmacokinetics in the Discovery and Development of New Medicines. IDrugs 2004, 7, 755–763. [Google Scholar] [PubMed]
- Di, L.; Fish, P.V.; Mano, T. Bridging Solubility between Drug Discovery and Development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical Particle Technologies: An Approach to Improve Drug Solubility, Dissolution and Bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Liu, H.; Guo, S.; Wei, S.; Liu, J.; Tian, B. Pharmacokinetics and Pharmacodynamics of Cyclodextrin-Based Oral Drug Delivery Formulations for Disease Therapy. Carbohydr. Polym. 2024, 329, 121763. [Google Scholar] [CrossRef]
- Kesharwani, R.; Jaiswal, P.; Patel, D.K.; Yadav, P.K. Lipid-Based Drug Delivery System (LBDDS): An Emerging Paradigm to Enhance Oral Bioavailability of Poorly Soluble Drugs. Biomed. Mater. Devices 2023, 1, 648–663. [Google Scholar] [CrossRef]
- Schittny, A.; Huwyler, J.; Puchkov, M. Mechanisms of Increased Bioavailability through Amorphous Solid Dispersions: A Review. Drug Deliv. 2020, 27, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.P.; Dugar, R.P. Application of Surfactants in Solid Dispersion Technology for Improving Solubility of Poorly Water Soluble Drugs. J. Drug Deliv. Sci. Technol. 2017, 41, 68–77. [Google Scholar] [CrossRef]
- Zarei, A.; Haghbakhsh, R.; Raeissi, S. Overview and Thermodynamic Modelling of Deep Eutectic Solvents as Co-Solvents to Enhance Drug Solubilities in Water. Eur. J. Pharm. Biopharm. 2023, 193, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Salt Formation to Improve Drug Solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.P.; Holm, R.; Diego, H.L. de Use of Pharmaceutical Salts and Cocrystals to Address the Issue of Poor Solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Sanches, B.M.A.; Ferreira, E.I. Is Prodrug Design an Approach to Increase Water Solubility? Int. J. Pharm. 2019, 568, 118498. [Google Scholar] [CrossRef] [PubMed]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.F.; dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The Expanding Role of Prodrugs in Contemporary Drug Design and Development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Subbaiah, M.A.M.; Rautio, J.; Meanwell, N.A. Prodrugs as Empowering Tools in Drug Discovery and Development: Recent Strategic Applications of Drug Delivery Solutions to Mitigate Challenges Associated with Lead Compounds and Drug Candidates. Chem. Soc. Rev. 2024, 53, 2099–2210. [Google Scholar] [CrossRef]
- Fralish, Z.; Chen, A.; Khan, S.; Zhou, P.; Reker, D. The Landscape of Small-Molecule Prodrugs. Nat. Rev. Drug Discov. 2024, 23, 365–380. [Google Scholar] [CrossRef]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of Passive and Carrier-Mediated Processes in Drug Transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Dobson, P.D.; Kell, D.B. Carrier-Mediated Cellular Uptake of Pharmaceutical Drugs: An Exception or the Rule? Nat. Rev. Drug Discov. 2008, 7, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Agrahari, V.; Khurana, V.; Pal, D.; Mitra, A.K. Transporter Effects on Cell Permeability in Drug Delivery. Expert Opin. Drug Deliv. 2017, 14, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a Drug’s Membrane Permeability: A Computational Model Validated With in Vitro Permeability Assay Data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef]
- Samineni, R.; Chimakurthy, J.; Konidala, S. Emerging Role of Biopharmaceutical Classification and Biopharmaceutical Drug Disposition System in Dosage Form Development: A Systematic Review. Turk. J. Pharm. Sci. 2022, 19, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.P.; Amidon, G.L. G.L. Amidon, H. Lennernas, V.P. Shah, and J.R. Crison. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability, Pharm Res 12, 413–420, 1995—Backstory of BCS. AAPS J. 2014, 16, 894–898. [Google Scholar] [CrossRef]
- Butnarasu, C.; Garbero, O.V.; Petrini, P.; Visai, L.; Visentin, S. Permeability Assessment of a High-Throughput Mucosal Platform. Pharmaceutics 2023, 15, 380. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of Drug Permeability and Prediction of Drug Absorption in Caco-2 Monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H. Regional Intestinal Drug Permeation: Biopharmaceutics and Drug Development. Eur. J. Pharm. Sci. 2014, 57, 333–341. [Google Scholar] [CrossRef]
- Dahlgren, D.; Roos, C.; Sjögren, E.; Lennernäs, H. Direct In Vivo Human Intestinal Permeability (Peff) Determined with Different Clinical Perfusion and Intubation Methods. J. Pharm. Sci. 2015, 104, 2702–2726. [Google Scholar] [CrossRef]
- Bartos, C.; Szabó-Révész, P.; Horváth, T.; Varga, P.; Ambrus, R. Comparison of Modern In Vitro Permeability Methods with the Aim of Investigation Nasal Dosage Forms. Pharmaceutics 2021, 13, 846. [Google Scholar] [CrossRef]
- Teksin, Z.S.; Seo, P.R.; Polli, J.E. Comparison of Drug Permeabilities and BCS Classification: Three Lipid-Component PAMPA System Method versus Caco-2 Monolayers. AAPS J. 2010, 12, 238–241. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Prodrugs for Improved Drug Delivery: Lessons Learned from Recently Developed and Marketed Products. Pharmaceutics 2020, 12, 1031. [Google Scholar] [CrossRef]
- Lomize, A.L.; Hage, J.M.; Schnitzer, K.; Golobokov, K.; LaFaive, M.B.; Forsyth, A.C.; Pogozheva, I.D. PerMM: A Web Tool and Database for Analysis of Passive Membrane Permeability and Translocation Pathways of Bioactive Molecules. J. Chem. Inf. Model. 2019, 59, 3094–3099. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Gu, Y.; Yu, Z.; Wang, Y.; Chen, L.; Lou, C.; Yang, C.; Li, W.; Liu, G.; Tang, Y. admetSAR3.0: A Comprehensive Platform for Exploration, Prediction and Optimization of Chemical ADMET Properties. Nucleic Acids Res. 2024, 52, W432–W438. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. ADMETopt: A Web Server for ADMET Optimization in Drug Design via Scaffold Hopping. J. Chem. Inf. Model. 2018, 58, 2051–2056. [Google Scholar] [CrossRef]
- Yi, J.; Shi, S.; Fu, L.; Yang, Z.; Nie, P.; Lu, A.; Wu, C.; Deng, Y.; Hsieh, C.; Zeng, X.; et al. OptADMET: A Web-Based Tool for Substructure Modifications to Improve ADMET Properties of Lead Compounds. Nat. Protoc. 2024, 19, 1105–1121. [Google Scholar] [CrossRef]
- Wei, Y.; Li, S.; Li, Z.; Wan, Z.; Lin, J. Interpretable-ADMET: A Web Service for ADMET Prediction and Optimization Based on Deep Neural Representation. Bioinformatics 2022, 38, 2863–2871. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Comer, J.; Herndon, C.; Leung, N.; Pavlova, A.; Swift, R.V.; Tung, C.; Rowley, C.N.; Amaro, R.E.; Chipot, C.; et al. Simulation-Based Approaches for Determining Membrane Permeability of Small Compounds. J. Chem. Inf. Model. 2016, 56, 721–733. [Google Scholar] [CrossRef]
- Bernardi, A.; Bennett, W.F.D.; He, S.; Jones, D.; Kirshner, D.; Bennion, B.J.; Carpenter, T.S. Advances in Computational Approaches for Estimating Passive Permeability in Drug Discovery. Membranes 2023, 13, 851. [Google Scholar] [CrossRef]
- Chen, H.-F. In Silico Log P Prediction for a Large Data Set with Support Vector Machines, Radial Basis Neural Networks and Multiple Linear Regression. Chem. Biol. Drug Des. 2009, 74, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Mannhold, R.; Rekker, R.F. The Hydrophobic Fragmental Constant Approach Forcalculating Log P in Octanol/Water and Aliphatic Hydrocarbon/Water Systems. Perspect. Drug Discov. Des. 2000, 18, 1–18. [Google Scholar] [CrossRef]
- Wolk, O.; Agbaria, R.; Dahan, A. Provisional In-Silico Biopharmaceutics Classification (BCS) to Guide Oral Drug Product Development. Drug Des. Dev. Ther. 2014, 8, 1563–1575. [Google Scholar] [CrossRef]
- Venable, R.M.; Krämer, A.; Pastor, R.W. Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef]
- Wang, Z.-J.; Deserno, M. A Systematically Coarse-Grained Solvent-Free Model for Quantitative Phospholipid Bilayer Simulations. J. Phys. Chem. B 2010, 114, 11207–11220. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shen, Z.; Li, Y. A Machine-Learning-Assisted Study of the Permeability of Small Drug-like Molecules across Lipid Membranes. Phys. Chem. Chem. Phys. 2020, 22, 19687–19696. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine Learning in Chemoinformatics and Drug Discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, K.; Hossain, M.E.; Aktar, A.; Mohiuddin, M.; Sarkar, K.K.; Biswas, B.; Aziz, M.A.; Abid, M.A.; Fukase, K. In Silico Analysis and Experimental Evaluation of Ester Prodrugs of Ketoprofen for Oral Delivery: With a View to Reduce Toxicity. Processes 2021, 9, 2221. [Google Scholar] [CrossRef]
- Clemente, C.M.; Onnainty, R.; Usseglio, N.; Granero, G.E.; Ravetti, S. Preformulation Studies of Novel Menthol Prodrugs with Antiparasitic Activity: Chemical Stability, In Silico, and In Vitro Permeability Assays. Drugs Drug Candidates 2023, 2, 770–780. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide–Drug Conjugates as Effective Prodrug Strategies for Targeted Delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, J.; Fishelson, Z.; Wang, C.; Zhang, S. Cell-Penetrating Peptide-Based Delivery of Macromolecular Drugs: Development, Strategies, and Progress. Biomedicines 2023, 11, 1971. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Liu, Q.; Fang, Y.; Zhu, Y.; Chen, F.; Zeng, W.; Ouyang, D.; Dong, J. Predicting Peptide Permeability Across Diverse Barriers: A Systematic Investigation. Mol. Pharm. 2024, 21, 4116–4127. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Q.; Li, B.; Lu, C.; Yang, S.; Long, J.; He, B.; Chen, H.; Huang, J. BBPpredict: A Web Service for Identifying Blood-Brain Barrier Penetrating Peptides. Front. Genet. 2022, 13, 845747. [Google Scholar] [CrossRef]
- Gupta, M.; Feng, J.; Bhisetti, G. Experimental and Computational Methods to Assess Central Nervous System Penetration of Small Molecules. Molecules 2024, 29, 1264. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Chen, Y.; Cai, X.; Xu, R. Predict Drug Permeability to Blood-Brain-Barrier from Clinical Phenotypes: Drug Side Effects and Drug Indications. Bioinformatics 2017, 33, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Alexiou, A.; Bilgrami, A.L.; Kamal, M.A.; Ashraf, G.M. DeePred-BBB: A Blood Brain Barrier Permeability Prediction Model With Improved Accuracy. Front. Neurosci. 2022, 16, 858126. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Saini, V.; Gupta, D.; Chawla, P.A.; Kumar, A. BBBper: A Machine Learning-Based Online Tool for Blood-Brain Barrier (BBB) Permeability Prediction. CNS Neurol. Disord. Drug Targets 2024. [Google Scholar] [CrossRef]
- Saxena, D.; Sharma, A.; Siddiqui, M.H.; Kumar, R. Blood Brain Barrier Permeability Prediction Using Machine Learning Techniques: An Update. Curr. Pharm. Biotechnol. 2019, 20, 1163–1171. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, F.; Zhao, Q.; Chen, W. A Merged Molecular Representation Deep Learning Method for Blood-Brain Barrier Permeability Prediction. Brief. Bioinform. 2022, 23, bbac357. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.T.C.; Yang, J.-S.; Liao, K.Y.K.; Tseng, W.C.W.; Lee, C.K.; Gill, M.; Compas, C.; See, S.; Tsai, F.-J. Predicting Blood–Brain Barrier Permeability of Molecules with a Large Language Model and Machine Learning. Sci. Rep. 2024, 14, 15844. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.S.; Goodwin, J.T. Solubility and Permeability Measurement and Applications in Drug Discovery. Comb. Chem. High Throughput Screen. 2010, 13, 101–111. [Google Scholar] [CrossRef]
- Yu, H.; Wang, Q.; Sun, Y.; Shen, M.; Li, H.; Duan, Y. A New PAMPA Model Proposed on the Basis of a Synthetic Phospholipid Membrane. PLoS ONE 2015, 10, e0116502. [Google Scholar] [CrossRef]
- Bermejo, M.; Avdeef, A.; Ruiz, A.; Nalda, R.; Ruell, J.A.; Tsinman, O.; González, I.; Fernández, C.; Sánchez, G.; Garrigues, T.M.; et al. PAMPA—A Drug Absorption in Vitro Model 7. Comparing Rat in Situ, Caco-2, and PAMPA Permeability of Fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [Google Scholar] [CrossRef]
- Mtewa, A.G.; Ngwira, K.; Lampiao, F.; Weisheit, A.; Tolo, C.U.; Ogwang, P.E.; Sesaazi, D.C. Fundamental Methods in Drug Permeability, pKa, LogP and LogDx Determination. J. Drug Res. Dev. 2019, 5, 1–6. [Google Scholar] [CrossRef]
- He, S.; Zhiti, A.; Barba-Bon, A.; Hennig, A.; Nau, W.M. Real-Time Parallel Artificial Membrane Permeability Assay Based on Supramolecular Fluorescent Artificial Receptors. Front. Chem. 2020, 8, 597927. [Google Scholar] [CrossRef] [PubMed]
- Gleiter, C.H.; Jägle, C.; Gresser, U.; Mörike, K. Candesartan. Cardiovasc. Drug Rev. 2004, 22, 263–284. [Google Scholar] [CrossRef]
- He, G.; Massarella, J.; Ward, P. Clinical Pharmacokinetics of the Prodrug Oseltamivir and Its Active Metabolite Ro 64-0802. Clin. Pharmacokinet. 1999, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Fukami, T.; Ogawa, H.; Taniguchi, T.; Nomura, Y.; Nakajima, M. Systematic Approach for Screening of Prodrugs: Evaluation Using Oseltamivir Analogues as Models. J. Pharm. Sci. 2021, 110, 925–934. [Google Scholar] [CrossRef]
- Balimane, P.V.; Patel, K.; Marino, A.; Chong, S. Utility of 96 Well Caco-2 Cell System for Increased Throughput of P-Gp Screening in Drug Discovery. Eur. J. Pharm. Biopharm. 2004, 58, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.A. Drug-Permeability and Transporter Assays in Caco-2 and MDCK Cell Lines. Future Med. Chem. 2011, 3, 2063–2077. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby Canine Kidney) Cells: A Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Markowska, M.; Oberle, R.; Juzwin, S.; Hsu, C.P.; Gryszkiewicz, M.; Streeter, A.J. Optimizing Caco-2 Cell Monolayers to Increase Throughput in Drug Intestinal Absorption Analysis. J. Pharmacol. Toxicol. Methods 2001, 46, 51–55. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, R.B.; Li, Y. Caco-2 Cell Permeability Assays to Measure Drug Absorption. Expert. Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef] [PubMed]
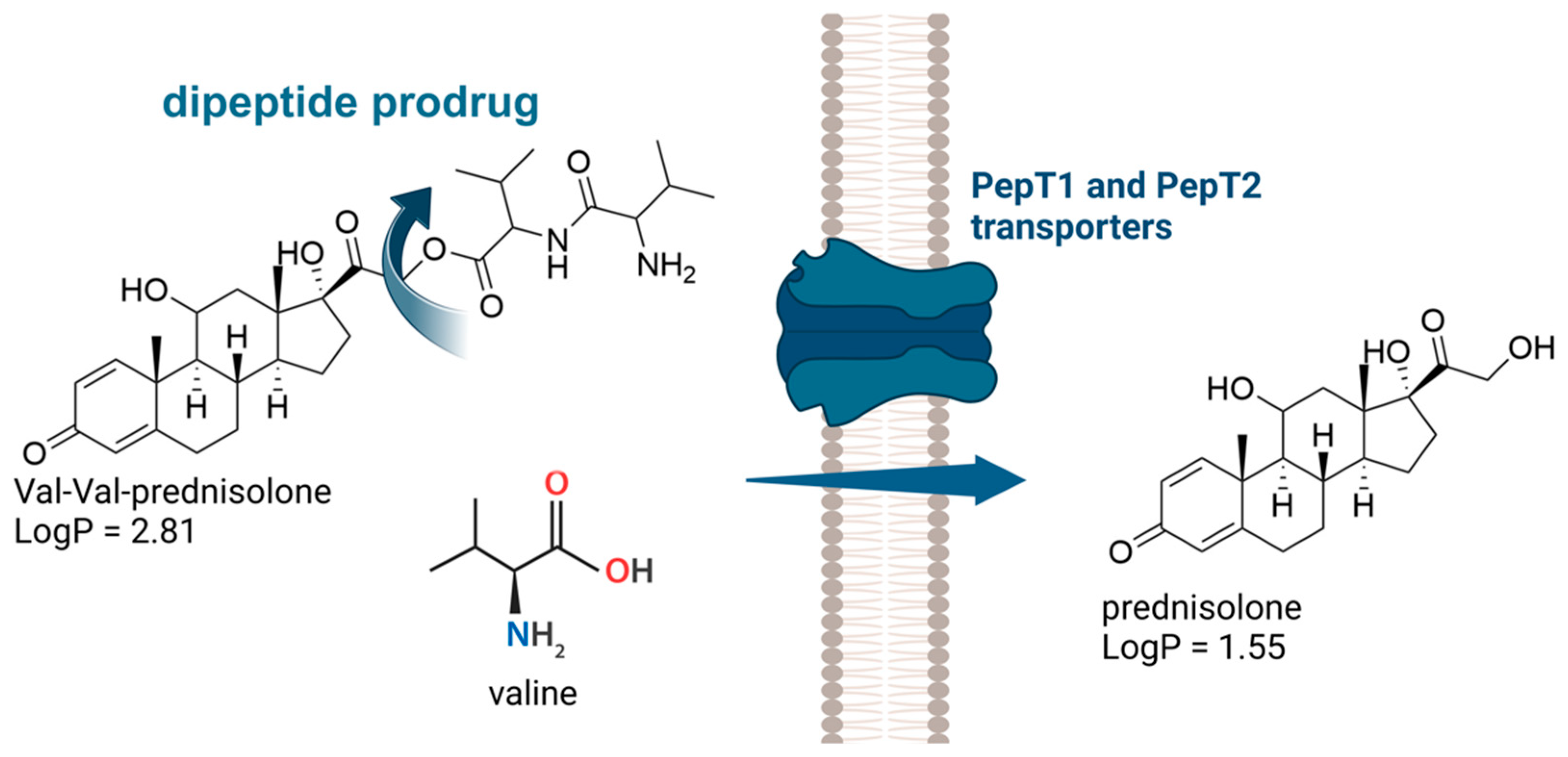
- Sheng, Y.; Yang, X.; Pal, D.; Mitra, A.K. Prodrug Approach to Improve Absorption of Prednisolone. Int. J. Pharm. 2015, 487, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.S.; Katragadda, S.; Gunda, S.; Mitra, A.K. In Vivo Ocular Pharmacokinetics of Acyclovir Dipeptide Ester Prodrugs by Microdialysis in Rabbits. Mol. Pharm. 2006, 3, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Pal, D.; Mitra, A.K. Circumvention of P-Gp and MRP2 Mediated Efflux of Lopinavir by a Histidine Based Dipeptide Prodrug. Int. J. Pharm. 2016, 512, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, J.; Gao, Y.; Jin, L.; Xu, Y.; Lian, H.; Sun, Y.; Sun, Y.; Liu, J.; Fan, R.; et al. A Carrier-Mediated Prodrug Approach To Improve the Oral Absorption of Antileukemic Drug Decitabine. Mol. Pharm. 2013, 10, 3195–3202. [Google Scholar] [CrossRef]
- Volpe, D.A. Application of Method Suitability for Drug Permeability Classification. AAPS J. 2010, 12, 670–678. [Google Scholar] [CrossRef]
- Schanker, L.S.; Tocco, D.J.; Brodie, B.B.; Hogben, C.A. Absorption of Drugs from the Rat Small Intestine. J. Pharmacol. Exp. Ther. 1958, 123, 81–88. [Google Scholar] [CrossRef]
- Stappaerts, J.; Brouwers, J.; Annaert, P.; Augustijns, P. In Situ Perfusion in Rodents to Explore Intestinal Drug Absorption: Challenges and Opportunities. Int. J. Pharm. 2015, 478, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Holenarsipur, V.K.; Gaud, N.; Sinha, J.; Sivaprasad, S.; Bhutani, P.; Subramanian, M.; Singh, S.P.; Arla, R.; Paruchury, S.; Sharma, T.; et al. Absorption and Cleavage of Enalapril, a Carboxyl Ester Prodrug, in the Rat Intestine: In Vitro, in Situ Intestinal Perfusion and Portal Vein Cannulation Models. Biopharm. Drug Dispos. 2015, 36, 385–397. [Google Scholar] [CrossRef]
- Li, Y.; Yang, M.; Zhao, Y.; Li, L.; Xu, W. Preparation and in Vitro Evaluation of Amphiphilic Paclitaxel Small Molecule Prodrugs and Enhancement of Oral Absorption. Eur. J. Med. Chem. 2021, 215, 113276. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Mannila, A.; Nevalainen, T.; Savolainen, J.; Järvinen, T.; Rautio, J. Large Neutral Amino Acid Transporter Enables Brain Drug Delivery via Prodrugs. J. Med. Chem. 2008, 51, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Pickering, D.S.; Spicer, J.A.; Denny, W.A.; Huttunen, K.M. Systemic and Brain Pharmacokinetics of Perforin Inhibitor Prodrugs. Mol. Pharm. 2016, 13, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, J.; Montaser, A.B.; Adla, S.K.; Leppänen, J.; Lehtonen, M.; Vellonen, K.S.; Laitinen, T.; Jalkanen, A.; Elmquist, W.F.; Timonen, J.; et al. Altering Distribution Profile of Palbociclib by Its Prodrugs. Eur. J. Pharm. Sci. 2024, 192, 106637. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M. Everted Gut Sac Model as a Tool in Pharmaceutical Research: Limitations and Applications. J. Pharm. Pharmacol. 2012, 64, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Barthe, L.; Woodley, J.F.; Kenworthy, S.; Houin, G. An Improved Everted Gut Sac as a Simple and Accurate Technique to Measure Paracellular Transport across the Small Intestine. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Gualdesi, M.S.; Briñón, M.C.; Quevedo, M.A. Intestinal Permeability of Lamivudine (3TC) and Two Novel 3TC Prodrugs. Experimental and Theoretical Analyses. Eur. J. Pharm. Sci. 2012, 47, 965–978. [Google Scholar] [CrossRef]
- Sodano, F.; Cristiano, C.; Rolando, B.; Marini, E.; Lazzarato, L.; Cuozzo, M.; Albrizio, S.; Russo, R.; Rimoli, M.G. Galactosylated Prodrugs: A Strategy to Improve the Profile of Nonsteroidal Anti-Inflammatory Drugs. Pharmaceuticals 2022, 15, 552. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.-P.; Chen, S.; Yu, B.-Y.; Zhu, D.-N.; Cordell, G.A.; Qiu, S.X. Prediction of Human Absorption of Natural Compounds by the Non-Everted Rat Intestinal Sac Model. Eur. J. Med. Chem. 2006, 41, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Antunes Viegas, D.; Rodrigues, M.; Francisco, J.; Falcão, A.; Alves, G.; Santos, A.O. Development and Application of an Ex Vivo Fosphenytoin Nasal Bioconversion/Permeability Evaluation Method. Eur. J. Pharm. Sci. 2016, 89, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Sitovs, A.; Mohylyuk, V. Ex Vivo Permeability Study of Poorly Soluble Drugs across Gastrointestinal Membranes: Acceptor Compartment Media Composition. Drug Discov. Today 2024, 29, 104214. [Google Scholar] [CrossRef] [PubMed]
- Dezani, A.B.; Pereira, T.M.; Caffaro, A.M.; Reis, J.M.; Serra, C.H.D.R. Determination of Lamivudine and Zidovudine Permeability Using a Different Ex Vivo Method in Franz Cells. J. Pharmacol. Toxicol. Methods 2013, 67, 194–202. [Google Scholar] [CrossRef] [PubMed]
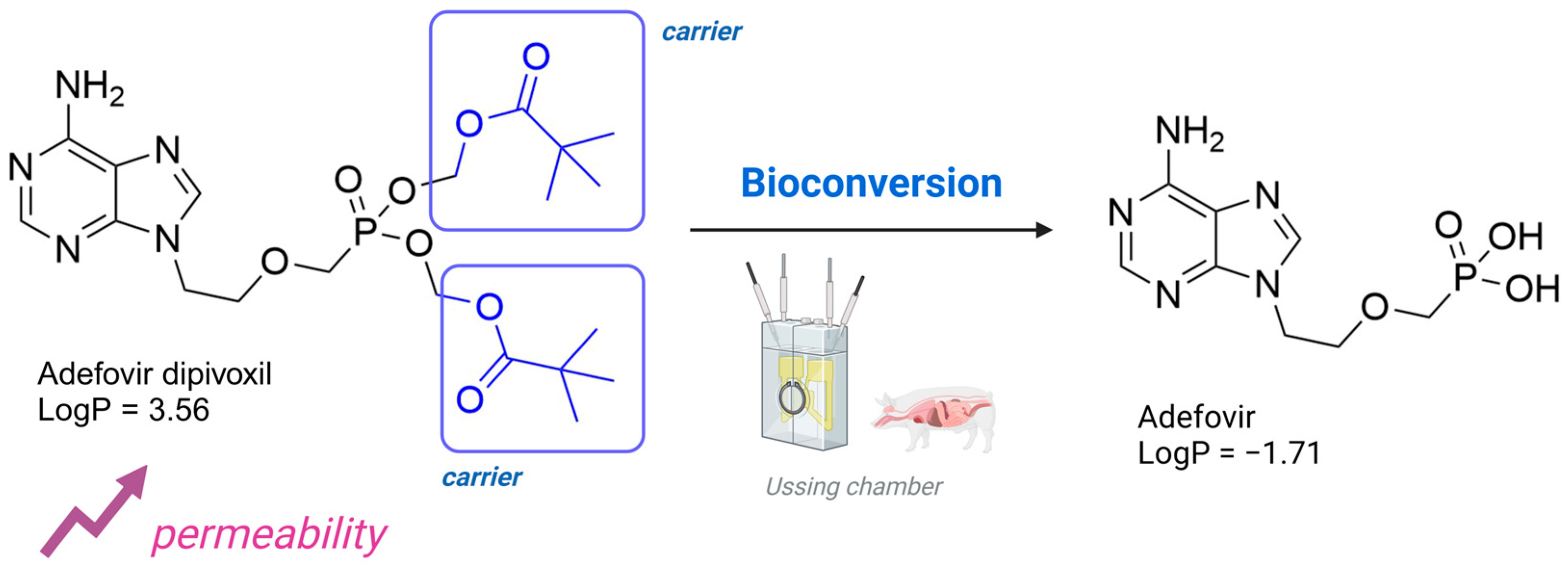
- Annaert, P.; Tukker, J.J.; van Gelder, J.; Naesens, L.; de Clercq, E.; van Den Mooter, G.; Kinget, R.; Augustijns, P. In Vitro, Ex Vivo, and in Situ Intestinal Absorption Characteristics of the Antiviral Ester Prodrug Adefovir Dipivoxil. J. Pharm. Sci. 2000, 89, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef]
- Schweckendiek, W. Treatment of psoriasis vulgaris. Med. Monatsschr. 1959, 13, 103–104. [Google Scholar] [PubMed]
- Matteo, P.; Federico, D.; Emanuela, M.; Giulia, R.; Tommaso, B.; Alfredo, G.; Anna, C.; Annamaria, O. New and Old Horizons for an Ancient Drug: Pharmacokinetics, Pharmacodynamics, and Clinical Perspectives of Dimethyl Fumarate. Pharmaceutics 2022, 14, 2732. [Google Scholar] [CrossRef]
- Landeck, L.; Asadullah, K.; Amasuno, A.; Pau-Charles, I.; Mrowietz, U. Dimethyl Fumarate (DMF) vs. Monoethyl Fumarate (MEF) Salts for the Treatment of Plaque Psoriasis: A Review of Clinical Data. Arch. Dermatol. Res. 2018, 310, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Mrowietz, U.; Christophers, E.; Altmeyer, P.; The German Fumaric Acid Ester Consensus Conference. Treatment of Severe Psoriasis with Fumaric Acid Esters: Scientific Background and Guidelines for Therapeutic Use. Br. J. Dermatol. 1999, 141, 424–429. [Google Scholar] [CrossRef]
- Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza; Bautista, E.; Chotpitayasunondh, T.; Gao, Z.; Harper, S.A.; Shaw, M.; Uyeki, T.M.; Zaki, S.R.; Hayden, F.G.; Hui, D.S.; et al. Clinical Aspects of Pandemic 2009 Influenza A (H1N1) Virus Infection. N. Engl. J. Med. 2010, 362, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.E. Pharmacokinetics of Oseltamivir: An Oral Antiviral for the Treatment and Prophylaxis of Influenza in Diverse Populations. J. Antimicrob. Chemother. 2010, 65 (Suppl. S2), ii5–ii10. [Google Scholar] [CrossRef]
- Li, W.; Escarpe, P.A.; Eisenberg, E.J.; Cundy, K.C.; Sweet, C.; Jakeman, K.J.; Merson, J.; Lew, W.; Williams, M.; Zhang, L.; et al. Identification of GS 4104 as an Orally Bioavailable Prodrug of the Influenza Virus Neuraminidase Inhibitor GS 4071. Antimicrob. Agents Chemother. 1998, 42, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Lew, W.; Chen, X.; Kim, C.U. Discovery and Development of GS 4104 (Oseltamivir): An Orally Active Influenza Neuraminidase Inhibitor. Curr. Med. Chem. 2000, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Yang, J.; Yang, D.; LeCluyse, E.L.; Black, C.; You, L.; Akhlaghi, F.; Yan, B. Anti-Influenza Prodrug Oseltamivir Is Activated by Carboxylesterase Human Carboxylesterase 1, and the Activation Is Inhibited by Antiplatelet Agent Clopidogrel. J. Pharmacol. Exp. Ther. 2006, 319, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.C.; Nakada, N.; Yamashita, N.; Tanaka, H. Oseltamivir Carboxylate, the Active Metabolite of Oseltamivir Phosphate (Tamiflu), Detected in Sewage Discharge and River Water in Japan. Environ. Health Perspect. 2010, 118, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Eugui, E.M. Mycophenolate Mofetil and Its Mechanisms of Action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Staatz, C.E.; Tett, S.E. Clinical Pharmacokinetics and Pharmacodynamics of Mycophenolate in Solid Organ Transplant Recipients. Clin. Pharmacokinet. 2007, 46, 13–58. [Google Scholar] [CrossRef]
- Bullingham, R.E.; Nicholls, A.J.; Kamm, B.R. Clinical Pharmacokinetics of Mycophenolate Mofetil. Clin. Pharmacokinet. 1998, 34, 429–455. [Google Scholar] [CrossRef] [PubMed]
- PubChem Mycophenolate Mofetil. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5281078 (accessed on 26 December 2024).
- Fujiyama, N.; Miura, M.; Kato, S.; Sone, T.; Isobe, M.; Satoh, S. Involvement of Carboxylesterase 1 and 2 in the Hydrolysis of Mycophenolate Mofetil. Drug Metab. Dispos. 2010, 38, 2210–2217. [Google Scholar] [CrossRef]
- Taskar, P.; Tatke, A.; Majumdar, S. Advances in the Use of Prodrugs for Drug Delivery to the Eye. Expert Opin. Drug Deliv. 2017, 14, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Antonijevic, N.M.; Zivkovic, I.D.; Jovanovic, L.M.; Matic, D.M.; Kocica, M.J.; Mrdovic, I.B.; Kanjuh, V.I.; Culafic, M.D. Dabigatran—Metabolism, Pharmacologic Properties and Drug Interactions. Curr. Drug Metab. 2017, 18, 622–635. [Google Scholar] [CrossRef]
- Fawzy, A.M.; Lip, G.Y.H. Pharmacokinetics and Pharmacodynamics of Oral Anticoagulants Used in Atrial Fibrillation. Expert Opin. Drug Metab. Toxicol. 2019, 15, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Paik, J. Dabigatran Etexilate: A Review in Pediatric Venous Thromboembolism. Pediatr. Drugs 2022, 24, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Brandstetter, H.; Turk, D.; Hoeffken, H.W.; Grosse, D.; Stürzebecher, J.; Martin, P.D.; Edwards, B.F.P.; Bode, W. Refined 2·3ÅX-Ray Crystal Structure of Bovine Thrombin Complexes Formed with the Benzamidine and Arginine-Based Thrombin Inhibitors NAPAP, 4-TAPAP and MQPA. J. Mol. Biol. 1992, 226, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Van Ryn, J.; Goss, A.; Hauel, N.; Wienen, W.; Priepke, H.; Nar, H.; Clemens, A. The Discovery of Dabigatran Etexilate. Front. Pharmacol. 2013, 4, 40985. [Google Scholar] [CrossRef] [PubMed]
- Schellens, J.H.M. Capecitabine. Oncologist 2007, 12, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.-S.; Huang, H.; Cai, L.; Zhao, L.; Peng, R.-J.; Lin, Y.; Tang, J.; Zeng, J.; Zhang, L.-H.; et al. Effect of Capecitabine Maintenance Therapy Using Lower Dosage and Higher Frequency vs Observation on Disease-Free Survival Among Patients With Early-Stage Triple-Negative Breast Cancer Who Had Received Standard Treatment: The SYSUCC-001 Randomized Clinical Trial. JAMA 2021, 325, 50. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.S.; Godara, A.; Byrne, M.M.; Saif, M.W. Capecitabine for the Treatment of Pancreatic Cancer. Expert Opin. Pharmacother. 2019, 20, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.; Al Doghaither, H.; Al-ghafari, A.; Pushparaj, P. 5-Fluorouracil and Capecitabine Therapies for the Treatment of Colorectal Cancer (Review). Oncol. Rep. 2023, 50, 175. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine Compared with Observation in Resected Biliary Tract Cancer (BILCAP): A Randomised, Controlled, Multicentre, Phase 3 Study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Reigner, B.; Blesch, K.; Weidekamm, E. Clinical Pharmacokinetics of Capecitabine. Clin. Pharmacokinet. 2001, 40, 85–104. [Google Scholar] [CrossRef] [PubMed]
- Zabiszak, M.; Frymark, J.; Ogawa, K.; Skrobańska, M.; Nowak, M.; Jastrzab, R.; Kaczmarek, M.T. Complexes of β-Lactam Antibiotics and Their Schiff-Base Derivatives as a Weapon in the Fight against Bacterial Resistance. Coord. Chem. Rev. 2023, 493, 215326. [Google Scholar] [CrossRef]
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules 2020, 25, 1543. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Smyth, T.P.; O’Donnell, M.E.; O’Connor, M.J.; St Ledger, J.O. β-Lactamase-Dependent Prodrugs—Recent Developments. Tetrahedron 2000, 56, 5699–5707. [Google Scholar] [CrossRef]
- Hakimelahi, G.H.; Shia, K.-S.; Xue, C.; Hakimelahi, S.; Moosavi-Movahedi, A.A.; Saboury, A.A.; Khalafi-Nezhad, A.; Soltani-Rad, M.N.; Osyetrov, V.; Wang, K.-P.; et al. Design, Synthesis, and Biological Evaluation of a Series of β-Lactam-Based Prodrugs. Bioorganic Med. Chem. 2002, 10, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.V.; Wuest, W.M. Signed, Sealed, Delivered: Conjugate and Prodrug Strategies as Targeted Delivery Vectors for Antibiotics. ACS Infect. Dis. 2019, 5, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Suárez, C.; Gudiol, F. Antibióticos Betalactámicos. Enfermedades Infecc. Y Microbiol. Clín. 2009, 27, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Gambo, S.B.; Mukhtar, A.A.; Labaran, H.B.; Labaran, H.B.; Mustapha, A.; Ibrahim, S.I.; Ali, M. Chemistry, Mode of Action, Bacterial Resistance, Classification and Adverse Effects of Beta-Lactam Antibiotics: A Review. Int. J. Dermatol. Res. 2023, 5, 11–16. [Google Scholar] [CrossRef]
- Varela, M.F.; Stephen, J.; Lekshmi, M.; Ojha, M.; Wenzel, N.; Sanford, L.M.; Hernandez, A.J.; Parvathi, A.; Kumar, S.H. Bacterial Resistance to Antimicrobial Agents. Antibiotics 2021, 10, 593. [Google Scholar] [CrossRef]
- Ruddle, C.C.; Smyth, T.P. Exploring the Chemistry of Penicillin as a β-Lactamase-Dependent Prodrug. Org. Biomol. Chem. 2007, 5, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.E.; Livermore, D.M.; Hooper, D.C.; Hope, W.W. Metallo-β-Lactamases: Structure, Function, Epidemiology, Treatment Options, and the Development Pipeline. Antimicrob. Agents Chemother. 2020, 64, e00397-20. [Google Scholar] [CrossRef]
- Bryskier, A. β-Lactam Prodrugs. In Antimicrobial Agents; Wiley: Hoboken, NJ, USA, 2005; pp. 348–376. ISBN 978-1-68367-191-6. [Google Scholar]
- Larsen, E.M.; Johnson, R.J. Microbial Esterases and Ester Prodrugs: An Unlikely Marriage for Combating Antibiotic Resistance. Drug Dev. Res. 2019, 80, 33–47. [Google Scholar] [CrossRef]
- Cuenca-Lopez, F.; Rivero, A.; Rivero-Juárez, A. Pharmacokinetics and Pharmacodynamics of Sofosbuvir and Ledipasvir for the Treatment of Hepatitis C. Expert. Opin. Drug Metab. Toxicol. 2017, 13, 105–112. [Google Scholar] [CrossRef]
- Ray, A.S.; Fordyce, M.W.; Hitchcock, M.J.M. Tenofovir Alafenamide: A Novel Prodrug of Tenofovir for the Treatment of Human Immunodeficiency Virus. Antivir. Res. 2016, 125, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Ghosh, M. Investigation of the Structure and Dynamics of Antiviral Drug Adefovir Dipivoxil by Site-Specific Spin–Lattice Relaxation Time Measurements and Chemical Shift Anisotropy Tensor Measurements. ACS Omega 2020, 5, 29373–29381. [Google Scholar] [CrossRef]
- Lin, R.-Z.; Sun, P.-J.; Tao, Q.; Yao, J.; Chen, J.-M.; Lu, T.-B. Mechanism Study on Stability Enhancement of Adefovir Dipivoxil by Cocrystallization: Degradation Kinetics and Structure-Stability Correlation. Eur. J. Pharm. Sci. 2016, 85, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Nakajima, T.; Shimazaki, A.; Kageyama, M.; Matsugi, T.; Matsumura, Y.; Gabelt, B.T.; Kaufman, P.L.; Hara, H. Pharmacological Characteristics of AFP-168 (Tafluprost), a New Prostanoid FP Receptor Agonist, as an Ocular Hypotensive Drug. Exp. Eye Res. 2004, 78, 767–776. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Ikäheimo, K.; Mannermaa, E.; Ropo, A. Pharmacokinetics, Efficacy, and Safety of the Preservative-Free Fixed Combination of Tafluprost 0.0015% and Timolol 0.5% in Healthy Volunteers: A Phase I Comparison vs. the Corresponding Preservative-Free Monotherapies. Clin. Pharmacokinet. 2016, 55, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Pantcheva, M.B.; Seibold, L.K.; Awadallah, N.S.; Kahook, M.Y. Tafluprost: A Novel Prostaglandin Analog for Treatment of Glaucoma. Adv. Ther. 2011, 28, 707–715. [Google Scholar] [CrossRef]
- Mandell, A.I.; Stentz, F.; Kitabchi, A.E. Dipivalyl Epinephrine: A New pro-Drug in the Treatment of Glaucoma. Ophthalmology 1978, 85, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Yao, K.; Zhang, Q.; Zhao, Y.; Liang, X.; Deng, C.; Chen, X.; Xiang, S.; Xiang, Y.; Wang, J. Epinephrine Reduces Intraocular Pressure by Regulating the Trabecular Meshwork Cytoskeleton and Extracellular Matrix Via β2-AR. Heliyon 2023.
- Reinecke, R.D.; Kuwabara, T. Corneal Deposits Secondary to Topical Epinephrine. Arch. Ophthalmol. 1963, 70, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A. Systemic Absorption of Topical Ocularly Applied Epinephrine and Dipivefrin. Arch. Ophthalmol. 1980, 98, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.A.; Kim, J.Y.; Ament, C.S.; Ferrufino-Ponce, Z.K.; Grabowska, A.; Cremers, S.L. Clinical Pseudophakic Cystoid Macular Edema. Risk Factors for Development and Duration after Treatment. J. Cataract. Refract. Surg. 2007, 33, 1550–1558. [Google Scholar] [CrossRef]
- Duan, P.; Liu, Y.; Li, J. The Comparative Efficacy and Safety of Topical Non-Steroidal Anti-Inflammatory Drugs for the Treatment of Anterior Chamber Inflammation after Cataract Surgery: A Systematic Review and Network Meta-Analysis. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 639–649. [Google Scholar] [CrossRef]
- Nardi, M.; Lobo, C.; Bereczki, A.; Cano, J.; Zagato, E.; Potts, S.; Sullins, G.; Notivol, R. Analgesic and Anti-Inflammatory Effectiveness of Nepafenac 0.1% for Cataract Surgery. Clin. Ophthalmol. 2007, 1, 527–533. [Google Scholar] [PubMed]
- Jones, B.M.; Neville, M.W. Nepafenac: An Ophthalmic Nonsteroidal Antiinflammatory Drug for Pain after Cataract Surgery. Ann. Pharmacother. 2013, 47, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Talpur, R.; Cox, K.; Duvic, M. Efficacy and Safety of Topical Tazarotene: A Review. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 195–210. [Google Scholar] [CrossRef]
- Bianchi, L.; Orlandi, A.; Campione, E.; Angeloni, C.; Costanzo, A.; Spagnoli, L.G.; Chimenti, S. Topical Treatment of Basal Cell Carcinoma with Tazarotene: A Clinicopathological Study on a Large Series of Cases. Br. J. Dermatol. 2004, 151, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Bardazzi, F.; Bianchi, F.; Parente, G.; Guareschi, E.; Landi, C. A Pilot Study on the Use of Topical Tazarotene to Treat Squamous Cell Carcinoma in Situ. J. Am. Acad. Dermatol. 2005, 52, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Cunningham, B.B.; Eichenfield, L.F.; Alió, A.B.; Buka, R.L. Treatment of Ichthyosiform Diseases with Topically Applied Tazarotene: Risk of Systemic Absorption. J. Am. Acad. Dermatol. 2007, 57, S123–S125. [Google Scholar] [CrossRef]
- Chandraratna, R.A.S. Tazarotene—First of a New Generation of Receptor-selective Retinoids. Br. J. Dermatol. 1996, 135, 18–25. [Google Scholar] [CrossRef]
- Lebwohl, M.G.; Breneman, D.L.; Goffe, B.S.; Grossman, J.R.; Ling, M.R.; Milbauer, J.; Pincus, S.H.; Sibbald, R.G.; Swinyer, L.J.; Weinstein, G.D.; et al. Tazarotene 0.1% Gel plus Corticosteroid Cream in the Treatment of Plaque Psoriasis. J. Am. Acad. Dermatol. 1998, 39, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New Insights into the Anti-Inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef]
- Rodriguez, C.; Camino, A.; Taly, A.; Peña, E.; Inatti, A.; Serrano, X. Betamethasone Dipropionate Loaded in Nanoliposomes vs Conventional Betamethasone Dipropionate: Comparative Study of Permeability and Penetrability In Vitro and Ex Vivo. J. Biosci. Med. 2024, 12, 140–156. [Google Scholar] [CrossRef]
- MC2 Therapeutics A Randomised, Open-Label, Maximal Use Trial, Evaluating the Pharmacokinetic Profile of Active Ingredients and Their Metabolites After Application of MC2-01 Cream Compared With Active Comparator in Subjects With Extensive Psoriasis Vulgaris; Clinicaltrials.Gov, 2019. Available online: https://www.kompetenznetz-leukaemie.de/uct_trial/pdf/uct_de/kurzprotokoll_.pdf?oid=:1:&id=1593 (accessed on 16 February 2025).
- DrugBank Online Betamethasone Dipropionate. Available online: https://go.drugbank.com/salts/DBSALT000851 (accessed on 27 December 2024).
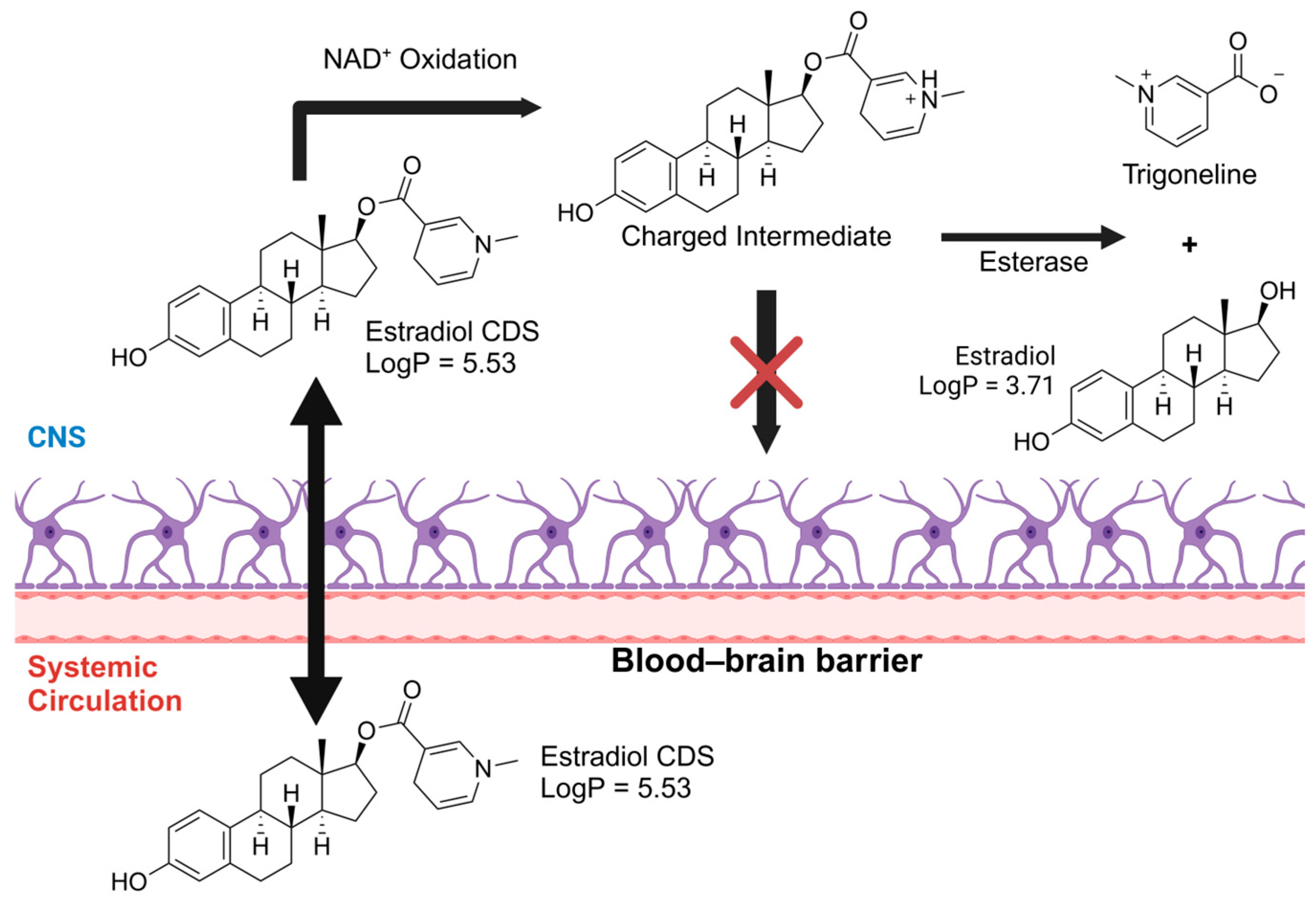
- Rautio, J.; Laine, K.; Gynther, M.; Savolainen, J. Prodrug Approaches for CNS Delivery. AAPS J. 2008, 10, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Mullersman, G.; Derendorf, H.; Brewster, M.E.; Estes, K.S.; Bodor, N. High-Performance Liquid Chromatographic Assay of a Central Nervous System (CNS)-Directed Estradiol Chemical Delivery System and Its Application after Intravenous Administration to Rats. Pharm. Res. 1988, 5, 172–177. [Google Scholar] [CrossRef]
- Synapse Delving into the Latest Updates on IDR-90104 with Synapse. Available online: https://synapse.patsnap.com/drug/53a45f918d3948b08a3264f813803fc9#overview (accessed on 27 December 2024).
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Martinelli, P. Pharmacokinetics of Levodopa. J. Neurol. 2010, 257, 253–261. [Google Scholar] [CrossRef]
- Baruzzi, A.; Contin, M.; Riva, R.; Procaccianti, G.; Albani, F.; Tonello, C.; Zoni, E.; Martinelli, P. Influence of Meal Ingestion Time on Pharmacokinetics of Orally Administered Levodopa in Parkinsonian Patients. Clin. Neuropharmacol. 1987, 10, 527–537. [Google Scholar] [CrossRef]
- Hawkins, R.A.; Mokashi, A.; Simpson, I.A. An Active Transport System in the Blood–Brain Barrier May Reduce Levodopa Availability. Exp. Neurol. 2005, 195, 267–271. [Google Scholar] [CrossRef]
- Han, S.-W.; Shin, J.-S. Aromatic L-Amino Acid Decarboxylases: Mechanistic Features and Microbial Applications. Appl. Microbiol. Biotechnol. 2022, 106, 4445–4458. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Uneven Pattern of Dopamine Loss in the Striatum of Patients with Idiopathic Parkinson’s Disease. N. Engl. J. Med. 1988, 318, 876–880. [Google Scholar] [CrossRef]
- Williams, D.G.; Hatch, D.J.; Howard, R.F. Codeine Phosphate in Paediatric Medicine. Br. J. Anaesth. 2001, 86, 413–421. [Google Scholar] [CrossRef]
- Moolenaar, F.; Grasmeijer, G.; Visser, J.; Meijer, D.K.F. Rectal versus Oral Absorption of Codeine Phosphate in Man. Biopharm. Drug Dispos. 1983, 4, 195–199. [Google Scholar] [CrossRef]
- Sanfilippo, G. Bollettino Della Società Italiana Di Biologia Sperimentale (in SafetyLit). Available online: https://www.safetylit.org/week/journalpage.php?jid=9782 (accessed on 27 December 2024).
- Kelly, E. Efficacy and Ligand Bias at the μ-Opioid Receptor. Br. J. Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Chen, L. The Physiological Role of Drug Transporters. Protein Cell 2015, 6, 334–350. [Google Scholar] [CrossRef]
- Nigam, S.K. What Do Drug Transporters Really Do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, G.; Hediger, M.A. Transporter-Mediated Drug Delivery. Molecules 2023, 28, 1151. [Google Scholar] [CrossRef]
- Szöllősi, D.; Rose-Sperling, D.; Hellmich, U.A.; Stockner, T. Comparison of Mechanistic Transport Cycle Models of ABC Exporters. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2018, 1860, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Schaller, L.; Lauschke, V.M. The Genetic Landscape of the Human Solute Carrier (SLC) Transporter Superfamily. Hum. Genet. 2019, 138, 1359–1377. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; et al. XP13512 [(±)-1-([(α-Isobutanoyloxyethoxy)Carbonyl] Aminomethyl)-1-Cyclohexane Acetic Acid], A Novel Gabapentin Prodrug: I. Design, Synthesis, Enzymatic Conversion to Gabapentin, and Transport by Intestinal Solute Transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef]
- Kageyama, T.; Nakamura, M.; Matsuo, A.; Yamasaki, Y.; Takakura, Y.; Hashida, M.; Kanai, Y.; Naito, M.; Tsuruo, T.; Minato, N.; et al. The 4F2hc/LAT1 Complex Transports l-DOPA across the Blood–Brain Barrier. Brain Res. 2000, 879, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D. Valacyclovir. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. ISBN 978-0-08-055232-3. [Google Scholar]
- MacDougall, C.; Guglielmo, B.J. Pharmacokinetics of Valaciclovir. J. Antimicrob. Chemother. 2004, 53, 899–901. [Google Scholar] [CrossRef]
- Khorassani, F.; Luther, K.; Talreja, O. Valbenazine and Deutetrabenazine: Vesicular Monoamine Transporter 2 Inhibitors for Tardive Dyskinesia. Am. J. Health-Syst. Pharm. 2020, 77, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.; Vijan, A.; Scott, F.L.; Jimenez, R.; Zhang, H.; Grigoriadis, D.E.; Loewen, G. Pharmacokinetic and Pharmacologic Characterization of the Dihydrotetrabenazine Isomers of Deutetrabenazine and Valbenazine. Clin. Pharmacol. Drug Dev. 2023, 12, 447–456. [Google Scholar] [CrossRef]
- Varma, M.V.; Ambler, C.M.; Ullah, M.; Rotter, C.J.; Sun, H.; Litchfield, J.; Fenner, K.S.; El-Kattan, A.F. Targeting Intestinal Transporters for Optimizing Oral Drug Absorption. Curr. Drug Metab. 2010, 11, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Estudante, M.; Morais, J.G.; Soveral, G.; Benet, L.Z. Intestinal Drug Transporters: An Overview. Adv. Drug Deliv. Rev. 2013, 65, 1340–1356. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, J.; Huang, Z.; Wang, M. Transporter-Mediated Tissue Targeting of Therapeutic Molecules in Drug Discovery. Bioorganic Med. Chem. Lett. 2015, 25, 993–997. [Google Scholar] [CrossRef]
- Puris, E.; Fricker, G.; Gynther, M. Targeting Transporters for Drug Delivery to the Brain: Can. We Do Better? Pharm. Res. 2022, 39, 1415–1455. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, H.; Naito, M.; Demizu, Y. Investigating the Cell Permeability of Proteolysis-Targeting Chimeras (PROTACs). Expert Opin. Drug Discov. 2023, 18, 357–361. [Google Scholar] [CrossRef] [PubMed]
- García Jiménez, D.; Rossi Sebastiano, M.; Vallaro, M.; Mileo, V.; Pizzirani, D.; Moretti, E.; Ermondi, G.; Caron, G. Designing Soluble PROTACs: Strategies and Preliminary Guidelines. J. Med. Chem. 2022, 65, 12639–12649. [Google Scholar] [CrossRef]
- Kuentz, M.; Bergström, C.A.S. Synergistic Computational Modeling Approaches as Team Players in the Game of Solubility Predictions. J. Pharm. Sci. 2021, 110, 22–34. [Google Scholar] [CrossRef]
- Chen, C.; Yang, Y.; Wang, Z.; Li, H.; Dong, C.; Zhang, X. Recent Advances in Pro-PROTAC Development to Address On-Target Off-Tumor Toxicity. J. Med. Chem. 2023, 66, 8428–8440. [Google Scholar] [CrossRef]
- He, X.; Weng, Z.; Zou, Y. Progress in the Controllability Technology of PROTAC. Eur. J. Med. Chem. 2024, 265, 116096. [Google Scholar] [CrossRef]
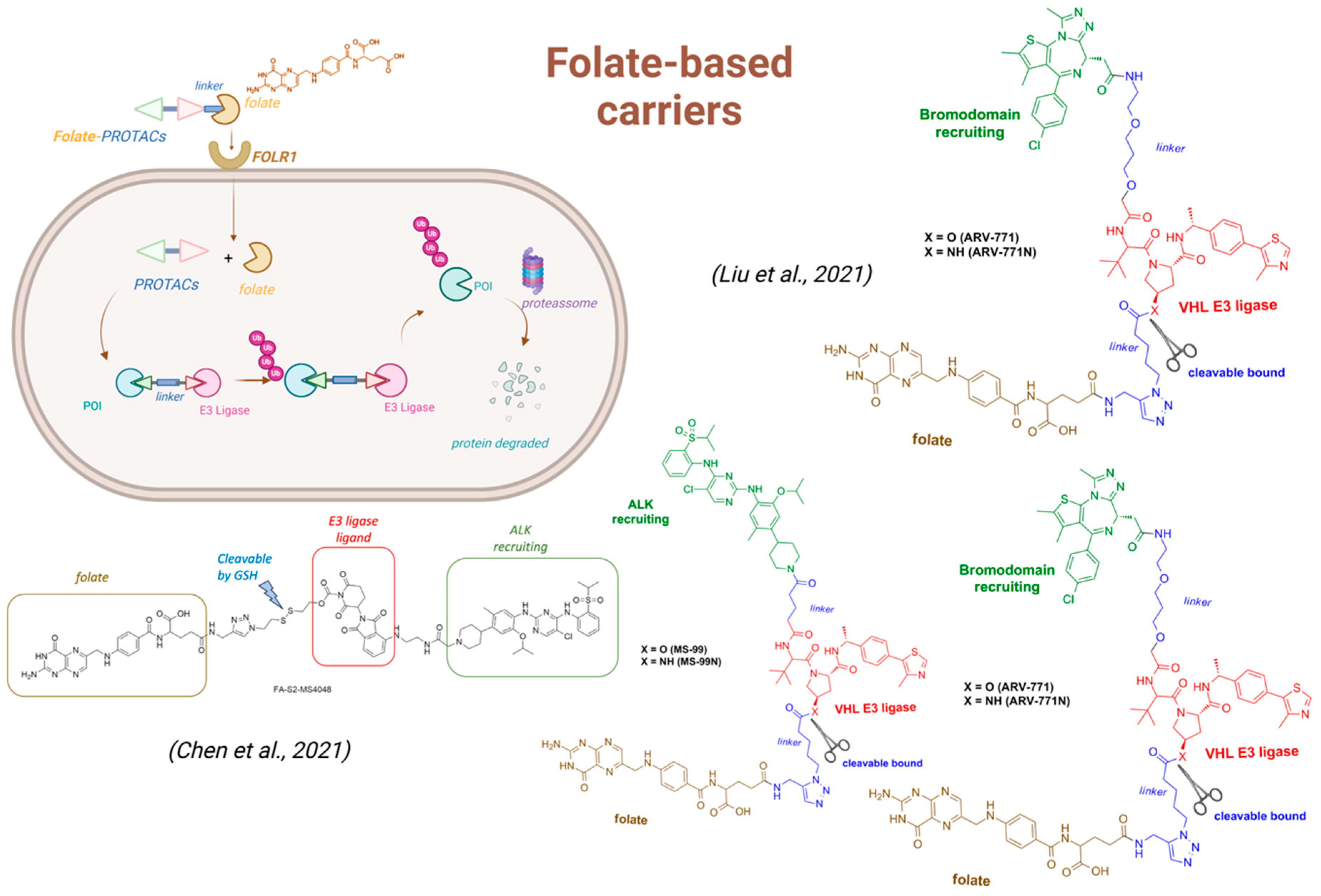
- Chen, H.; Liu, J.; Kaniskan, H.Ü.; Wei, W.; Jin, J. Folate-Guided Protein Degradation by Immunomodulatory Imide Drug-Based Molecular Glues and Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 12273–12285. [Google Scholar] [CrossRef]
- Wang, S.; Feng, Z.; Qu, C.; Yu, S.; Zhang, H.; Deng, R.; Luo, D.; Pu, C.; Zhang, Y.; Li, R. Novel Amphiphilic PROTAC with Enhanced Pharmacokinetic Properties for ALK Protein Degradation. J. Med. Chem. 2024, 67, 9842–9856. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Wallace-Povirk, A.; Ning, C.; Frühauf, J.; Tong, N.; Gangjee, A.; Matherly, L.H.; Hou, Z. Folate Transporter Dynamics and Therapy with Classic and Tumor-Targeted Antifolates. Sci. Rep. 2021, 11, 6389. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Liu, Y.; Shen, Y.; Meng, F.; Kaniskan, H.Ü.; Jin, J.; Wei, W. Cancer Selective Target Degradation by Folate-Caged PROTACs. J. Am. Chem. Soc. 2021, 143, 7380–7387. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as Therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
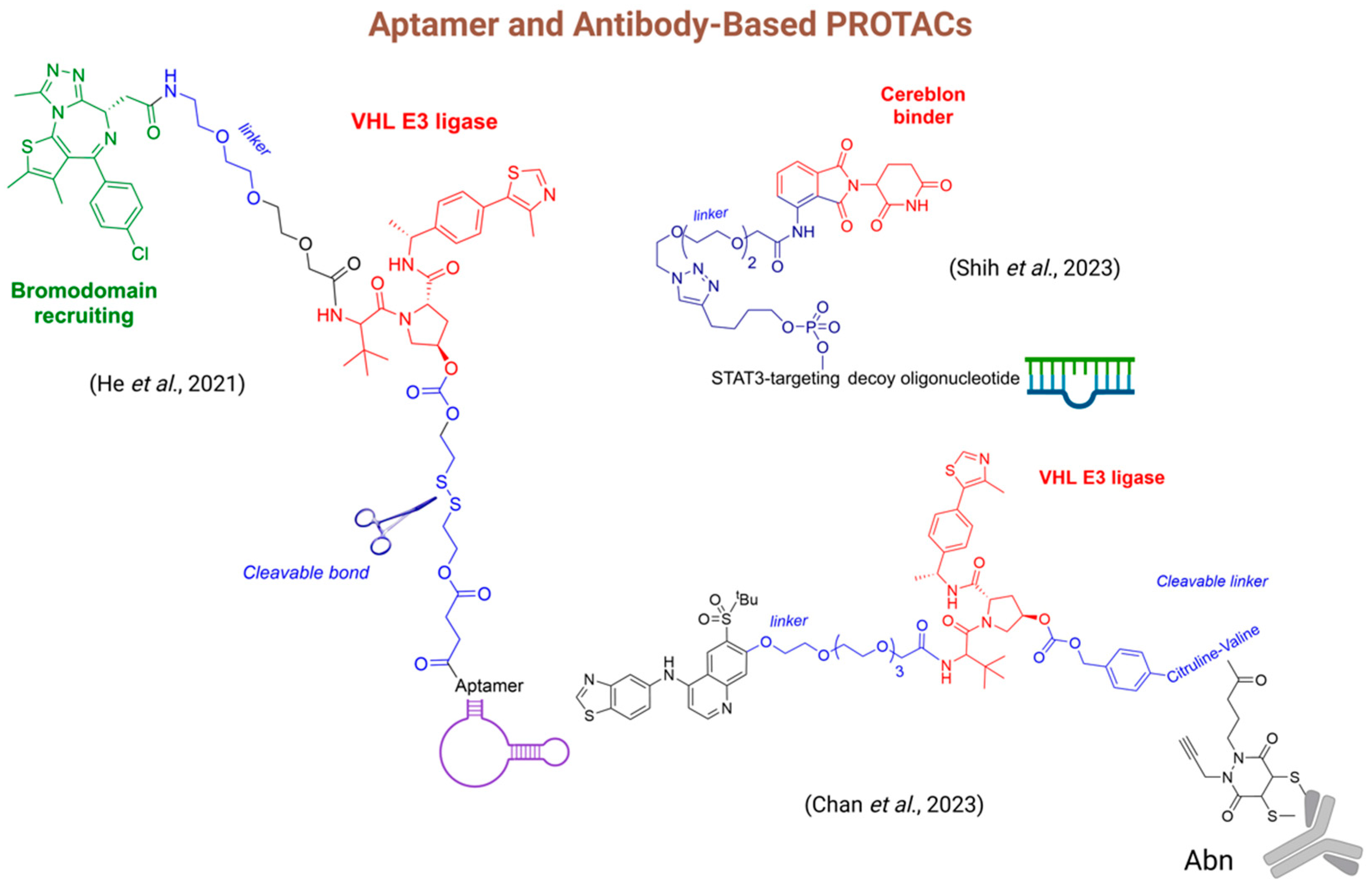
- He, J.; Peng, T.; Peng, Y.; Ai, L.; Deng, Z.; Wang, X.-Q.; Tan, W. Molecularly Engineering Triptolide with Aptamers for High Specificity and Cytotoxicity for Triple-Negative Breast Cancer. J. Am. Chem. Soc. 2020, 142, 2699–2703. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-PROTAC Conjugates (APCs) for Tumor-Specific Targeting in Breast Cancer. Angew. Chem. Int. Ed. Engl. 2021, 60, 23299–23305. [Google Scholar] [CrossRef]
- Shih, P.-C.; Naganuma, M.; Tsuji, G.; Demizu, Y.; Naito, M. Development of Decoy Oligonucleotide-Warheaded Chimeric Molecules Targeting STAT3. Bioorganic Med. Chem. 2023, 95, 117507. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Sathyamurthi, P.S.; Queisser, M.A.; Mullin, M.; Shrives, H.; Coe, D.M.; Burley, G.A. Antibody-Proteolysis Targeting Chimera Conjugate Enables Selective Degradation of Receptor-Interacting Serine/Threonine-Protein Kinase 2 in HER2+ Cell Lines. Bioconjug. Chem. 2023, 34, 2049–2054. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Wang, X.; Liu, H.; Zhang, Y.; Xie, T.; Zhang, H.; Li, X.; Peng, T.; Sun, X.; et al. Development of a Novel PROTAC Using the Nucleic Acid Aptamer as a Targeting Ligand for Tumor Selective Degradation of Nucleolin. Mol. Ther. Nucleic Acids 2022, 30, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Bukari, B.; Samarasinghe, R.M.; Noibanchong, J.; Shigdar, S.L. Non-Invasive Delivery of Therapeutics into the Brain: The Potential of Aptamers for Targeted Delivery. Biomedicines 2020, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Fu, W.; Lin, S.; Tian, T.; Li, S.; Shao, X.; Zhang, Y.; Zhang, T.; Tang, Z.; Zhou, Y.; et al. Targeted and Effective Glioblastoma Therapy via Aptamer-Modified Tetrahedral Framework Nucleic Acid-Paclitaxel Nanoconjugates That Can Pass the Blood Brain Barrier. Nanomedicine 2019, 21, 102061. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar] [PubMed]
- Dragovich, P.S.; Adhikari, P.; Blake, R.A.; Blaquiere, N.; Chen, J.; Cheng, Y.-X.; den Besten, W.; Han, J.; Hartman, S.J.; He, J.; et al. Antibody-Mediated Delivery of Chimeric Protein Degraders Which Target Estrogen Receptor Alpha (ERα). Bioorganic Med. Chem. Lett. 2020, 30, 126907. [Google Scholar] [CrossRef] [PubMed]
- Vartak, R.; Deore, B.; Sanhueza, C.A.; Patel, K. Cetuximab-Based PROteolysis Targeting Chimera for Effectual Downregulation of NSCLC with Varied EGFR Mutations. Int. J. Biol. Macromol. 2023, 252, 126413. [Google Scholar] [CrossRef]
- Hong, K.B.; An, H. Degrader–Antibody Conjugates: Emerging New Modality. J. Med. Chem. 2023, 66, 140–148. [Google Scholar] [CrossRef]
- Dragovich, P.S. Degrader-Antibody Conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.; Chen, W.; Wu, Y.; Xing, D. New-Generation Advanced PROTACs as Potential Therapeutic Agents in Cancer Therapy. Mol. Cancer 2024, 23, 110. [Google Scholar] [CrossRef]
- Guo, Y.; Li, X.; Xie, Y.; Wang, Y. What Influences the Activity of Degrader−Antibody Conjugates (DACs). Eur. J. Med. Chem. 2024, 268, 116216. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Steinman, L.; Kim, D.T.; Fathman, C.G.; Rothbard, J.B. Polyarginine Enters Cells More Efficiently than Other Polycationic Homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.-H. Cell-Penetrating Peptide Sequence and Modification Dependent Uptake and Subcellular Distribution of Green Florescent Protein in Different Cell Lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef]
- Gori, A.; Lodigiani, G.; Colombarolli, S.G.; Bergamaschi, G.; Vitali, A. Cell Penetrating Peptides: Classification, Mechanisms, Methods of Study, and Applications. ChemMedChem 2023, 18, e202300236. [Google Scholar] [CrossRef]
- Dobchev, D.A.; Mager, I.; Tulp, I.; Karelson, G.; Tamm, T.; Tamm, K.; Janes, J.; Langel, U.; Karelson, M. Prediction of Cell-Penetrating Peptides Using Artificial Neural Networks. Curr. Comput. Aided Drug Des. 2010, 6, 79–89. [Google Scholar] [CrossRef]
- He, S.; Fang, Y.; Wu, M.; Zhang, P.; Gao, F.; Hu, H.; Sheng, C.; Dong, G. Enhanced Tumor Targeting and Penetration of Proteolysis-Targeting Chimeras through iRGD Peptide Conjugation: A Strategy for Precise Protein Degradation in Breast Cancer. J. Med. Chem. 2023, 66, 16828–16842. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Y.; Zhang, J.; Li, Y.; Wang, J.; Liu, N.; Zhang, J.; Pan, X. Discovery of Novel Penetrating Peptides Able to Target Human Leukemia and Lymphoma for Enhanced PROTAC Delivery. Eur. J. Med. Chem. 2024, 277, 116734. [Google Scholar] [CrossRef]
- Dai, M.-Y.; Shi, Y.-Y.; Wang, A.-J.; Liu, X.-L.; Liu, M.; Cai, H.-B. High-Potency PD-1/PD-L1 Degradation Induced by Peptide-PROTAC in Human Cancer Cells. Cell Death Dis. 2022, 13, 924. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, H.; Ohoka, N.; Takyo, M.; Ito, T.; Tsuchiya, K.; Kurohara, T.; Fukuhara, K.; Inoue, T.; Naito, M.; Demizu, Y. Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation. Int. J. Mol. Sci. 2021, 22, 8772. [Google Scholar] [CrossRef] [PubMed]
- Juan, A.; Del Mar Noblejas-López, M.; Arenas-Moreira, M.; Alonso-Moreno, C.; Ocaña, A. Options to Improve the Action of PROTACs in Cancer: Development of Controlled Delivery Nanoparticles. Front. Cell Dev. Biol. 2022, 9, 805336. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tandon, I.; Heelan, W.; Wang, Y.; Tang, W.; Hu, Q. Proteolysis-Targeting Chimera (PROTAC) Delivery System: Advancing Protein Degraders towards Clinical Translation. Chem. Soc. Rev. 2022, 51, 5330–5350. [Google Scholar] [CrossRef]
- Gao, J.; Hou, B.; Zhu, Q.; Yang, L.; Jiang, X.; Zou, Z.; Li, X.; Xu, T.; Zheng, M.; Chen, Y.-H.; et al. Engineered Bioorthogonal POLY-PROTAC Nanoparticles for Tumour-Specific Protein Degradation and Precise Cancer Therapy. Nat. Commun. 2022, 13, 4318. [Google Scholar] [CrossRef]
- Zhang, C.; Zeng, Z.; Cui, D.; He, S.; Jiang, Y.; Li, J.; Huang, J.; Pu, K. Semiconducting Polymer Nano-PROTACs for Activatable Photo-Immunometabolic Cancer Therapy. Nat. Commun. 2021, 12, 2934. [Google Scholar] [CrossRef]
- Zhang, C.; He, S.; Zeng, Z.; Cheng, P.; Pu, K. Smart Nano-PROTACs Reprogram Tumor Microenvironment for Activatable Photo-metabolic Cancer Immunotherapy. Angew. Chem. Int. Ed. 2022, 61, e202114957. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fan, J.; Wang, R.; Wang, X.; Li, N.; You, Q.; Jiang, Z. Ligation to Scavenging Strategy Enables On-Demand Termination of Targeted Protein Degradation. J. Am. Chem. Soc. 2023, 145, 7218–7229. [Google Scholar] [CrossRef]
- Zhong, J.; Zhao, R.; Wang, Y.; Su, Y.; Lan, X. Nano-PROTACs: State of the Art and Perspectives. Nanoscale 2024, 16, 4378–4391. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Luo, T.; Wang, M. Delivering on Cell-Selective Protein Degradation Using Chemically Tailored PROTACs. ChemBioChem 2023, 24, e202300413. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Tong, W.; Wei, A.; Mu, X. Progress of Proteolysis-Targeting Chimeras (PROTACs) Delivery System in Tumor Treatment. Int. J. Biol. Macromol. 2024, 275, 133680. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dong, Q.-Q.; Ni, Y.-K.; Xu, X.-L.; Chen, C.-X.; Chen, W. Nano-Proteolysis Targeting Chimeras (Nano-PROTACs) in Cancer Therapy. Int. J. Nanomed. 2024, 19, 5739–5761. [Google Scholar] [CrossRef]
- Yang, W.; Saboo, S.; Zhou, L.; Askin, S.; Bak, A. Early Evaluation of Opportunities in Oral Delivery of PROTACs to Overcome Their Molecular Challenges. Drug Discov. Today 2024, 29, 103865. [Google Scholar] [CrossRef]
- Kirkby, M.; Sabri, A.B.; Scurr, D.J.; Moss, G.P. Dendrimer-Mediated Permeation Enhancement of Chlorhexidine Digluconate: Determination of in Vitro Skin Permeability and Visualisation of Dermal Distribution. Eur. J. Pharm. Biopharm. 2021, 159, 77–87. [Google Scholar] [CrossRef]
- Da Silva Santos, S.; Igne Ferreira, E.; Giarolla, J. Dendrimer Prodrugs. Molecules 2016, 21, 686. [Google Scholar] [CrossRef]
- Yan, L.-T.; Yu, X. Enhanced Permeability of Charged Dendrimers across Tense Lipid Bilayer Membranes. ACS Nano 2009, 3, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical Cocrystals: A Review of Preparations, Physicochemical Properties and Applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Wang, X.; Du, S.; Zhang, R.; Jia, X.; Yang, T.; Zhang, X. Drug-Drug Cocrystals: Opportunities and Challenges. Asian J. Pharm. Sci. 2021, 16, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Samie, A.; Alavian, H. A Perspective on the Permeability of Cocrystals/Organic Salts of Oral Drugs. Mol. Pharm. 2024, 21, 4860–4911. [Google Scholar] [CrossRef]
- Panzade, P.S.; Shendarkar, G.R. Pharmaceutical Cocrystal: A Game Changing Approach for the Administration of Old Drugs in New Crystalline Form. Drug Dev. Ind. Pharm. 2020, 46, 1559–1568. [Google Scholar] [CrossRef]
- Panzade, P.S.; Shendarkar, G.R. Pharmaceutical Cocrystal: An Antique and Multifaceted Approach. Curr. Drug Deliv. 2017, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xie, C.; Lu, H.; Guo, N.; Lou, Y.; Su, W.; Hao, H. Cocrystal and Its Application in the Field of Active Pharmaceutical Ingredients and Food Ingredients. Curr. Pharm. Des. 2018, 24, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Fala, L. Entresto (Sacubitril/Valsartan): First-in-Class Angiotensin Receptor Neprilysin Inhibitor FDA Approved for Patients with Heart Failure. Am. Health Drug Benefits 2015, 8, 330–334. [Google Scholar]
- Ayalasomayajula, S.; Langenickel, T.; Pal, P.; Boggarapu, S.; Sunkara, G. Clinical Pharmacokinetics of Sacubitril/Valsartan (LCZ696): A Novel Angiotensin Receptor-Neprilysin Inhibitor. Clin. Pharmacokinet. 2017, 56, 1461–1478. [Google Scholar] [CrossRef]
- Cheney, M.L.; Weyna, D.R.; Shan, N.; Hanna, M.; Wojtas, L.; Zaworotko, M.J. Coformer Selection in Pharmaceutical Cocrystal Development: A Case Study of a Meloxicam Aspirin Cocrystal That Exhibits Enhanced Solubility and Pharmacokinetics. J. Pharm. Sci. 2011, 100, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-M.; Yu, M.-C.; Wang, L.-Y.; Li, Y.-T.; Wu, Z.-Y.; Yan, C.-W. A Supramolecular Adduct of Tegafur and Syringic Acid: The First Tegafur-Nutraceutical Cocrystal with Perfected in Vitro and in Vivo Characteristics as Well as Synergized Anticancer Activities. New J. Chem. 2020, 44, 15994–16005. [Google Scholar] [CrossRef]
- Mannava, M.K.C.; Garai, A.; Nangia, A.K. Diffusion and Flux Improvement of Drugs through Complexation. Mol. Pharm. 2023, 20, 2293–2316. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Solubility 1 | Permeability 2 | Examples of Drugs |
|---|---|---|---|
| I | High | High | Acyclovir, captopril, abacavir |
| II | Low | High | Atorvastatin, diclofenac, ciprofloxacin |
| III | High | Low | Cimetidine, atenolol, amoxicillin |
| IV | Low | Low | Furosemide, chlorthalidone, methotrexate |
| Structure | Name (Code) | LogP 1 | Current Development Stage |
|---|---|---|---|
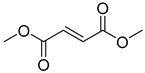 | Dimethyl fumarate (1) | 1.51 | Market prodrug |
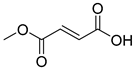 | Monomethyl fumarate (2) | 0.77 | Drug |
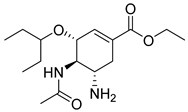 | Oseltamivir (3) | 1.23 | Market prodrug |
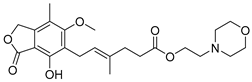 | Mofetil mycophenolate (4) | 2.68 | Market prodrug |
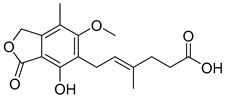 | Mycophenolic acid (5) | 3.01 | Drug |
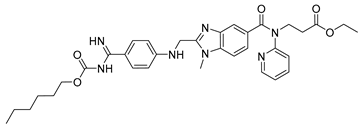 | Dabigatran etexilate (6) | 4.86 | Market prodrug |
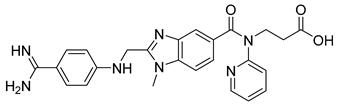 | Dabigatran (7) | 2.04 | Drug |
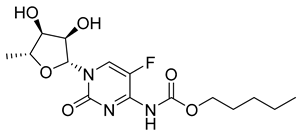 | Capecitabine (8) | 1.43 | Market prodrug |
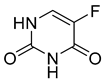 | 5-fluorouracil (9) | −0.08 | Market prodrug |
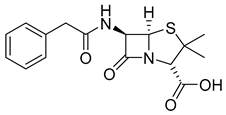 | Penicillin (10) | 0.93 | Drug |
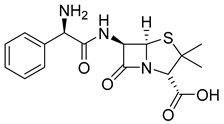 | Ampicillin (11) | 0.15 | Drug |
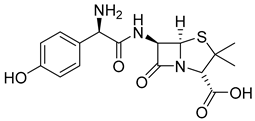 | Amoxicillin (12) | −0.13 | Drug |
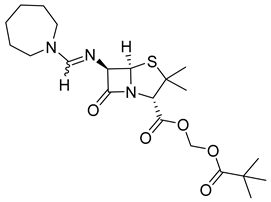 | Pivmecillinam (13) | 3.16 | Market prodrug |
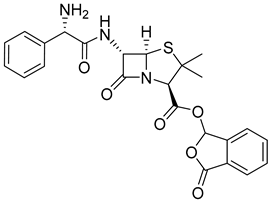 | Talampicillin (14) | 1.67 | Market prodrug |
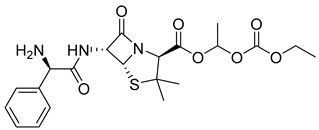 | Bacampicillin (15) | 1.22 | Market prodrug |
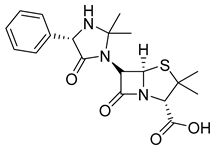 | Hetacillin (16) | 1.63 | Market prodrug |
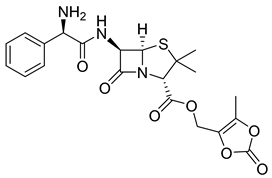 | Lenampicillin (17) | 0.97 | Market prodrug |
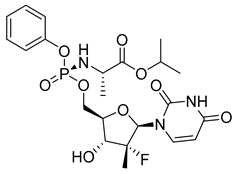 | Sofosbuvir (18) | 1.81 | Market prodrug |
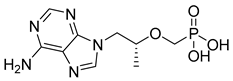 | Tenofovir (19) | −1.30 | Drug |
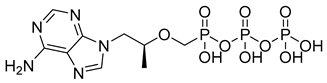 | Tenofovir diphosphate (20) | −1.84 | Market prodrug |
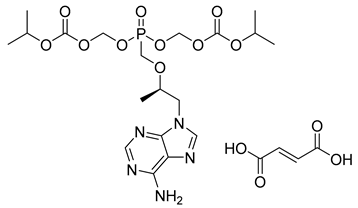 | Tenofovir disoproxil fumarate (21) | 3.02 | Market prodrug |
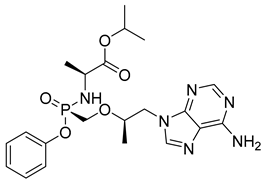 | Tenofovir alafenamide (22) | 2.05 | Market prodrug |
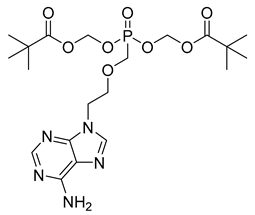 | Adefovir dipivoxil (23) | 3.56 | Market prodrug |
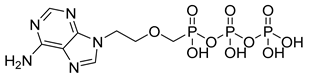 | Adefovir diphosphate (24) | −2.28 | Market prodrug |
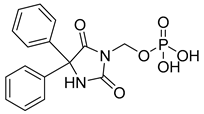 | Fosphenytoin (25) | 1.81 | Market prodrug |
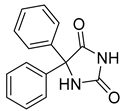 | Phenytoin (26) | 2.28 | Drug |
| Structure | Name (Code) | LogP 1 | Current Development Stage |
|---|---|---|---|
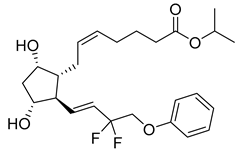 | Tafluprost (27) | 4.11 | Market prodrug |
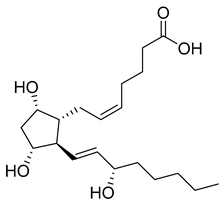 | Prostaglandin F2a (28) | 2.20 | Drug |
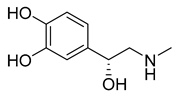 | Epinephrine (29) | 0.33 | Drug |
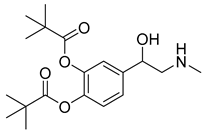 | Dipivefrin (30) | 3.61 | Market prodrug |
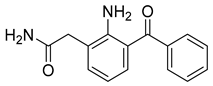 | Nepafenac (31) | 1.90 | Market prodrug |
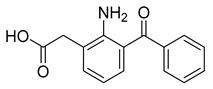 | Amfenac (32) | 2.77 | Drug |
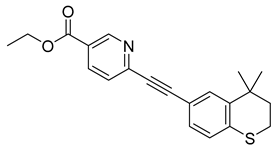 | Tazarotene (33) | 5.30 | Market prodrug |
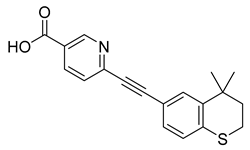 | Tazarotenic acid (34) | 4.33 | Drug |
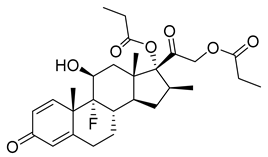 | Betamethasone dipropionate (35) | 3.86 | Market prodrug |
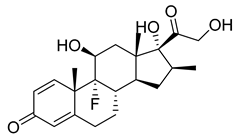 | Betamethasone (36) | 1.75 | Drug |
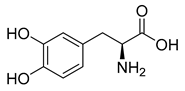 | Levodopa (37) | 0.58 | Drug |
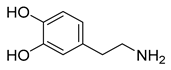 | Dopamine (38) | 0.85 | Active metabolite |
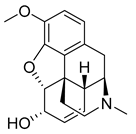 | Codeine (39) | 1.27 | Market prodrug |
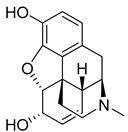 | Morphine (40) | 1.23 | Drug |
| Structure | Name (Code) | LogP 1 | Current Development Stage |
|---|---|---|---|
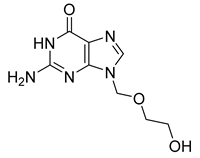 | Acyclovir (41) | −1.85 | Drug |
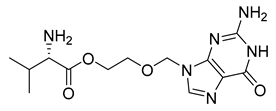 | Valacyclovir (42) | −0.92 | Market prodrug |
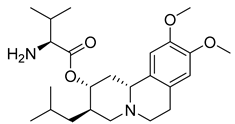 | Valbenazine (43) | 3.29 | Market prodrug |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, M.M.; Gini, A.L.R.; Moura, J.A.; Scarim, C.B.; Chin, C.M.; dos Santos, J.L. Prodrug Approach as a Strategy to Enhance Drug Permeability. Pharmaceuticals 2025, 18, 297. https://doi.org/10.3390/ph18030297
de Souza MM, Gini ALR, Moura JA, Scarim CB, Chin CM, dos Santos JL. Prodrug Approach as a Strategy to Enhance Drug Permeability. Pharmaceuticals. 2025; 18(3):297. https://doi.org/10.3390/ph18030297
Chicago/Turabian Stylede Souza, Mateus Mello, Ana Luísa Rodriguez Gini, Jhonnathan Alves Moura, Cauê Benito Scarim, Chung Man Chin, and Jean Leandro dos Santos. 2025. "Prodrug Approach as a Strategy to Enhance Drug Permeability" Pharmaceuticals 18, no. 3: 297. https://doi.org/10.3390/ph18030297
APA Stylede Souza, M. M., Gini, A. L. R., Moura, J. A., Scarim, C. B., Chin, C. M., & dos Santos, J. L. (2025). Prodrug Approach as a Strategy to Enhance Drug Permeability. Pharmaceuticals, 18(3), 297. https://doi.org/10.3390/ph18030297