

Physiologically Based Pharmacokinetic Modeling of Tofacitinib: Predicting Drug Exposure and Optimizing Dosage in Special Populations and Drug–Drug Interaction Scenarios


Abstract
:1. Introduction
2. Results
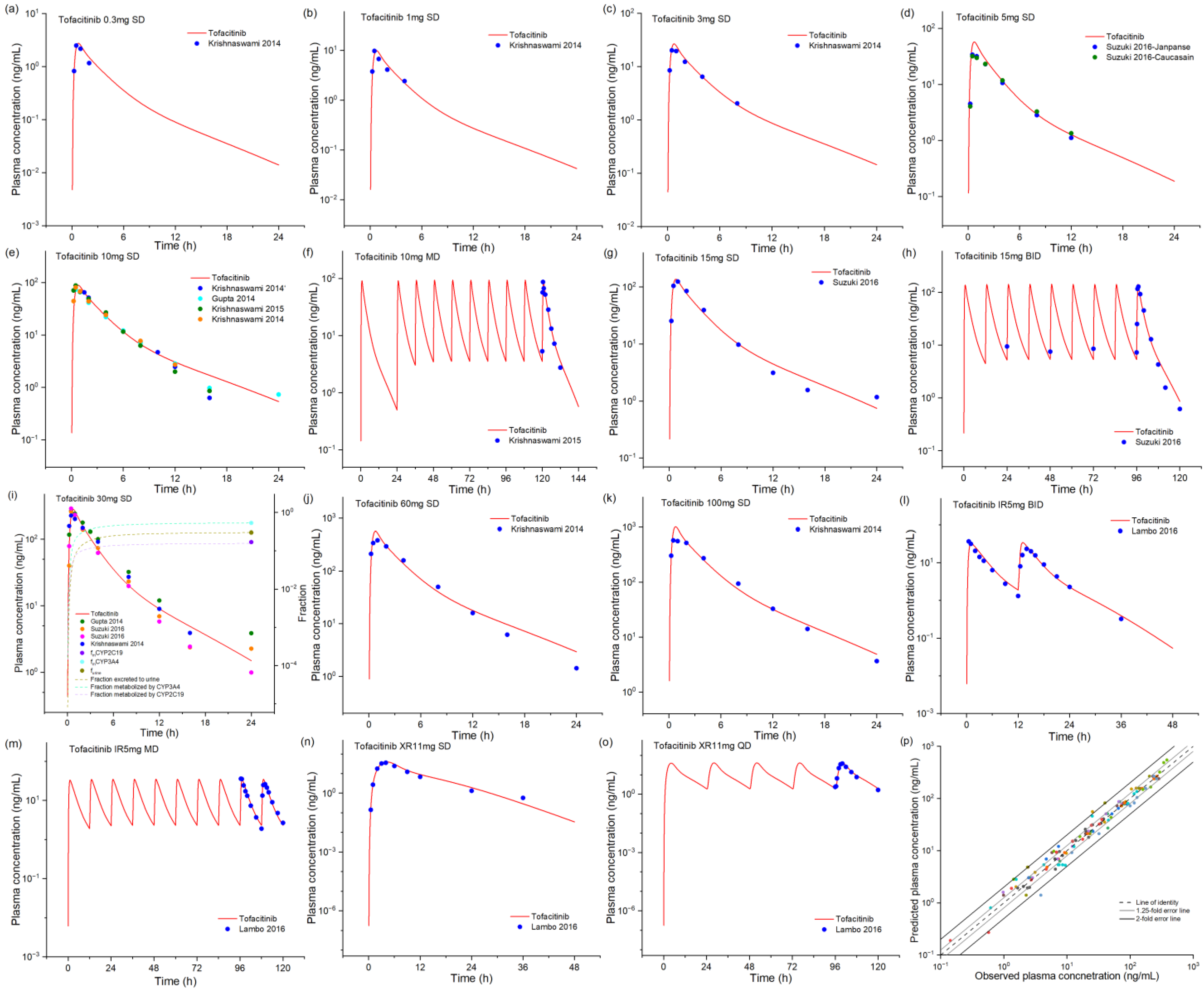
2.1. Tofacitinib Base Model Development and Validation
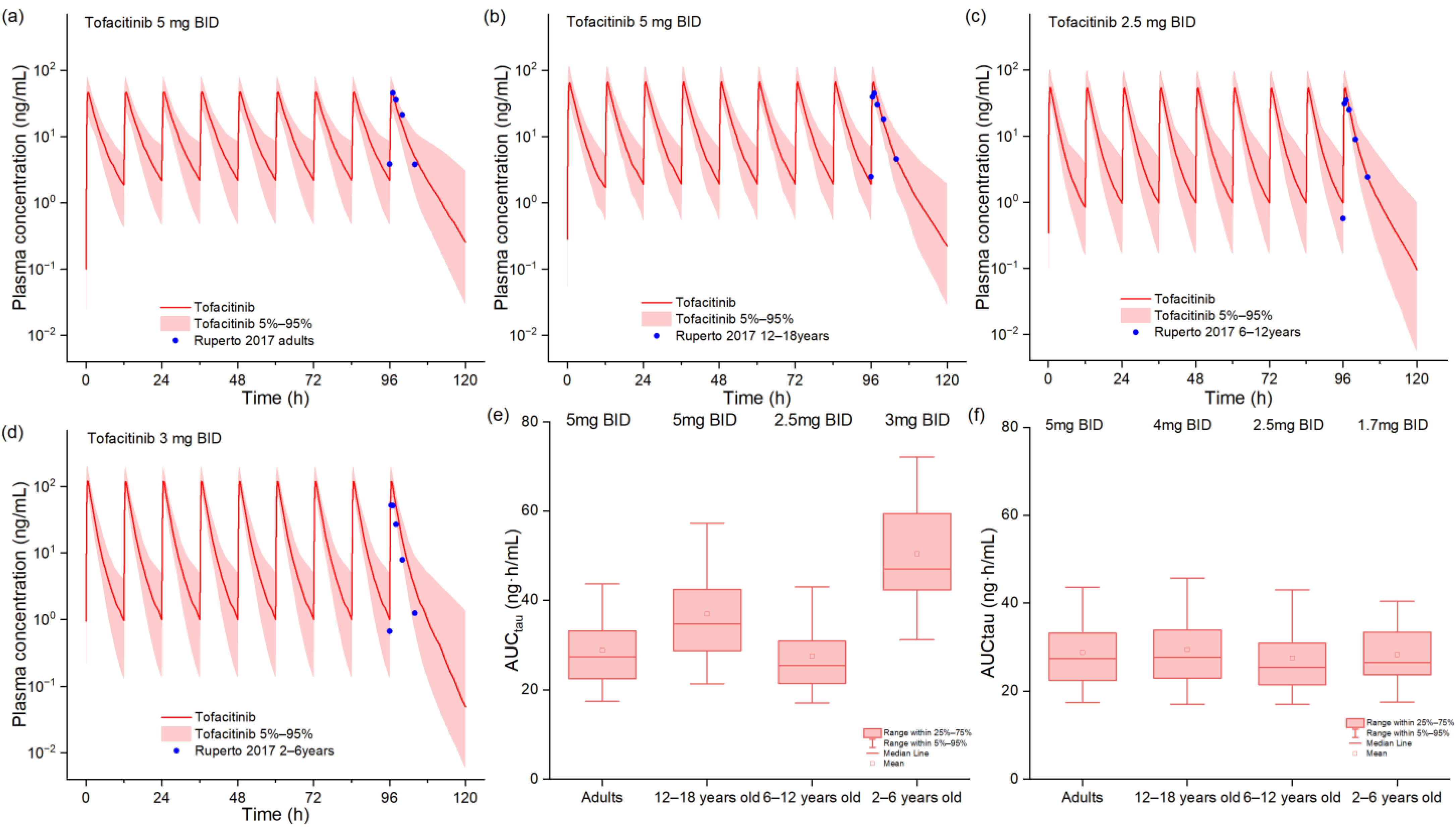
2.2. PBPK Model in Pediatric Patients
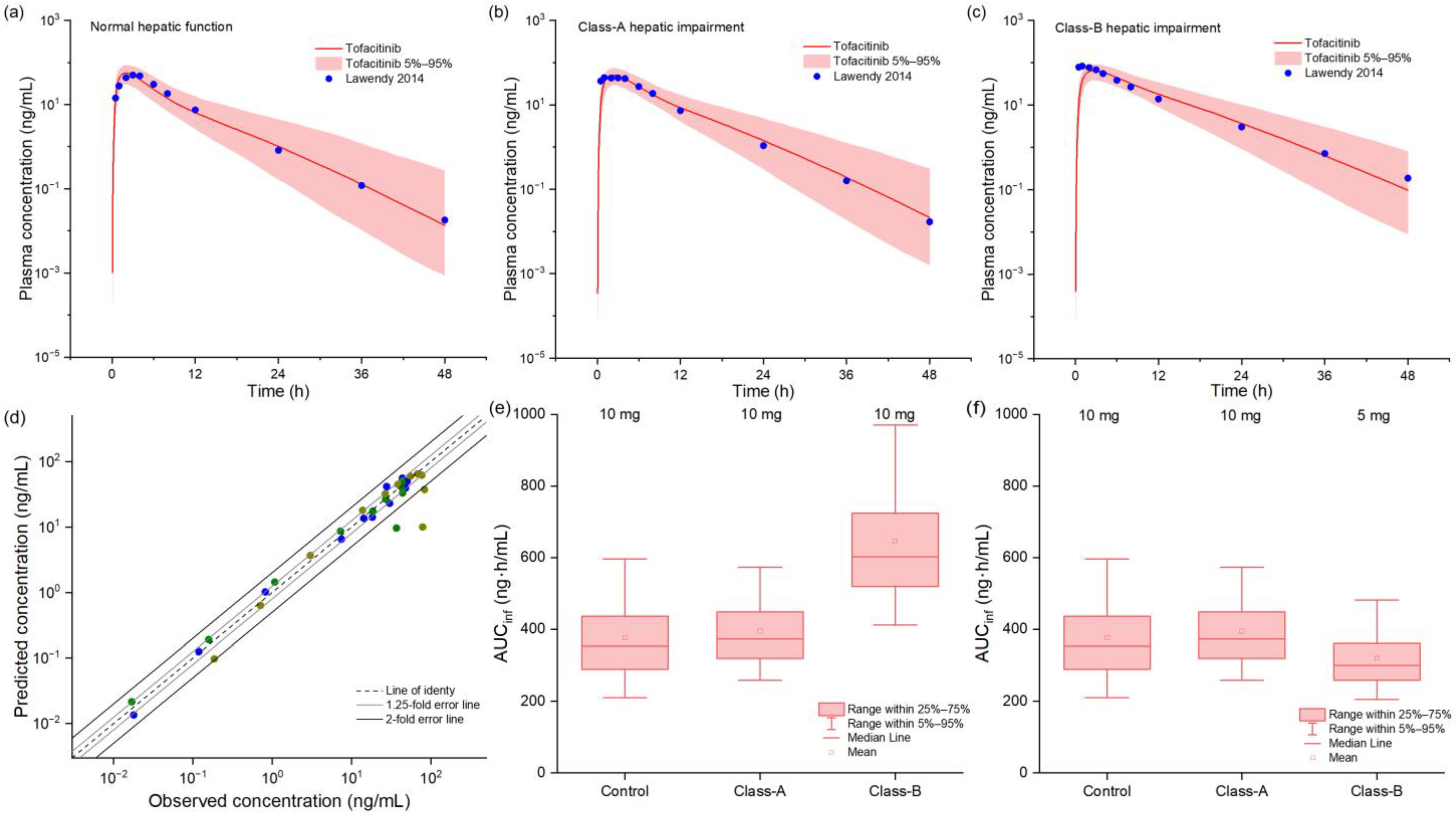
2.3. PBPK Model in Hepatic Impairment Populations
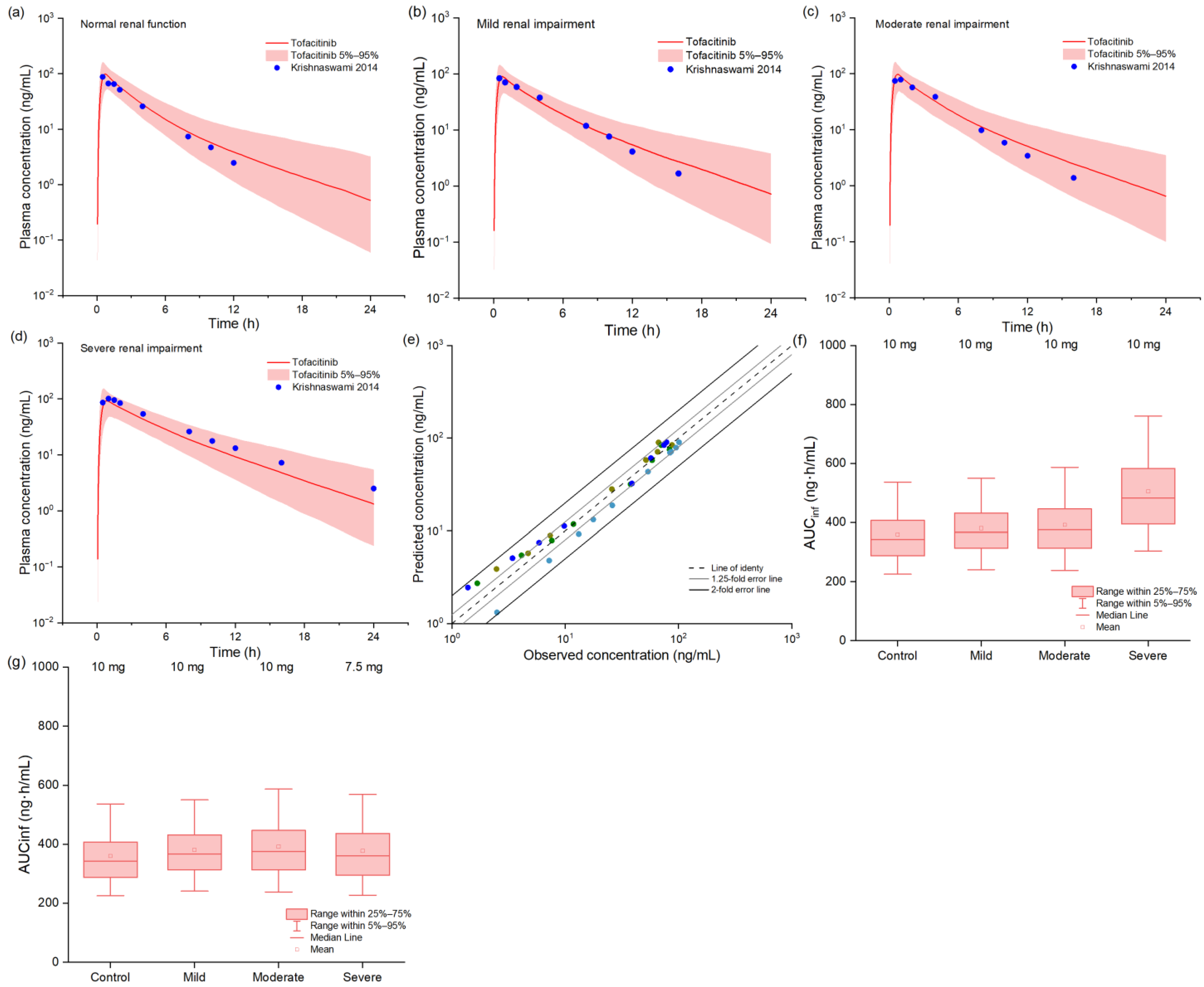
2.4. PBPK Model in Renal Impairment Populations
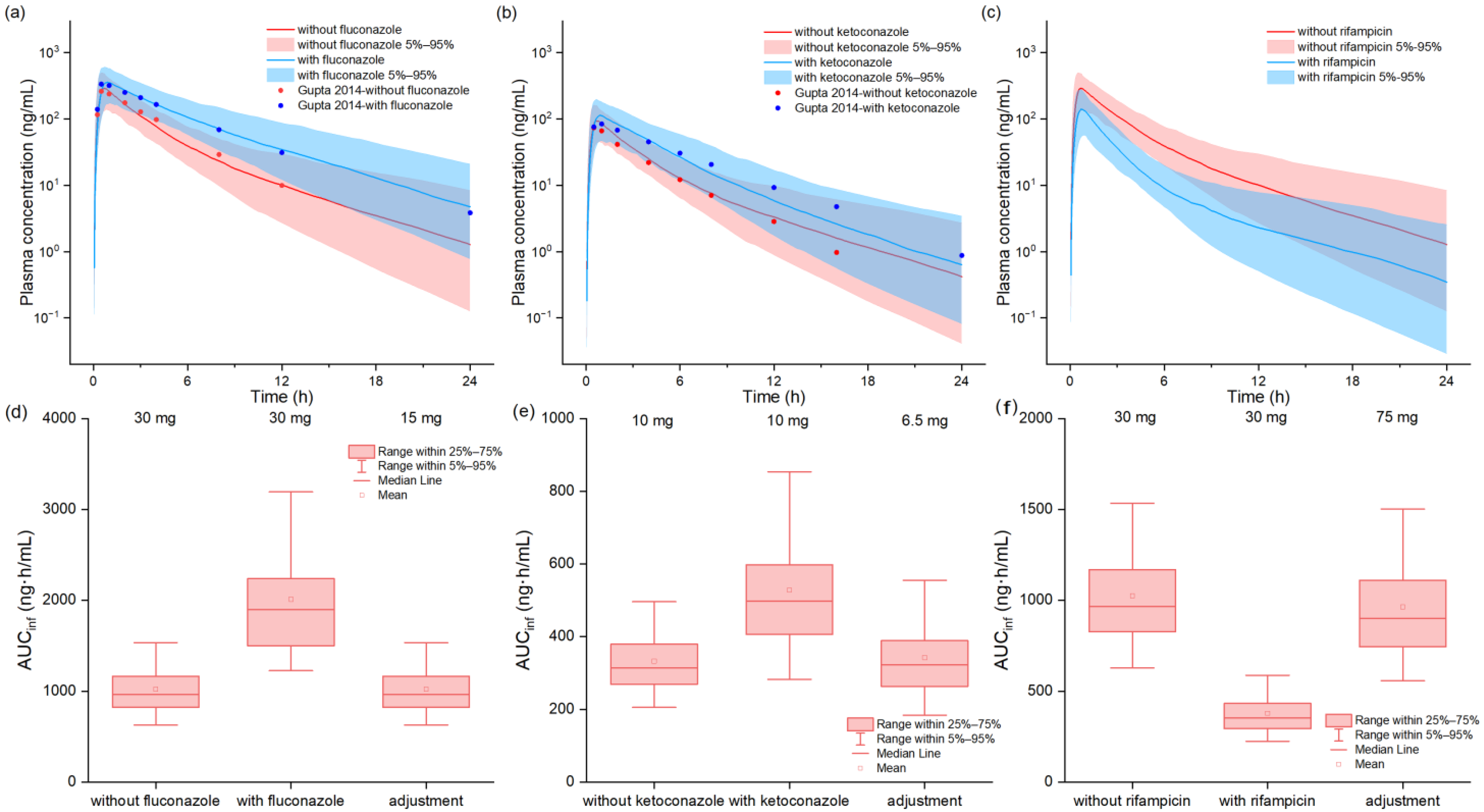
2.5. Drug–Drug Interactions Model of Tofacitinib with Perpetrators
3. Discussion
4. Materials and Methods
4.1. PBPK Modeling Platform and Related Software
4.2. PBPK Base Model Development and Evaluation of Tofacitinib
4.3. Extrapolation to Pediatric Patients
4.4. Extrapolation to the Hepatic Impairment Populations
4.5. Extrapolation to the Renal Impairment Populations
4.6. Drug–Drug Interactions Model of Tofacitinib with Fluconazole, Ketoconazole, and Rifampicin
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PK | Pharmacokinetics |
| DDI | Drug–drug interaction |
| PBPK | Physiologically based pharmacokinetic |
| CYP | Cytochrome P450 |
| Cmax | Peak plasma concentration |
| AUC | Area under the curve |
| JAK | Janus kinase |
| RA | Rheumatoid arthritis |
| IMID | Immune-mediated inflammatory disease |
| IR | Immediate release |
| XR | Extended release |
| JIA | Juvenile idiopathic arthritis |
| JPsA | Juvenile psoriatic arthritis |
| Tmax | Time to peak plasma concentration |
| AUCinf | Area under the curve from 0 to infinity |
| FE | Fold error |
| GMFE | Geometric mean fold error |
| CmaxR | Peak plasma concentration ratio |
| AUCR | Area under the curve ratio |
| HI | Hepatic impairment |
| RI | Renal impairment |
| AUCtau | Area under the curve in the dosing interval |
| Km | Michaelis–Menten constant |
| Kcat | Catalytic rate constant |
| eGFR | Estimated glomerular filtration rate |
References
- Taylor, P.C. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology 2019, 58, i17–i26. [Google Scholar] [CrossRef]
- Angelini, J.; Talotta, R.; Roncato, R.; Fornasier, G.; Barbiero, G.; Dal Cin, L.; Brancati, S.; Scaglione, F. JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future. Biomolecules 2020, 10, 1002. [Google Scholar] [CrossRef]
- Harrington, R.; Harkins, P.; Conway, R. Janus Kinase Inhibitors in Rheumatoid Arthritis: An Update on the Efficacy and Safety of Tofacitinib, Baricitinib and Upadacitinib. J. Clin. Med. 2023, 12, 6690. [Google Scholar] [CrossRef]
- Nash, P.; Kerschbaumer, A.; Dörner, T.; Dougados, M.; Fleischmann, R.M.; Geissler, K.; McInnes, I.; Pope, J.E.; van der Heijde, D.; Stoffer-Marx, M.; et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: A consensus statement. Ann. Rheum. Dis. 2021, 80, 71–87. [Google Scholar] [CrossRef]
- Dhillon, S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, P.; Niu, W.; Fang, R.; Chen, H.; An, Y.; Wang, W.; Jiang, C.; Ye, J. Preparation and evaluation of dissolving tofacitinib microneedles for effective management of rheumatoid arthritis. Eur. J. Pharm. Sci. 2023, 188, 106518. [Google Scholar] [CrossRef]
- Berekmeri, A.; Mahmood, F.; Wittmann, M.; Helliwell, P. Tofacitinib for the treatment of psoriasis and psoriatic arthritis. Expert. Rev. Clin. Immunol. 2018, 14, 719–730. [Google Scholar] [CrossRef]
- Gorantla, S.; Saha, R.N.; Singhvi, G. Spectrophotometric method to quantify tofacitinib in lyotropic liquid crystalline nanoparticles and skin layers: Application in ex vivo dermal distribution studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 255, 119719. [Google Scholar] [CrossRef]
- Talasila, S.; Lee, E.; Teichner, E.M.; Siegfried, E.C.; Jackson Cullison, S.R. Analysis of publicly available adverse events reported for pediatric patients treated with Janus kinase inhibitors. Pediatr. Dermatol. 2024, 41, 1040–1046. [Google Scholar] [CrossRef]
- Krishnaswami, S.; Boy, M.; Chow, V.; Chan, G. Safety, tolerability, and pharmacokinetics of single oral doses of tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin. Pharmacol. Drug Dev. 2015, 4, 83–88. [Google Scholar] [CrossRef]
- Bannwarth, B.; Kostine, M.; Poursac, N. A pharmacokinetic and clinical assessment of tofacitinib for the treatment of rheumatoid arthritis. Expert. Opin. Drug Metab. Toxicol. 2013, 9, 753–761. [Google Scholar] [CrossRef] [PubMed]
- FDA. Clinical Pharmacology and Biopharmaceutics Review(s). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203214Orig1s000ClinPharmR.pdf (accessed on 31 July 2024).
- Veeravalli, V.; Dash, R.P.; Thomas, J.A.; Babu, R.J.; Madgula, L.M.V.; Srinivas, N.R. Critical Assessment of Pharmacokinetic Drug-Drug Interaction Potential of Tofacitinib, Baricitinib and Upadacitinib, the Three Approved Janus Kinase Inhibitors for Rheumatoid Arthritis Treatment. Drug Saf. 2020, 43, 711–725. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tsuchiwata, S.; Chang, C.; Nicholas, T.; Su, C.; Le, V.H.; Kushner, J.; Kulisek, N. Bridging Efficacy of Tofacitinib Immediate-Release to Extended-Release Formulations for Treatment of Ulcerative Colitis: Application of a Model-Informed Drug Development Approach. Clin. Pharmacol. Drug Dev. 2022, 11, 976–986. [Google Scholar] [CrossRef]
- Gupta, P.; Chow, V.; Wang, R.; Kaplan, I.; Chan, G.; Alvey, C.; Ni, G.; Ndongo, M.N.; LaBadie, R.R.; Krishnaswami, S. Evaluation of the effect of fluconazole and ketoconazole on the pharmacokinetics of tofacitinib in healthy adult subjects. Clin. Pharmacol. Drug Dev. 2014, 3, 72–77. [Google Scholar] [CrossRef]
- Lamba, M.; Wang, R.; Kaplan, I.; Salageanu, J.; Tarabar, S.; Krishnaswami, S. The effect of rifampin on the pharmacokinetics of tofacitinib (CP-690,550) in healthy volunteers. In Clinical Pharmacology & Therapeutics; Nature Publishing Group: New York, NY, USA, 2012; p. S35. [Google Scholar]
- Brunner, H.I.; Akikusa, J.D.; Al-Abadi, E.; Bohnsack, J.F.; Boteanu, A.L.; Chedeville, G.; Cuttica, R.; De La Pena, W.; Jung, L.; Kasapcopur, O.; et al. Safety and efficacy of tofacitinib for the treatment of patients with juvenile idiopathic arthritis: Preliminary results of an open-label, long-term extension study. Ann. Rheum. Dis. 2024, 83, 1561–1571. [Google Scholar] [CrossRef]
- Koubek, D.N.; Pulk, R.A.; Rosano, J.V. A Review of Therapeutic Escalation for Pediatric Patients Admitted for Inflammatory Bowel Disease Flares. J. Pediatr. Pharmacol. Ther. 2023, 28, 649–657. [Google Scholar] [CrossRef]
- Malkawi, W.A.; AlRafayah, E.; AlHazabreh, M.; AbuLaila, S.; Al-Ghananeem, A.M. Formulation Challenges and Strategies to Develop Pediatric Dosage Forms. Children 2022, 9, 488. [Google Scholar] [CrossRef]
- Ryan, N.; Cooper, S.; Dominik, A.; Quinn, S.; Broderick, A.; Bourke, B.; Hussey, S. Outcomes of Tofacitinib Use in an Irish Pediatric Cohort. JPGN Rep. 2023, 4, e332. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.I.; Zuber, Z.; Tzaribachev, N.; Kingsbury, D.J.; Foeldvari, I.; Horneff, G.; Smolewska, E.; Vehe, R.K.; Hazra, A.; et al. Pharmacokinetic and safety profile of tofacitinib in children with polyarticular course juvenile idiopathic arthritis: Results of a phase 1, open-label, multicenter study. Pediatr. Rheumatol. Online J. 2017, 15, 86. [Google Scholar] [CrossRef]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Wagner, C.; Pan, Y.; Hsu, V.; Sinha, V.; Zhao, P. Predicting the Effect of CYP3A Inducers on the Pharmacokinetics of Substrate Drugs Using Physiologically Based Pharmacokinetic (PBPK) Modeling: An Analysis of PBPK Submissions to the US FDA. Clin. Pharmacokinet. 2016, 55, 475–483. [Google Scholar] [CrossRef]
- Schwab, M.; Billing, H.; Mörike, K. Drug Use, Dosing, and Toxicity in Renal Disease. In Pediatric Kidney Disease; Geary, D.F., Schaefer, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1927–1947. [Google Scholar] [CrossRef]
- Verbeeck, R.K.; Musuamba, F.T. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur. J. Clin. Pharmacol. 2009, 65, 757–773. [Google Scholar] [CrossRef]
- Lawendy, N.; Lamba, M.; Chan, G.; Wang, R.; Alvey, C.W.; Krishnaswami, S. The effect of mild and moderate hepatic impairment on the pharmacokinetics of tofacitinib, an orally active Janus kinase inhibitor. Clin. Pharmacol. Drug Dev. 2014, 3, 421–427. [Google Scholar] [CrossRef]
- Krishnaswami, S.; Chow, V.; Boy, M.; Wang, C.; Chan, G. Pharmacokinetics of tofacitinib, a janus kinase inhibitor, in patients with impaired renal function and end-stage renal disease. J. Clin. Pharmacol. 2014, 54, 46–52. [Google Scholar] [CrossRef]
- Suzuki, M.; Tse, S.; Hirai, M.; Kurebayashi, Y. Application of Physiologically-Based Pharmacokinetic Modeling for the Prediction of Tofacitinib Exposure in Japanese. Kobe J. Med. Sci. 2017, 62, E150–E161. [Google Scholar]
- Krishnaswami, S.; Wang, T.; Yuan, Y.; Alvey, C.W.; Checchio, T.; Peterson, M.; Shi, H.; Riese, R. Single- and multiple-dose pharmacokinetics of tofacitinib in healthy Chinese volunteers. Clin. Pharmacol. Drug Dev. 2015, 4, 395–399. [Google Scholar] [CrossRef]
- Lamba, M.; Wang, R.; Fletcher, T.; Alvey, C.; Kushner Jt Stock, T.C. Extended-Release Once-Daily Formulation of Tofacitinib: Evaluation of Pharmacokinetics Compared With Immediate-Release Tofacitinib and Impact of Food. J. Clin. Pharmacol. 2016, 56, 1362–1371. [Google Scholar] [CrossRef]
- Freriksen, J.J.M.; van der Heijden, J.E.M.; de Hoop-Sommen, M.A.; Greupink, R.; de Wildt, S.N. Physiologically Based Pharmacokinetic (PBPK) Model-Informed Dosing Guidelines for Pediatric Clinical Care: A Pragmatic Approach for a Special Population. Paediatr. Drugs 2023, 25, 5–11. [Google Scholar] [CrossRef]
- Marsousi, N.; Desmeules, J.A.; Rudaz, S.; Daali, Y. Usefulness of PBPK Modeling in Incorporation of Clinical Conditions in Personalized Medicine. J. Pharm. Sci. 2017, 106, 2380–2391. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.; Unadkat, J.D.; Zhang, X.; Wu, D.; Heimbach, T. Applications, Challenges, and Outlook for PBPK Modeling and Simulation: A Regulatory, Industrial and Academic Perspective. Pharm. Res. 2022, 39, 1701–1731. [Google Scholar] [CrossRef]
- Malhotra, B.; Dickins, M.; Alvey, C.; Jumadilova, Z.; Li, X.; Duczynski, G.; Gandelman, K. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br. J. Clin. Pharmacol. 2011, 72, 263–269. [Google Scholar] [CrossRef]
- Karonen, T.; Laitila, J.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Fluconazole but not the CYP3A4 inhibitor, itraconazole, increases zafirlukast plasma concentrations. Eur. J. Clin. Pharmacol. 2012, 68, 681–688. [Google Scholar] [CrossRef]
- Tse, S.; Dowty, M.E.; Menon, S.; Gupta, P.; Krishnaswami, S. Application of Physiologically Based Pharmacokinetic Modeling to Predict Drug Exposure and Support Dosing Recommendations for Potential Drug-Drug Interactions or in Special Populations: An Example Using Tofacitinib. J. Clin. Pharmacol. 2020, 60, 1617–1628. [Google Scholar] [CrossRef]
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Chan, G.L.; Krishnaswami, S.; Prakash, C. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef]
- Niwa, T.; Yamazaki, H. Comparison of cytochrome P450 2C subfamily members in terms of drug oxidation rates and substrate inhibition. Curr. Drug Metab. 2012, 13, 1145–1159. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef]
- Saganuwan, S.A. Application of modified Michaelis—Menten equations for determination of enzyme inducing and inhibiting drugs. BMC Pharmacol. Toxicol. 2021, 22, 57. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Q.; Wang, H.; Song, W.; Wang, J.; Mamun, A.A.; Geng, P.; Zhou, Y.; Wang, S. Effect of P. corylifolia on the pharmacokinetic profile of tofacitinib and the underlying mechanism. Front. Pharmacol. 2024, 15, 1351882. [Google Scholar] [CrossRef]
- Kathuria, H.; Handral, H.K.; Cha, S.; Nguyen, D.T.P.; Cai, J.; Cao, T.; Wu, C.; Kang, L. Enhancement of Skin Delivery of Drugs Using Proposome Depends on Drug Lipophilicity. Pharmaceutics 2021, 13, 1457. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, T.; Yang, H.; Ma, W.Y.; He, Q.; Xu, M.; Yang, Y. Physiologically-Based Pharmacokinetic Modeling of Tenofovir Disoproxil Fumarate in Pregnant Women. Curr. Drug Metab. 2023, 23, 1115–1123. [Google Scholar] [CrossRef]
- Saeheng, T.; Na-Bangchang, K.; Karbwang, J. Utility of physiologically based pharmacokinetic (PBPK) modeling in oncology drug development and its accuracy: A systematic review. Eur. J. Clin. Pharmacol. 2018, 74, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Cohen, S.; Wilkinson, B.E.; Connell, C.A.; French, J.L.; Gomez-Reino, J.; Gruben, D.; Kanik, K.S.; Krishnaswami, S.; Pascual-Ramos, V.; et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012, 64, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Salerno, S.N.; Edginton, A.; Gerhart, J.G.; Laughon, M.M.; Ambalavanan, N.; Sokol, G.M.; Hornik, C.D.; Stewart, D.; Mills, M.; Martz, K.; et al. Physiologically-Based Pharmacokinetic Modeling Characterizes the CYP3A-Mediated Drug-Drug Interaction Between Fluconazole and Sildenafil in Infants. Clin. Pharmacol. Ther. 2021, 109, 253–262. [Google Scholar] [CrossRef]
- Marok, F.Z.; Wojtyniak, J.G.; Fuhr, L.M.; Selzer, D.; Schwab, M.; Weiss, J.; Haefeli, W.E.; Lehr, T. A Physiologically Based Pharmacokinetic Model of Ketoconazole and Its Metabolites as Drug-Drug Interaction Perpetrators. Pharmaceutics 2023, 15, 679. [Google Scholar] [CrossRef]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | CmaxR | AUCR | ||
|---|---|---|---|---|
| Predicted/Mean | Observed/Mean(90%CI) | Predicted/Mean | Observed/Mean(90%CI) | |
| Fluconazole | 1.36 | 1.27 (1.12~1.44) | 1.95 | 1.79 (1.64~1.96) |
| Ketoconazole | 1.33 | 1.16 (1.05~1.29) | 1.63 | 2.03 (1.91~2.16) |
| Rifampicin | 0.52 | 0.26 (0.23~0.31) | 0.37 | 0.16 (0.14~0.18) |
| Parameters | Tofacitinib | Reference/Source |
|---|---|---|
| lipophilicity | 1.15 | [42] |
| plasma fraction unbound | 0.61 | [12] |
| MW (molecular weight) | 312.40 g/mol | [12] |
| pKa | 5.07 | [12] |
| solubility | 2.9 mg/mL | [12] |
| specific intestinal permeability | 7.24 × 10−6 cm/min | optimized |
| partition coefficients calculation | diverse | Rodgers and Rowland |
| cellular permeability | 4.37 × 10−4 cm/min | PK-Sim standard |
| formulation | normal dissolved, IR Weibull, XR Weibull, fed Weibull | |
| 50% dissolution time | IR 0.29 h, XR 3.22 h, fed 1.25 h | optimized |
| dissolution shape | IR 0.59, XR 2.42, fed 1.01 | optimized |
| GFR fraction | 1.0 | assumed |
| Km,CYP3A4 | 10.61 μmol/L | [41] |
| Kcat,CYP3A4 | 0.49 L/min | optimized |
| fm,CYP3A4 | 0.53 | [37] |
| CLspec,CYP2C19 | 0.06 L/min | optimized |
| fm,CYP2C19 | 0.17 | [37] |
| TSspec | 0.53 L/min | optimized |
| furine | 0.3 | [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Wang, Z.; Zhang, Q.; Zhang, W.; Zheng, L.; Hu, W. Physiologically Based Pharmacokinetic Modeling of Tofacitinib: Predicting Drug Exposure and Optimizing Dosage in Special Populations and Drug–Drug Interaction Scenarios. Pharmaceuticals 2025, 18, 425. https://doi.org/10.3390/ph18030425
Cao Z, Wang Z, Zhang Q, Zhang W, Zheng L, Hu W. Physiologically Based Pharmacokinetic Modeling of Tofacitinib: Predicting Drug Exposure and Optimizing Dosage in Special Populations and Drug–Drug Interaction Scenarios. Pharmaceuticals. 2025; 18(3):425. https://doi.org/10.3390/ph18030425
Chicago/Turabian StyleCao, Zhihai, Zilong Wang, Qian Zhang, Wei Zhang, Liang Zheng, and Wei Hu. 2025. "Physiologically Based Pharmacokinetic Modeling of Tofacitinib: Predicting Drug Exposure and Optimizing Dosage in Special Populations and Drug–Drug Interaction Scenarios" Pharmaceuticals 18, no. 3: 425. https://doi.org/10.3390/ph18030425
APA StyleCao, Z., Wang, Z., Zhang, Q., Zhang, W., Zheng, L., & Hu, W. (2025). Physiologically Based Pharmacokinetic Modeling of Tofacitinib: Predicting Drug Exposure and Optimizing Dosage in Special Populations and Drug–Drug Interaction Scenarios. Pharmaceuticals, 18(3), 425. https://doi.org/10.3390/ph18030425