

Exploring Macrocyclic Chemical Space: Strategies and Technologies for Drug Discovery


Abstract

1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Treatment | Activity | Reference |
|---|---|---|---|
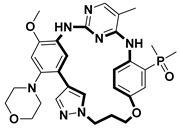 | Anticancer | EGFRT790M/C797S inhibitor | [66] |
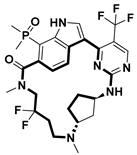 | Anticancer | CDK7 inhibitor | [67] |
 | Prevention of dengue virus infection | EPHA2/A4 and GAK inhibitor | [68] |
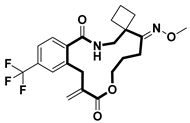 | Virus inhibitor | H1N1 inhibitor | [69] |
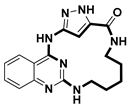 | Anticancer | MST3 inhibitor | [70] |
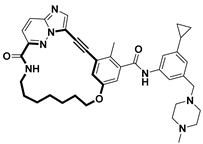 | Anticancer | TRK inhibitor | [71] |
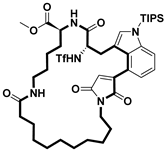 | Virus inhibitor | SARS-CoV-2 inhibitor | [72] |
 | Virus inhibitor | SARS-CoV-2-main protease inhibitor | [73] |
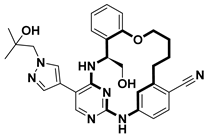 | Anticancer | HPK1 inhibitor | [74] |
2. Modular Biomimetic Assembly Strategies for the Synthesis of Pseudonatural Macrocyclic Compounds
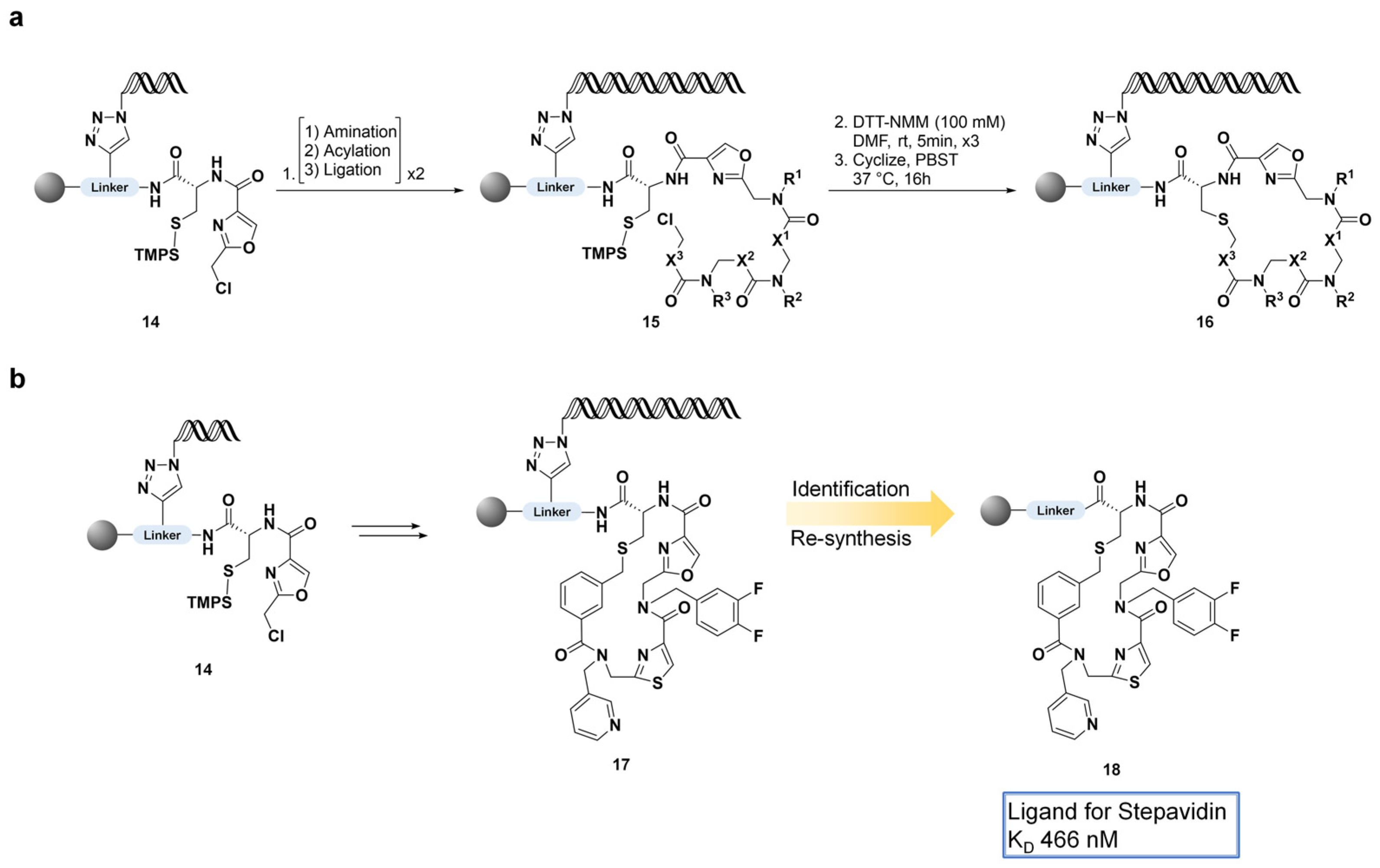
3. DNA-Encoded Macrocycle Libraries (DELs)
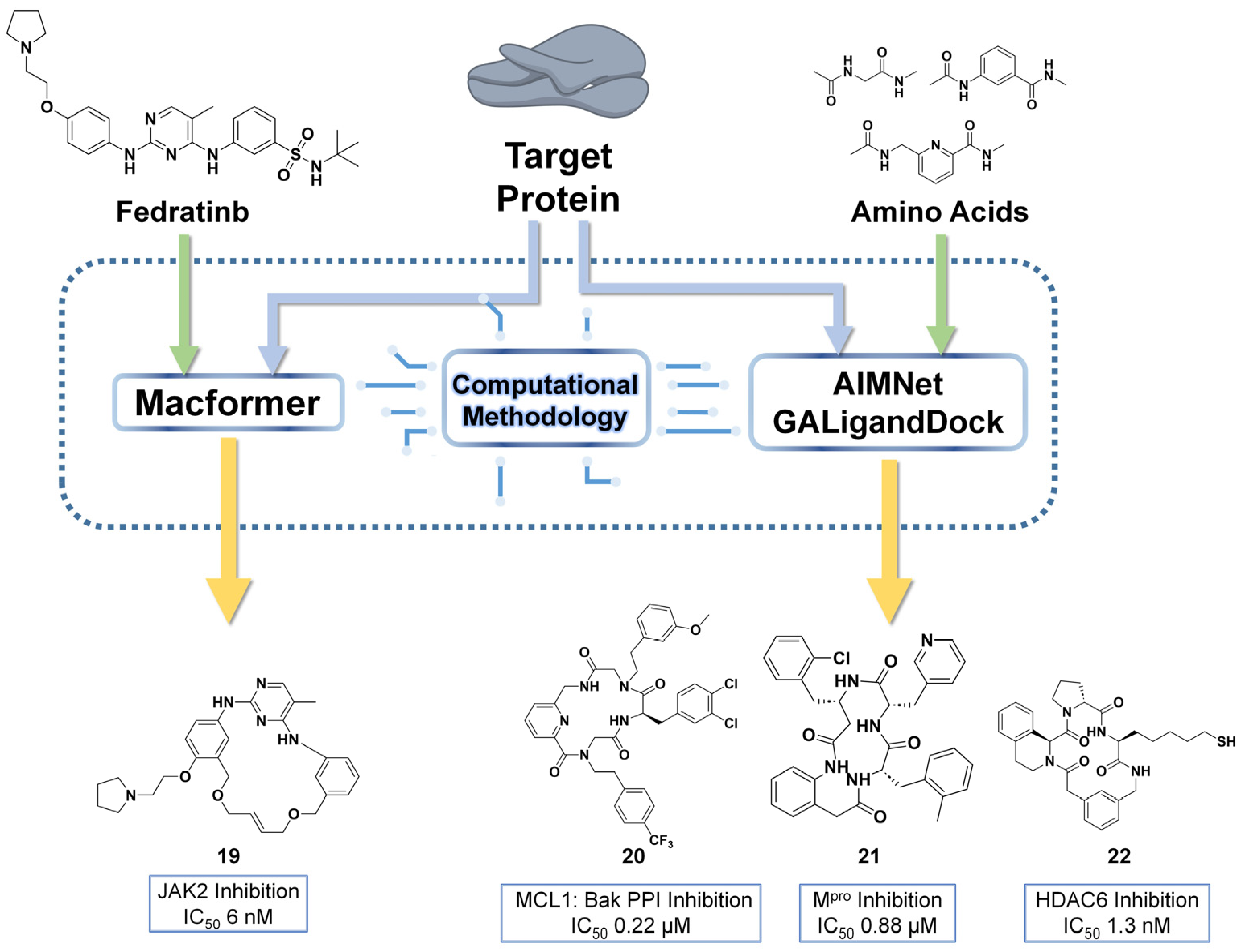
4. Computational Design of Macrocycles
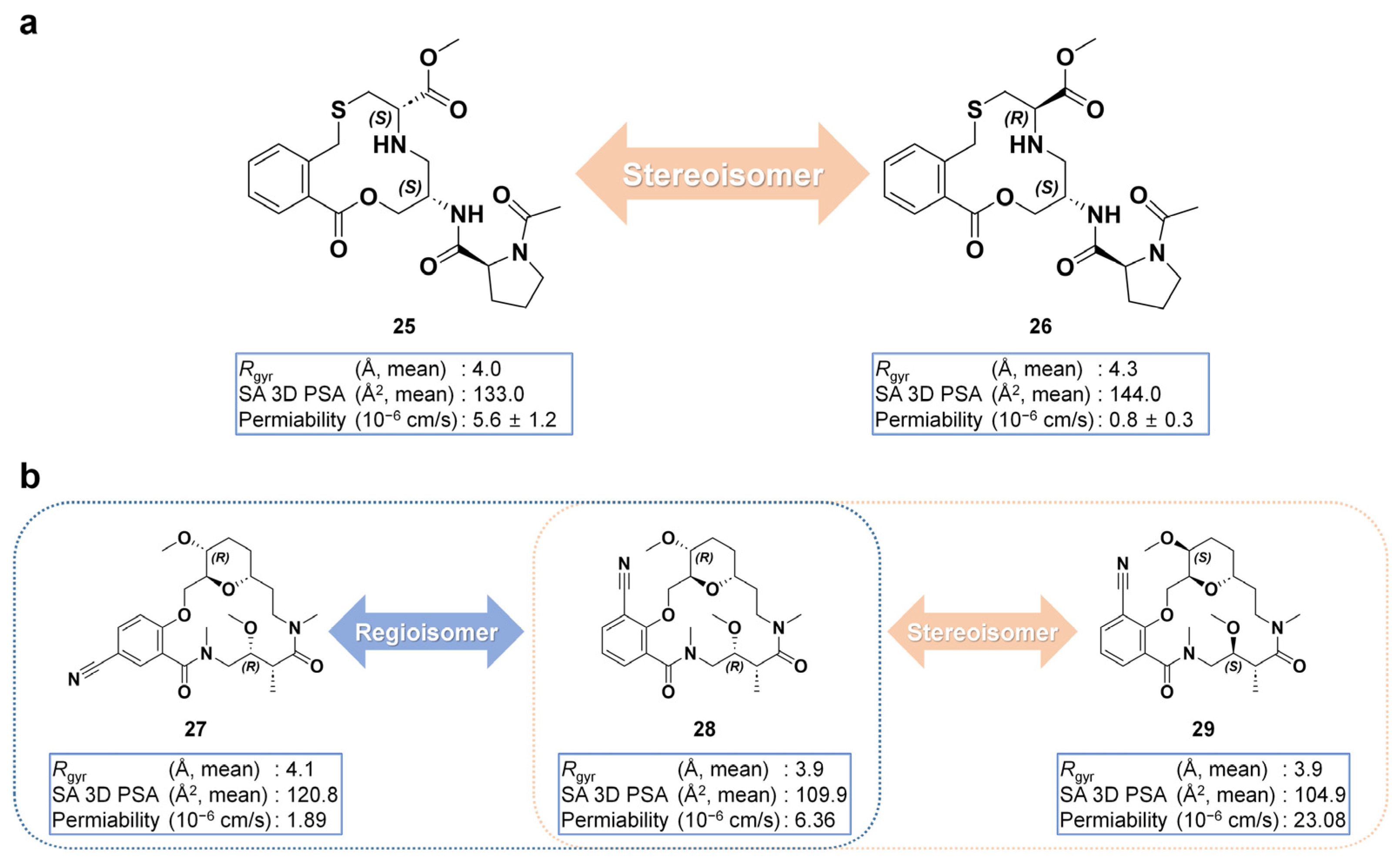
5. Cell Permeability and Bioavailability of Macrocycles
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kang, E.J.; Lee, E. Total Synthesis of Oxacyclic Macrodiolide Natural Products. Chem. Rev. 2005, 105, 4348–4378. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Sasaki, M.; Hattori, I.; Sakai, M.; Tanino, K. Total Synthesis of Norzoanthamine. Science 2004, 305, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Kaki, R.; Takeda, Y.; Hiyama, T.; Nagai, N.; Yamagishi, H.; Furutani, H. 1,4-Bis(diarylamino)-2,5-bis(4-cyanophenylethenyl)benzenes: Fluorophores Exhibiting Efficient Red and Near-Infrared Emissions in Solid State. Angew. Chem. Int. Ed. 2012, 51, 4095–4099. [Google Scholar] [CrossRef]
- Sloman, D.L.; Bacon, J.W.; Porco, J.A., Jr. Total Synthesis and Absolute Stereochemical Assignment of Kibdelone C. J. Am. Chem. Soc. 2011, 133, 9952–9955. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.-L.; Zhou, J. Catalytic Asymmetric Synthesis of Oxindoles Bearing a Tetrasubstituted Stereocenter at the C-3 Position. Adv. Synth. Catal. 2010, 352, 1381–1407. [Google Scholar] [CrossRef]
- Brown, D.G.; Boström, J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef]
- Zhuo, C.-X.; Zhang, W.; You, S.-L. Catalytic Asymmetric Dearomatization Reactions. Angew. Chem. Int. Ed. 2012, 51, 12662–12686. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.A.; Luhavaya, H.; Li, J.; Dahesh, S.; Nizet, V.; Yamanaka, K.; Moore, B.S. Isolation and structure elucidation of lipopeptide antibiotic taromycin B from the activated taromycin biosynthetic gene cluster. J. Antibiot. 2018, 71, 333–338. [Google Scholar] [CrossRef]
- Byun, W.S.; Bae, E.S.; Cui, J.; Park, H.J.; Oh, D.-C.; Lee, S.K. Antitumor Activity of Pulvomycin via Targeting Activated-STAT3 Signaling in Docetaxel-Resistant Triple-Negative Breast Cancer Cells. Biomedicines 2021, 9, 436. [Google Scholar] [CrossRef] [PubMed]
- Smits, T.H.M.; Duffy, B.; Blom, J.; Ishimaru, C.A.; Stockwell, V.O.; Pantocin, A. A peptide-derived antibiotic involved in biological control by plant-associated Pantoea species. Arch. Microbiol. 2019, 201, 713–722. [Google Scholar] [CrossRef]
- Guo, C.; Mandalapu, D.; Ji, X.; Gao, J.; Zhang, Q. Chemistry and Biology of Teixobactin. Chem.-Eur. J. 2018, 24, 5406–5422. [Google Scholar] [CrossRef]
- Weiss, C.; Figueras, E.; Borbely, A.N.; Sewald, N. Cryptophycins: Cytotoxic cyclodepsipeptides with potential for tumor targeting. J. Pept. Sci. 2017, 23, 514–531. [Google Scholar] [CrossRef]
- Gajski, G.; Garaj-Vrhovac, V. Melittin: A lytic peptide with anticancer properties. Environ. Toxicol. Pharmacol. 2013, 36, 697–705. [Google Scholar] [CrossRef]
- Kanda, Y.; Nakamura, H.; Umemiya, S.; Puthukanoori, R.K.; Murthy Appala, V.R.; Gaddamanugu, G.K.; Paraselli, B.R.; Baran, P.S. Two-Phase Synthesis of Taxol. J. Am. Chem. Soc. 2020, 142, 10526–10533. [Google Scholar] [CrossRef]
- Corbett, K.M.; Ford, L.; Warren, D.B.; Pouton, C.W.; Chalmers, D.K. Cyclosporin Structure and Permeability: From A to Z and Beyond. J. Med. Chem. 2021, 64, 13131–13151. [Google Scholar] [CrossRef]
- Ong, S.C.; Gaston, R.S. Thirty Years of Tacrolimus in Clinical Practice. Transplantation 2021, 105, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Robertson, A.A.B.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L.; Vaishnav, P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Kärkäs, M.D.; Porco, J.A., Jr.; Stephenson, C.R.J. Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 116, 9683–9747. [Google Scholar] [CrossRef]
- Cantrell, C.L.; Dayan, F.E.; Duke, S.O. Natural Products As Sources for New Pesticides. J. Nat. Prod. 2012, 75, 1231–1242. [Google Scholar] [CrossRef]
- Garcia Jimenez, D.; Poongavanam, V.; Kihlberg, J. Macrocycles in Drug Discovery—Learning from the Past for the Future. J. Med. Chem. 2023, 66, 5377–5396. [Google Scholar] [CrossRef]
- Marsault, E.; Peterson, M.L. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. [Google Scholar] [CrossRef]
- Terrett, N.K. Methods for the synthesis of macrocycle libraries for drug discovery. Drug Discov. Today Technol. 2010, 7, e97–e104. [Google Scholar] [CrossRef]
- Doak, B.C.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem. 2016, 59, 2312–2327. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Floriano, R.S.; Isaacs, L.; Rowan, E.G.; Wheate, N.J. The ex vivo neurotoxic, myotoxic and cardiotoxic activity of cucurbituril-based macrocyclic drug delivery vehicles. Toxicol. Res. 2014, 3, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Aydin, C.; Gildemeister, R.; Romano, K.P.; Cao, H.; Özen, A.; Soumana, D.; Newton, A.; Petropoulos, C.J.; Huang, W.; et al. Evaluating the Role of Macrocycles in the Susceptibility of Hepatitis C Virus NS3/4A Protease Inhibitors to Drug Resistance. ACS Chem. Biol. 2013, 8, 1469–1478. [Google Scholar] [CrossRef]
- Swenson, C.S.; Mandava, G.; Thomas, D.M.; Moellering, R.E. Tackling Undruggable Targets with Designer Peptidomimetics and Synthetic Biologics. Chem. Rev. 2024, 124, 13020–13093. [Google Scholar] [CrossRef]
- Naylor, M.R.; Bockus, A.T.; Blanco, M.-J.; Lokey, R.S. Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets. Curr. Opin. Chem. Biol. 2017, 38, 141–147. [Google Scholar] [CrossRef]
- Colas, K.; Bindl, D.; Suga, H. Selection of Nucleotide-Encoded Mass Libraries of Macrocyclic Peptides for Inaccessible Drug Targets. Chem. Rev. 2024, 124, 12213–12241. [Google Scholar] [CrossRef]
- Jin, X.; Ding, N.; Guo, H.-Y.; Hu, Q. Macrocyclic-based strategy in drug design: From lab to the clinic. Eur. J. Med. Chem. 2024, 277, 116733. [Google Scholar] [CrossRef]
- Meeprasert, A.; Hannongbua, S.; Rungrotmongkol, T. Key Binding and Susceptibility of NS3/4A Serine Protease Inhibitors against Hepatitis C Virus. J. Chem. Inf. Model. 2014, 54, 1208–1217. [Google Scholar] [CrossRef]
- Viarengo-Baker, L.A.; Brown, L.E.; Rzepiela, A.A.; Whitty, A. Defining and navigating macrocycle chemical space. Chem. Sci. 2021, 12, 4309–4328. [Google Scholar] [CrossRef]
- Magpusao, A.N.; Rutledge, K.; Hamlin, T.A.; Lawrence, J.-M.; Mercado, B.Q.; Leadbeater, N.E.; Peczuh, M.W. Rules of Macrocycle Topology: A [13]-Macrodilactone Case Study. Chem.-Eur. J. 2016, 22, 6001–6011. [Google Scholar] [CrossRef]
- Fang, P.; Pang, W.-K.; Xuan, S.; Chan, W.-L.; Leung, K.C.-F. Recent advances in peptide macrocyclization strategies. Chem. Soc. Rev. 2024, 53, 11725–11771. [Google Scholar] [CrossRef] [PubMed]
- Amrhein, J.A.; Knapp, S.; Hanke, T. Synthetic Opportunities and Challenges for Macrocyclic Kinase Inhibitors. J. Med. Chem. 2021, 64, 7991–8009. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Wang, M.; Horsman, M.; Boddy, C.N. 6-Deoxyerythronolide B Synthase Thioesterase-Catalyzed Macrocyclization Is Highly Stereoselective. Org. Lett. 2012, 14, 2278–2281. [Google Scholar] [CrossRef]
- Anderl, F.; Größl, S.; Wirtz, C.; Fürstner, A. Total Synthesis of Belizentrin Methyl Ester: Report on a Likely Conquest. Angew. Chem. Int. Ed. 2018, 57, 10712–10717. [Google Scholar] [CrossRef]
- Sengupta, S.; Mehta, G. Macrocyclization via C–H functionalization: A new paradigm in macrocycle synthesis. Org. Biomol. Chem. 2020, 18, 1851–1876. [Google Scholar] [CrossRef]
- Fürstner, A. Lessons from Natural Product Total Synthesis: Macrocyclization and Postcyclization Strategies. Accounts Chem. Res. 2021, 54, 861–874. [Google Scholar] [CrossRef]
- Doyle, M.P.; Peterson, C.S.; Protopopova, M.N.; Marnett, A.B.; Parker, D.L.; Ene, D.G.; Lynch, V. Macrocycle Formation by Catalytic Intramolecular Cyclopropanation. A New General Methodology for the Synthesis of Macrolides. J. Am. Chem. Soc. 1997, 119, 8826–8837. [Google Scholar] [CrossRef]
- Saridakis, I.; Kaiser, D.; Maulide, N. Unconventional Macrocyclizations in Natural Product Synthesis. ACS Cent. Sci. 2020, 6, 1869–1889. [Google Scholar] [CrossRef]
- Feng, W.; Yamato, K.; Yang, L.; Ferguson, J.S.; Zhong, L.; Zou, S.; Yuan, L.; Zeng, X.C.; Gong, B. Efficient Kinetic Macrocyclization. J. Am. Chem. Soc. 2009, 131, 2629–2637. [Google Scholar] [CrossRef]
- Yuan, L.; Feng, W.; Yamato, K.; Sanford, A.R.; Xu, D.; Guo, H.; Gong, B. Highly Efficient, One-Step Macrocyclizations Assisted by the Folding and Preorganization of Precursor Oligomers. J. Am. Chem. Soc. 2004, 126, 11120–11121. [Google Scholar] [CrossRef]
- Forsythe, P.; Paterson, S. Ciclosporin 10 years on: Indications and efficacy. Vet. Rec. 2014, 174, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Fujimura, T.; Iwasaki, K.; Sakuma, S.; Fujitsu, T.; Nakamura, K.; Shimomura, K.; Kuno, T.; Tanaka, C.; Kobayashi, M. Interaction of Tacrolimus (FK506) and Its Metabolites with FKBP and Calcineurin. Biochem. Biophys. Res. Commun. 1994, 202, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Bolanowski, M.; Kałużny, M.; Witek, P.; Jawiarczyk-Przybyłowska, A. Pasireotide—A novel somatostatin receptor ligand after 20 years of use. Rev. Endocr. Metab. Disord. 2022, 23, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, K.; Qi, S.; Kraus, J.; Ly, B.; Srinivasan, K.; Tariga, H.; Croston, G.; La, E.; Wiśniewska, H.; Ortiz, C.; et al. Discovery of Potent, Selective, and Short-Acting Peptidic V2 Receptor Agonists. J. Med. Chem. 2019, 62, 4991–5005. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Setmelanotide: First Approval. Drugs 2021, 81, 397–403. [Google Scholar] [CrossRef]
- Duggan, S. Vosoritide: First Approval. Drugs 2021, 81, 2057–2062. [Google Scholar] [CrossRef]
- Lamb, Y.N. Pacritinib: First Approval. Drugs 2022, 82, 831–838. [Google Scholar] [CrossRef]
- Dhillon, S. Repotrectinib: First Approval. Drugs 2024, 84, 239–246. [Google Scholar] [CrossRef]
- Syed, Y.Y. Rezafungin: First Approval. Drugs 2023, 83, 833–840. [Google Scholar] [CrossRef]
- Beckmann HS, G.; Nie, F.; Hagerman, C.E.; Johansson, H.; Tan, Y.S.; Wilcke, D.; Spring, D.R. A strategy for the diversity-oriented synthesis of macrocyclic scaffolds using multidimensional coupling. Nat. Chem. 2013, 5, 861–867. [Google Scholar] [CrossRef]
- Collins, S.; Bartlett, S.; Nie, F.; Sore, H.F.; Spring, D.R. Diversity-Oriented Synthesis of Macrocycle Libraries for Drug Discovery and Chemical Biology. Synthesis 2016, 48, 1457–1473. [Google Scholar] [CrossRef]
- Grossmann, A.; Bartlett, S.; Janecek, M.; Hodgkinson, J.T.; Spring, D.R. Diversity-Oriented Synthesis of Drug-Like Macrocyclic Scaffolds Using an Orthogonal Organo- and Metal Catalysis Strategy. Angew. Chem. Int. Ed. 2014, 53, 13093–13097. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, S.H.; Iampolska, A.D.; Radchenko, D.S.; Vashchenko, B.V.; Voitenko, Z.V.; Grygorenko, O.O. A Diversity-Oriented Approach to Large Libraries of Artificial Macrocycles. Eur. J. Org. Chem. 2021, 2021, 2313–2330. [Google Scholar] [CrossRef]
- Ciardiello, J.J.; Galloway, W.R.J.D.; O’Connor, C.J.; Sore, H.F.; Stokes, J.E.; Wu, Y.; Spring, D.R. An expedient strategy for the diversity-oriented synthesis of macrocyclic compounds with natural product-like characteristics. Tetrahedron 2016, 72, 3567–3578. [Google Scholar] [CrossRef]
- Ciardiello, J.J.; Stewart, H.L.; Sore, H.F.; Galloway, W.R.J.D.; Spring, D.R. A novel complexity-to-diversity strategy for the diversity-oriented synthesis of structurally diverse and complex macrocycles from quinine. Bioorg. Med. Chem. 2017, 25, 2825–2843. [Google Scholar] [CrossRef]
- Hu, S.; Tong, L.; Qin, Q.; Wen, J.; Li, Y.; Feng, F.; Wu, K.; Zhou, Y.; Shang, J.; Wang, J.; et al. Design, Synthesis, and Biological Evaluation of Novel Diaminopyrimidine Macrocycles as Fourth Generation Reversible EGFR Inhibitors That Overcome Clinical Resistance to Osimertinib Mediated by C797S Mutation. J. Med. Chem. 2024, 67, 20531–20558. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, Y.; Liu, J.; Jiang, T.; Yu, X.; Zhang, H.l; Liang, T.; Peng, J.; Cai, X.; Lan, X.; et al. Discovery of a Novel Macrocyclic Noncovalent CDK7 Inhibitor for Cancer Therapy. J. Med. Chem. 2024, 67, 20580–20594. [Google Scholar] [CrossRef]
- Gerninghaus, J.; Zhubi, R.; Krämer, A.; Karim, M.; Tran, D.H.N.; Joerger, A.C.; Schreiber, C.; Berger, L.M.; Berger, B.-T.; Ehret, T.A.L.; et al. Back-Pocket Optimization of 2-Aminopyrimidine-Based Macrocycles Leads to Potent EPHA2/GAK Kinase Inhibitors. J. Med. Chem. 2024, 67, 12534–12552. [Google Scholar] [CrossRef]
- Xu, D.; Gong, Y.; Zhang, L.; Xiao, F.; Wang, X.; Qin, J.; Tan, L.; Yang, T.; Lin, Z.; Xu, Z.; et al. Modular Biomimetic Strategy Enables Discovery and SAR Exploration of Oxime Macrocycles as Influenza A Virus (H1N1) Inhibitors. J. Med. Chem. 2024, 67, 8201–8224. [Google Scholar] [CrossRef]
- Amrhein, J.A.; Berger, L.M.; Balourdas, D.-I.; Joerger, A.C.; Menge, A.; Krämer, A.; Frischkorn, J.M.; Berger, B.-T.; Elson, L.; Kaiser, A.; et al. Synthesis of Pyrazole-Based Macrocycles Leads to a Highly Selective Inhibitor for MST3. J. Med. Chem. 2024, 67, 674–690. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Wang, Y.; Xiang, S.; Zhou, H.; Song, S.; Song, X.; Tu, Z.; Zhou, Y.; Ding, K.; et al. Structure-Based Optimization of the Third Generation Type II Macrocycle TRK Inhibitors with Improved Activity against Solvent-Front, xDFG, and Gatekeeper Mutations. J. Med. Chem. 2023, 66, 12950–12965. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, J.; Zhang, X.; Zhang, R.; Wang, Q.; Wang, L.; Zhang, L.; Xie, X.; Li, C.; Zhou, Y.; et al. Synthesis of maleimide-braced peptide macrocycles and their potential anti-SARS-CoV-2 mechanisms. Chem. Commun. 2023, 59, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Mostafa, A.; Mostafa, Y.A.; Abou-Ghadir, O.F.; Abdelazeem, A.H.; Gouda, A.M.; Kutkat, O.; Abo Shama, N.M.; Shehata, M.; Gomaa, H.A.M.; et al. Discovery of novel oxazole-based macrocycles as anti-coronaviral agents targeting SARS-CoV-2 main protease. Bioorg. Chem. 2021, 116, 105363. [Google Scholar] [CrossRef]
- Wang, M.-S.; Wang, Z.-Z.; Li, Z.-L.; Gong, Y.; Duan, C.-X.; Cheng, Q.-H.; Huang, W.; Yang, G.-F. Discovery of Macrocycle-Based HPK1 Inhibitors for T-Cell-Based Immunotherapy. J. Med. Chem. 2023, 66, 611–626. [Google Scholar] [CrossRef]
- Mulligan, V.K. The emerging role of computational design in peptide macrocycle drug discovery. Expert. Opin. Drug Discov. 2020, 15, 833–852. [Google Scholar] [CrossRef]
- Jain, A.N.; Cleves, A.E.; Gao, Q.; Wang, X.; Liu, Y.; Sherer, E.C.; Reibarkh, M.Y. Complex macrocycle exploration: Parallel, heuristic, and constraint-based conformer generation using ForceGen. J. Comput.-Aided Mol. Des. 2019, 33, 531–558. [Google Scholar] [CrossRef]
- Yu, H.S.; Deng, Y.; Wu, Y.; Sindhikara, D.; Rask, A.R.; Kimura, T.; Abel, R.; Wang, L. Accurate and Reliable Prediction of the Binding Affinities of Macrocycles to Their Protein Targets. J. Chem. Theory Comput. 2017, 13, 6290–6300. [Google Scholar] [CrossRef]
- Allen, S.E.; Dokholyan, N.V.; Bowers, A.A. Dynamic Docking of Conformationally Constrained Macrocycles: Methods and Applications. ACS Chem. Biol. 2016, 11, 10–24. [Google Scholar] [CrossRef]
- Alogheli, H.; Olanders, G.; Schaal, W.; Brandt, P.; Karlén, A. Docking of Macrocycles: Comparing Rigid and Flexible Docking in Glide. J. Chem. Inf. Model. 2017, 57, 190–202. [Google Scholar] [CrossRef]
- Tian, J.-H.; Zheng, Z.; Pan, Y.-C.; Wang, Y.; Guo, D.-S. Macrocycle-based differential sensing: Design strategies and applications. Responsive Mater. 2025, 3, e20240036. [Google Scholar] [CrossRef]
- Zhai, S.; Tan, Y.; Zhu, C.; Zhang, C.; Gao, Y.; Mao, Q.; Zhang, Y.; Duan, H.; Yin, Y. PepExplainer: An explainable deep learning model for selection-based macrocyclic peptide bioactivity prediction and optimization. Eur. J. Med. Chem. 2024, 275, 116628. [Google Scholar] [CrossRef] [PubMed]
- Nangia, A.K. Importance of Structural Databases, Molecular Pharmacophores, Supramolecular Heterosynthons, and Artificial Intelligence–Machine Learning–Neural Network Tools in Drug Discovery. Cryst. Growth Des. 2024, 24, 6888–6910. [Google Scholar] [CrossRef]
- Yu, Y.; Huang, J.; He, H.; Han, J.; Ye, G.; Xu, T.; Sun, X.; Chen, X.; Ren, X.; Li, C.; et al. Accelerated Discovery of Macrocyclic CDK2 Inhibitor QR-6401 by Generative Models and Structure-Based Drug Design. ACS Med. Chem. Lett. 2023, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Chen, I.J.; Chan, A.W.E.; Foloppe, N. Modelling the binding mode of macrocycles: Docking and conformational sampling. Bioorg. Med. Chem. 2020, 28, 115143. [Google Scholar] [CrossRef]
- Over, B.; Matsson, P.; Tyrchan, C.; Artursson, P.; Doak, B.C.; Foley, M.A.; Hilgendorf, C.; Johnston, S.E.; Lee, M.D.; Lewis, R.J.; et al. Structural and conformational determinants of macrocycle cell permeability. Nat. Chem. Biol. 2016, 12, 1065–1074. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Doak, B.C.; Backlund, M.; Poongavanam, V.; Over, B.; Ermondi, G.; Caron, G.; Matsson, P.; Kihlberg, J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. [Google Scholar] [CrossRef]
- Song, B.; Guo, X.; Yang, L.; Yu, H.; Zong, X.; Liu, X.; Wang, H.; Xu, Z.; Lin, Z.; Yang, W. Rhodium(III)-Catalyzed C-H/O2 Dual Activation and Macrocyclization: Synthesis and Evaluation of Pyrido[2,1-a]isoindole Grafted Macrocyclic Inhibitors for Influenza H1N1. Angew. Chem. Int. Ed. 2023, 62, e202218886. [Google Scholar] [CrossRef]
- Bi, T.; Cui, Y.; Liu, S.; Yu, H.; Qiu, W.; Hou, K.-Q.; Zou, J.; Yu, Z.; Zhang, F.; Xu, Z.; et al. Ligand-Enabled Pd-Catalyzed sp3 C−H Macrocyclization: Synthesis and Evaluation of Macrocyclic Sulfonamide for the Treatment of Parkinson’s Disease. Angew. Chem. Int. Ed. 2024, 63, e202412296. [Google Scholar] [CrossRef]
- Koesema, E.; Roy, A.; Paciaroni, N.G.; Coito, C.; Tokmina-Roszyk, M.; Kodadek, T. Synthesis and Screening of a DNA-Encoded Library of Non-Peptidic Macrocycles. Angew. Chem. Int. Ed. 2022, 61, e202116999. [Google Scholar] [CrossRef]
- Diao, Y.; Liu, D.; Ge, H.; Zhang, R.; Jiang, K.; Bao, R.; Zhu, X.; Bi, H.; Liao, W.; Chen, Z.; et al. Macrocyclization of linear molecules by deep learning to facilitate macrocyclic drug candidates discovery. Nat. Commun. 2023, 14, 4552. [Google Scholar] [CrossRef]
- Salveson, P.J.; Moyer, A.P.; Said, M.Y.; Gӧkçe, G.; Li, X.; Kang, A.; Nguyen, H.; Bera, A.K.; Levine, P.M.; Bhardwaj, G.; et al. Expansive discovery of chemically diverse structured macrocyclic oligoamides. Science 2024, 384, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Begnini, F.; Poongavanam, V.; Atilaw, Y.; Erdelyi, M.; Schiesser, S.; Kihlberg, J. Cell Permeability of Isomeric Macrocycles: Predictions and NMR Studies. ACS Med. Chem. Lett. 2021, 12, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Quan, H.; Xu, Z.; Wang, H.; Xia, Y.; Lou, L.; Yang, W. A modular biomimetic strategy for the synthesis of macrolide P-glycoprotein inhibitors via Rh-catalyzed C-H activation. Nat. Commun. 2020, 11, 2151. [Google Scholar] [CrossRef] [PubMed]
- Rivera, D.G.; Ojeda-Carralero, G.M.; Reguera, L.; Van der Eycken, E.V. Peptide macrocyclization by transition metal catalysis. Chem. Soc. Rev. 2020, 49, 2039–2059. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Pasini, D. The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures. Molecules 2013, 18, 9512–9530. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I. General Method for Synthesis of Functionalized Macrocycles and Catenanes Utilizing “Click” Chemistry. J. Am. Chem. Soc. 2008, 130, 12872–12873. [Google Scholar] [CrossRef]
- Bodine, K.D.; Gin, D.Y.; Gin, M.S. Synthesis of Readily Modifiable Cyclodextrin Analogues via Cyclodimerization of an Alkynyl−Azido Trisaccharide. J. Am. Chem. Soc. 2004, 126, 1638–1639. [Google Scholar] [CrossRef]
- Ojima, I.; Lin, S.; Inoue, T.; Miller, M.L.; Borella, C.P.; Geng, X.; Walsh, J.J. Macrocycle Formation by Ring-Closing Metathesis. Application to the Syntheses of Novel Macrocyclic Taxoids. J. Am. Chem. Soc. 2000, 122, 5343–5353. [Google Scholar] [CrossRef]
- Lee, C.W.; Grubbs, R.H. Formation of Macrocycles via Ring-Closing Olefin Metathesis. J. Org. Chem. 2001, 66, 7155–7158. [Google Scholar] [CrossRef]
- Mannocci, L.; Leimbacher, M.; Wichert, M.; Scheuermann, J.; Neri, D. 20 years of DNA-encoded chemical libraries. Chem. Commun. 2011, 47, 12747–12753. [Google Scholar] [CrossRef] [PubMed]
- Gironda-Martínez, A.; Donckele, E.J.; Samain, F.; Neri, D. DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci. 2021, 4, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Yuen, L.H.; Franzini, R.M. Achievements, Challenges, and Opportunities in DNA-Encoded Library Research: An Academic Point of View. ChemBioChem 2017, 18, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Y.; Li, X. Strategies for developing DNA-encoded libraries beyond binding assays. Nat. Chem. 2022, 14, 129–140. [Google Scholar] [CrossRef]
- Paulsen, J.L.; Yu, H.S.; Sindhikara, D.; Wang, L.; Appleby, T.; Villaseñor, A.G.; Schmitz, U.; Shivakumar, D. Evaluation of Free Energy Calculations for the Prioritization of Macrocycle Synthesis. J. Chem. Inf. Model. 2020, 60, 3489–3498. [Google Scholar] [CrossRef]
- Ono, T.; Tabata, K.V.; Goto, Y.; Saito, Y.; Suga, H.; Noji, H.; Morimoto, J.; Sando, S. Label-free quantification of passive membrane permeability of cyclic peptides across lipid bilayers: Penetration speed of cyclosporin A across lipid bilayers. Chem. Sci. 2023, 14, 345–349. [Google Scholar] [CrossRef]
- Sethio, D.; Poongavanam, V.; Xiong, R.; Tyagi, M.; Duy Vo, D.; Lindh, R.; Kihlberg, J. Simulation Reveals the Chameleonic Behavior of Macrocycles. J. Chem. Inf. Model. 2023, 63, 138–146. [Google Scholar] [CrossRef]
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21, 712–717. [Google Scholar] [CrossRef]
- Dinsmore, C.J.; Bogusky, M.J.; Culberson, J.C.; Bergman, J.M.; Homnick, C.F.; Zartman, C.B.; Mosser, S.D.; Schaber, M.D.; Robinson, R.G.; Koblan, K.S.; et al. Conformational Restriction of Flexible Ligands Guided by the Transferred NOE Experiment: Potent Macrocyclic Inhibitors of Farnesyltransferase. J. Am. Chem. Soc. 2001, 123, 2107–2108. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Davies, N.L.; James, K. Comparison of diffusion coefficients for matched pairs of macrocyclic and linear molecules over a drug-like molecular weight range. Org. Biomol. Chem. 2011, 9, 7727–7733. [Google Scholar] [CrossRef]
- Okumu, F.W.; Pauletti, G.M.; Vander Velde, D.G.; Siahaan, T.J.; Borchardt, R.T. Effect of Restricted Conformational Flexibility on the Permeation of Model Hexapeptides Across Caco-2 Cell Monolayers. Pharm. Res. 1997, 14, 169–175. [Google Scholar] [CrossRef]
- Kwon, Y.-U.; Kodadek, T. Quantitative Comparison of the Relative Cell Permeability of Cyclic and Linear Peptides. Chem. Biol. 2007, 14, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Parker, J.; Tong, J.; Kodadek, T. Synthesis of Membrane-Permeable Macrocyclic Peptides via Imidazopyridinium Grafting. J. Am. Chem. Soc. 2024, 146, 14633–14644. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Eng, H.; Farley, K.A.; Goetz, G.H.; Huang, Y.; Jiao, Z.; Kalgutkar, A.S.; Kablaoui, N.M.; Khunte, B.; Liras, S.; et al. Comparative pharmacokinetic profile of cyclosporine (CsA) with a decapeptide and a linear analogue. Org. Biomol. Chem. 2017, 15, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, A.; Blaise, É.; Boudreault, P.-L.; Comeau, C.; Doucet, A.; Giarrusso, M.; Collin, M.-P.; Neubauer, T.; Kölling, F.; Göller, A.H.; et al. Structure–Permeability Relationship of Semipeptidic Macrocycles—Understanding and Optimizing Passive Permeability and Efflux Ratio. J. Med. Chem. 2020, 63, 6774–6783. [Google Scholar] [CrossRef]
- Zhang, H.; Ginn, J.; Zhan, W.; Liu, Y.J.; Leung, A.; Toita, A.; Okamoto, R.; Wong, T.-T.; Imaeda, T.; Hara, R.; et al. Design, Synthesis, and Optimization of Macrocyclic Peptides as Species-Selective Antimalaria Proteasome Inhibitors. J. Med. Chem. 2022, 65, 9350–9375. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, F.; Zhang, Y.; Song, C. Macrocycles and macrocyclization in anticancer drug discovery: Important pieces of the puzzle. Eur. J. Med. Chem. 2024, 268, 116234. [Google Scholar] [CrossRef]
- Abdelraouf, K.; Braggs Kirk, H.; Yin, T.; Truong Luan, D.; Hu, M.; Tam Vincent, H. Characterization of Polymyxin B-Induced Nephrotoxicity: Implications for Dosing Regimen Design. Antimicrob. Agents Chemother. 2012, 56, 4625–4629. [Google Scholar] [CrossRef]
- Shakour, H.; Mumtaz, R.; Abu-Samra, A. A Diagnostic Dilemma: Daptomycin-Induced Drug Reaction With Eosinophilia and Systemic Symptoms (DRESS) Syndrome and Eosinophilic Pneumonia With Concurrent COVID-19. Cureus 2025, 17, e77736. [Google Scholar] [CrossRef]



| Structure | Name | Treatment | Target | FDA Approved Year |
|---|---|---|---|---|
 | Cyclosporin [51] | Immunosuppressant | Cyclophilin-1 | 1983 |
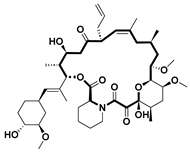 | Tacrolimus [52] | Immunosuppressant | FKBP-12 | 1994 |
 | Pasireotide [53] | Orphan drug | Somatostatin receptor | 2012 |
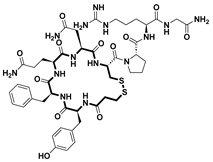 | Desmopressin [54] | Nocturia treatment | Vasopressin V2 receptors | 2018 |
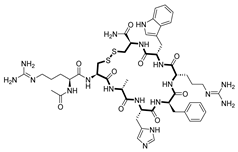 | Setmelanotide [55] | Chronic weight management | Melanocortin-4 receptor | 2021 |
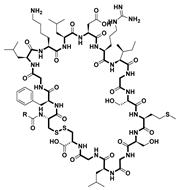 | Vosoritide [56] | Achondroplasia treatment | Natriuretic peptide receptor B (NPR-B) | 2022 |
 | Pacritinib [57] | Anticancer | JAK2 | 2022 |
 | Repotrectinib [58] | Anticancer | Poto-oncogene tyrosine- protein kinase ROS1 and TRKs | 2023 |
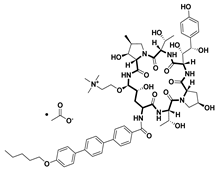 | Rezafungin acetate [59] | Antifungal drug | 1,3-β-D-glucan synthase | 2023 |
| Strategies | Entry 1 | Entry 2 | Reference | ||
|---|---|---|---|---|---|
| Structure | Permeability | Structure | Permeability | ||
| Functional group modification | 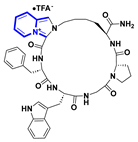 | 5.94 a | 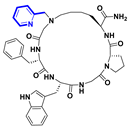 | 7.58 a | [113] |
| Cyclization | 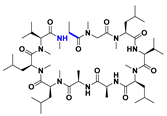 | 0.6 b | 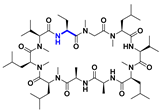 | 5.4 b | [114] |
| Backbone constitution |  | 4.6 a |  | 5.6 a | [115] |
| Stereoinversion | 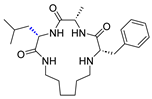 | 21.5 c |  | 53.5 c | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.; Baek, E.; Kim, J. Exploring Macrocyclic Chemical Space: Strategies and Technologies for Drug Discovery. Pharmaceuticals 2025, 18, 617. https://doi.org/10.3390/ph18050617
Kim T, Baek E, Kim J. Exploring Macrocyclic Chemical Space: Strategies and Technologies for Drug Discovery. Pharmaceuticals. 2025; 18(5):617. https://doi.org/10.3390/ph18050617
Chicago/Turabian StyleKim, Taegwan, Eunbee Baek, and Jonghoon Kim. 2025. "Exploring Macrocyclic Chemical Space: Strategies and Technologies for Drug Discovery" Pharmaceuticals 18, no. 5: 617. https://doi.org/10.3390/ph18050617
APA StyleKim, T., Baek, E., & Kim, J. (2025). Exploring Macrocyclic Chemical Space: Strategies and Technologies for Drug Discovery. Pharmaceuticals, 18(5), 617. https://doi.org/10.3390/ph18050617