

Methionine Dependency and Restriction in Cancer: Exploring the Pathogenic Function and Therapeutic Potential


Abstract
:1. Introduction
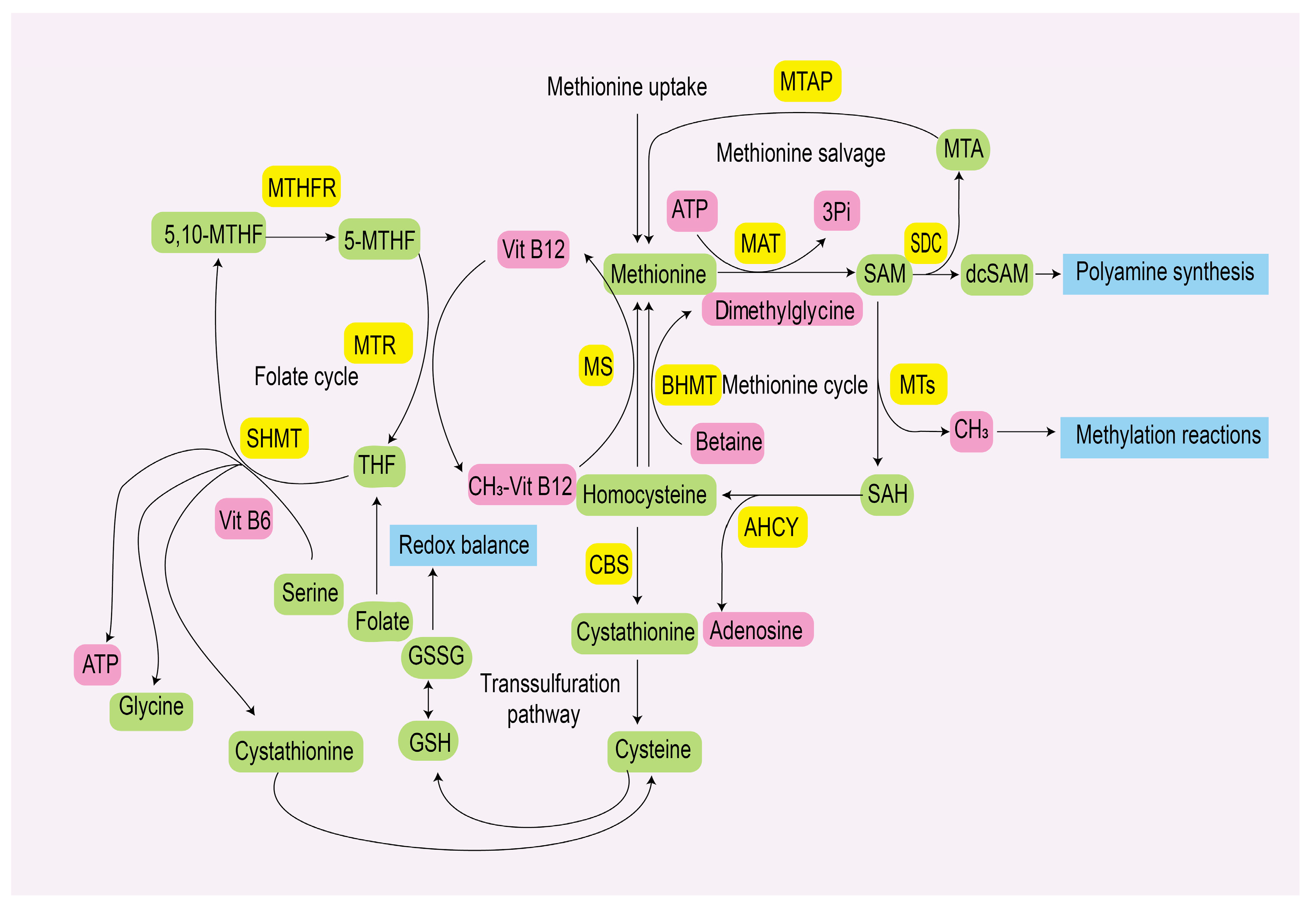
2. Methionine Metabolism
2.1. Methionine Metabolic Pathway
2.2. Methionine Uptake Pathway
3. Methionine Metabolism in Cancer Cells
3.1. Methionine Metabolism and Epigenetic Modification
3.1.1. DNA Methylation
3.1.2. RNA Methylation
3.1.3. Histone Methylation
3.1.4. Other Methionine-Related Modifications
3.2. Methionine Metabolism and Ferroptosis
3.3. Methionine Metabolism and Synthetic Lethality
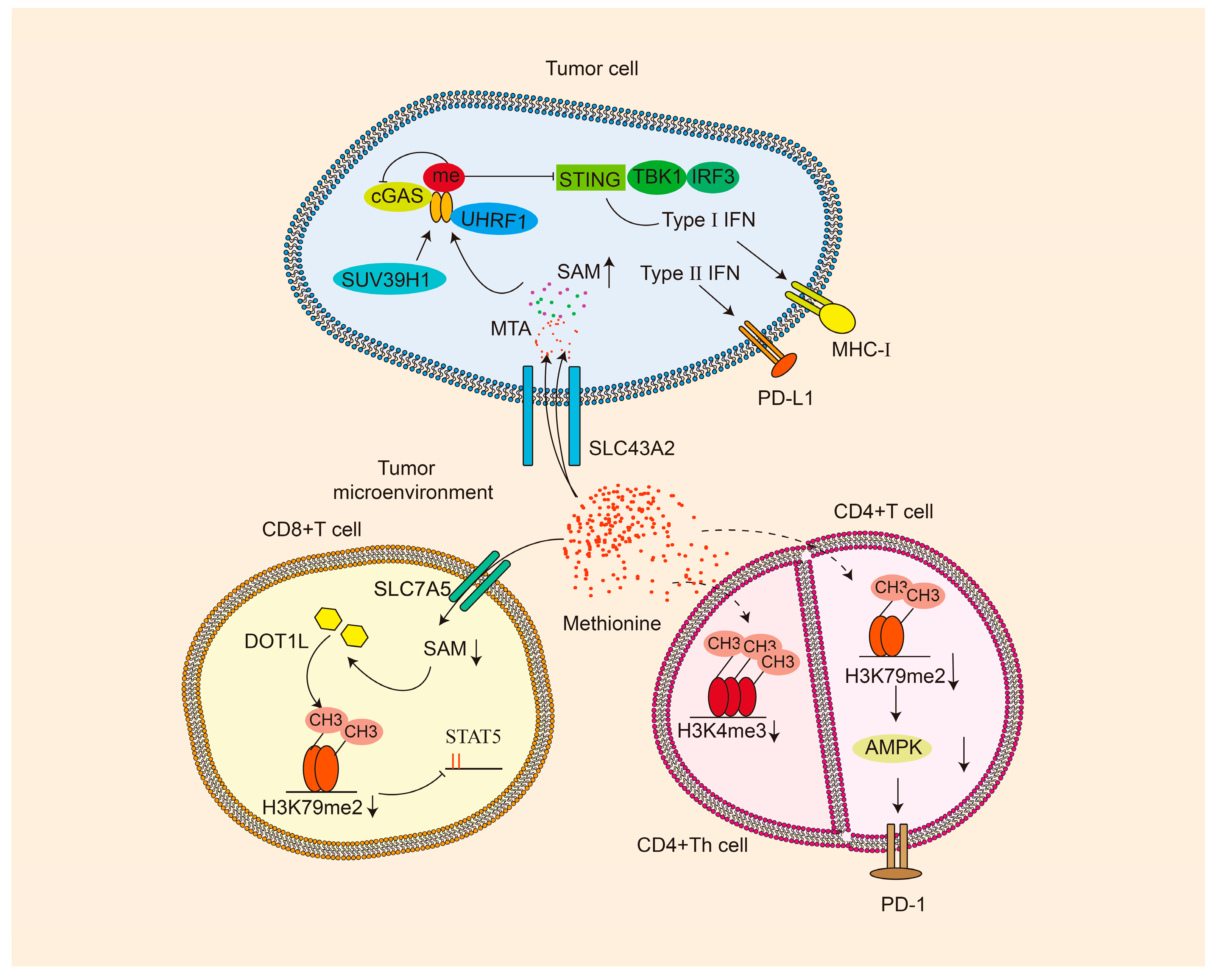
4. Methionine and Tumor Microenvironment
5. Therapeutic Strategies Targeting Methionine in Cancer

{kind=link}
{kind=link}
{kind=link}
| Treatment | Compounds | Clinical Stage | Applications | Reference |
|---|---|---|---|---|
| Direct methionine restriction | MRD/DMR | phase II Clinical | MRD combined with cystemustine treatment for melanoma or glioma | [160] |
| rMETases | phase I Clinical | High-stage cancer patients | [161] | |
| hCGL | Preclinical | Prostate cancer | [162] | |
| MGL S3 | Preclinical | Neuroblastoma, breast cancer, non-small cell lung carcinoma, colon cancer and epidermoid carcinoma | [163,164] | |
| Erymet | Preclinical | Colorectal carcinoma, glioblastoma, gastric and pancreatic cancers | [83,165] | |
| SYNB1353 | Preclinical | / | [166] | |
| SGN1 | phase I/IIa Clinical | Advanced solid tumor patients | [167] | |
| Indirect methionine restriction | Cycloleucine | Preclinical | / | [168,169] |
| PF-9366 (MAT2A inhibitor) | Preclinical | Lung carcinoma, leukemia | [15,170] | |
| FIDAS-3/FIDAS-5 (MAT2A inhibitor) | Preclinical | Colon cancer, multiple myeloma | [171,172] | |
| AGI-24512/AGI-25696/AG-270/AGI-41998 (MAT2A inhibitor) | Preclinical/phase I Clinical | Advanced solid tumors or lymphoma with MTAP loss | [117,121,173,174] | |
| IDE397 (MAT2A inhibitor) | phase I/II Clinical | Solid tumors harboring MTAP deletion | [175,176] | |
| BCH (SLC7A5 and SLC43A2 inhibitor) | Preclinical | Lung cancer, gastrointestinal cancer | [94,177] | |
| KMH-233 (SLC7A5 inhibitor) | Preclinical | Breast cancer cells | [178] | |
| JPH203 (SLC7A5 inhibitor) | Preclinical/phase I Clinical | Prostate and thyroid cancer | [179,180,181] |
5.1. Direct Methionine Restriction
5.1.1. Methionine Restriction Diet
5.1.2. Methionine γ Lyase
5.2. Indirect Methionine Restriction
5.2.1. MAT2A Inhibitors
Cycloleucine
PF-9366
FIDAS-3, FIDAS-5
Other Small Molecule Compounds
5.2.2. SLC Transporters Inhibitors
5.3. Adaptive Resistance of Methionine Restriction Therapy
5.4. Clinical Translation of Methionine Restriction Therapy
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SAM | S-adenosyl-methionine |
| TME | Tumor microenvironment |
| MATs | Methionine adenosyltransferases |
| SAH | S-Adenosylhomocysteine |
| 5-MTHF | 5-methyltetrahydrofolate |
| MTA | Methylthioadenosine |
| MTAP | Methylthioadenosine phosphorylase |
| 5,10-MTHF | 5,10-methylene tetrahydrofolate |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| DNMTs | DNA methyltransferases |
| CpG | Cytosine–phosphate–guanine |
| DAPK1 | Death-associated protein kinase 1 |
| PUMA | P53 upregulated modulator of apoptosis |
| BNIP3 | BCL2 interacting protein 3 |
| lncRNA | Long non-coding RNA |
| PET | Positron emission tomography |
| WTAP | Wilms tumor 1-associated protein |
| SLC | Solute carriers |
| PD-L1 | Programmed cell death ligand 1 |
| VISTA | V-domain immunoglobulin suppressor of T cell activation |
| ATAD2 | ATPase family AAA domain-containing protein 2 |
| PRMT | Protein arginine methyltransferase |
| ACSL3 | Acyl-CoA synthetase long chain family member 3 |
| TNBC | Triple-negative breast cancer |
| PARP | Poly (ADP-ribose) polymerase |
| CSCs | Tumor stem cells |
| ROS | Reactive oxygen species |
| MCR | Methionine/cystine restriction |
| CHAC1 | Cation transport regulator homolog 1 |
| MR | Methionine restriction |
| MAPK14 | Mitogen-activated protein kinase P38 |
| MK2 | MAPK-activated protein kinase 2 |
| GSH | Glutathione |
| NF-κB | Nuclear factor kappa B |
| PD-1 | Programmed cell death protein 1 |
| CAR-T | Chimeric antigen receptor T-cell |
| cGAS | Cyclic GMP-AMP synthase |
| UHRF1 | Ubiquitin-like with plant homeodomain and RING finger domains 1 |
| STING | Stimulator of interferon genes |
| MGL | Methionine γ lyase |
| hCGL | Human enzyme cystathionine-γ-lyase |
| MRD | Methionine restriction diet |
| rMETase | Recombinant methioninase |
References
- Sugimura, T.; Birnbaum, S.M.; Winitz, M.; Greenstein, J.P. Quantitative nutritional studies with water-soluble, chemically defined diets. VIII. The forced feeding of diets each lacking in one essential amino acid. Arch. Biochem. Biophys. 1959, 81, 448–455. [Google Scholar] [CrossRef]
- Aoki, Y.; Han, Q.; Tome, Y.; Yamamoto, J.; Kubota, Y.; Masaki, N.; Obara, K.; Hamada, K.; Wang, J.D.; Inubushi, S.; et al. Reversion of methionine addiction of osteosarcoma cells to methionine independence results in loss of malignancy, modulation of the epithelial-mesenchymal phenotype and alteration of histone-H3 lysine-methylation. Front. Oncol. 2022, 12, 1009548. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.; Erbe, R.W. High in vivo rates of methionine biosynthesis in transformed human and malignant rat cells auxotrophic for methionine. Proc. Natl. Acad. Sci. USA 1976, 73, 1523–1527. [Google Scholar] [CrossRef]
- Tisdale, M.J. Effect of methionine replacement by homocysteine on the growth of cells. Cell Biol. Int. Rep. 1980, 4, 563–570. [Google Scholar] [CrossRef]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Halpern, B.C.; Clark, B.R.; Hardy, D.N.; Halpern, R.M.; Smith, R.A. The effect of replacement of methionine by homocystine on survival of malignant and normal adult mammalian cells in culture. Proc. Natl. Acad. Sci. USA 1974, 71, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Y.; Li, X.; Wang, Y.; Zhang, Z.; Wang, Z.; Zhang, X. Participation of protein metabolism in cancer progression and its potential targeting for the management of cancer. Amino Acids 2023, 55, 1223–1246. [Google Scholar] [CrossRef]
- Chen, X.; Yi, C.; Yang, M.-J.; Sun, X.; Liu, X.; Ma, H.; Li, Y.; Li, H.; Wang, C.; He, Y.; et al. Metabolomics study reveals the potential evidence of metabolic reprogramming towards the Warburg effect in precancerous lesions. J. Cancer 2021, 12, 1563–1574. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, Y.; Zhou, Y.; Liu, Q.; Chen, X.; Liu, X.; Grune, T.; Shi, L.; Hou, M.; Liu, Z. Effects of methionine intake on cognitive function in mild cognitive impairment patients and APP/PS1 Alzheimer’s Disease model mice: Role of the cystathionine-β-synthase/H2S pathway. Redox Biol. 2023, 59, 102595. [Google Scholar] [CrossRef]
- Parkhitko, A.A.; Jouandin, P.; Mohr, S.E.; Perrimon, N. Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 2019, 18, e13034. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Jiang, L.; Zhu, Y.; Yang, S.; Qiu, H.; Cheng, J.; Liang, Q.; Tu, Z.C.; Ye, C. Methionine restriction constrains lipoylation and activates mitochondria for nitrogenic synthesis of amino acids. Nat. Commun. 2023, 14, 2504. [Google Scholar] [CrossRef]
- Wang, L.; Hu, B.; Pan, K.; Chang, J.; Zhao, X.; Chen, L.; Lin, H.; Wang, J.; Zhou, G.; Xu, W.; et al. SYVN1-MTR4-MAT2A Signaling Axis Regulates Methionine Metabolism in Glioma Cells. Front. Cell Dev. Biol. 2021, 9, 633259. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Kaiser, S.E.; Bolaños, B.; Nowlin, D.; Grantner, R.; Karlicek-Bryant, S.; Feng, J.L.; Jenkinson, S.; Freeman-Cook, K.; Dann, S.G.; et al. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 2017, 13, 785–792. [Google Scholar] [CrossRef]
- Zhao, L.; Su, H.; Liu, X.; Wang, H.; Feng, Y.; Wang, Y.; Chen, H.; Dai, L.; Lai, S.; Xu, S.; et al. mTORC1-c-Myc pathway rewires methionine metabolism for HCC progression through suppressing SIRT4 mediated ADP ribosylation of MAT2A. Cell Biosci. 2022, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef]
- Villa, E.; Sahu, U.; O’Hara, B.P.; Ali, E.S.; Helmin, K.A.; Asara, J.M.; Gao, P.; Singer, B.D.; Ben-Sahra, I. mTORC1 stimulates cell growth through SAM synthesis and m6A mRNA-dependent control of protein synthesis. Mol. Cell 2021, 81, 2076–2093.e9. [Google Scholar] [CrossRef]
- Aoki, Y.; Han, Q.; Kubota, Y.; Masaki, N.; Obara, K.; Tome, Y.; Bouvet, M.; Nishida, K.; Hoffman, R.M. Oncogenes and Methionine Addiction of Cancer: Role of c-MYC. Cancer Genom. Proteom. 2023, 20, 165–170. [Google Scholar] [CrossRef]
- Strekalova, E.; Malin, D.; Weisenhorn, E.M.M.; Russell, J.D.; Hoelper, D.; Jain, A.; Coon, J.J.; Lewis, P.W.; Cryns, V.L. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res. Treat. 2019, 175, 39–50. [Google Scholar] [CrossRef]
- Alam, M.; Shima, H.; Matsuo, Y.; Long, N.C.; Matsumoto, M.; Ishii, Y.; Sato, N.; Sugiyama, T.; Nobuta, R.; Hashimoto, S.; et al. mTORC1-independent translation control in mammalian cells by methionine adenosyltransferase 2A and S-adenosylmethionine. J. Biol. Chem. 2022, 298, 102084. [Google Scholar] [CrossRef]
- Sun, L.L.; He, H.Y.; Li, W.; Jin, W.L.; Wei, Y.J. The solute carrier transporters (SLCs) family in nutrient metabolism and ferroptosis. Biomark. Res. 2024, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Yan, Y.; He, M.; Li, J.; Wang, L.; Jia, W.; Yang, L.; Jiang, J.; Chen, Y.; Li, F.; et al. SLC43A2 and NFκB signaling pathway regulate methionine/cystine restriction-induced ferroptosis in esophageal squamous cell carcinoma via a feedback loop. Cell Death Dis. 2023, 14, 347. [Google Scholar] [CrossRef] [PubMed]
- Palacín, M.; Nunes, V.; Font-Llitjós, M.; Jiménez-Vidal, M.; Fort, J.; Gasol, E.; Pineda, M.; Feliubadaló, L.; Chillarón, J.; Zorzano, A. The genetics of heteromeric amino acid transporters. Physiology 2005, 20, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Howden, A.J.; Brenes, A.; Spinelli, L.; Hukelmann, J.L.; Macintyre, A.N.; Liu, X.; Thomson, S.; Taylor, P.M.; Rathmell, J.C.; et al. Antigen receptor control of methionine metabolism in T cells. eLife 2019, 8, e44210. [Google Scholar] [CrossRef]
- Huovinen, R.; Leskinen-Kallio, S.; Någren, K.; Lehikoinen, P.; Ruotsalainen, U.; Teräs, M. Carbon-11-methionine and PET in evaluation of treatment response of breast cancer. Br. J. Cancer 1993, 67, 787–791. [Google Scholar] [CrossRef]
- Jin, X.; Liu, L.; Liu, D.; Wu, J.; Wang, C.; Wang, S.; Wang, F.; Yu, G.; Jin, X.; Xue, Y.W.; et al. Unveiling the methionine cycle: A key metabolic signature and NR4A2 as a methionine-responsive oncogene in esophageal squamous cell carcinoma. Cell Death Differ. 2024, 31, 558–573. [Google Scholar] [CrossRef]
- Bergström, M.; Collins, V.P.; Ehrin, E.; Ericson, K.; Eriksson, L.; Greitz, T.; Halldin, C.; von Holst, H.; Långström, B.; Lilja, A.; et al. Discrepancies in brain tumor extent as shown by computed tomography and positron emission tomography using [68Ga]EDTA, [11C]glucose, and [11C]methionine. J. Comput. Assist. Tomogr. 1983, 7, 1062–1066. [Google Scholar] [CrossRef]
- Li, J.; Ni, B.; Yu, X.; Wang, C.; Li, L.; Zhou, Y.; Gu, Y.; Huang, G.; Hou, J.; Liu, J.; et al. Metabolic kinetic modeling of [(11)C]methionine based on total-body PET in multiple myeloma. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 2683–2691. [Google Scholar] [CrossRef]
- Jansson, T.; Westlin, J.E.; Ahlström, H.; Lilja, A.; Långström, B.; Bergh, J. Positron emission tomography studies in patients with locally advanced and/or metastatic breast cancer: A method for early therapy evaluation? J. Clin. Oncol. 1995, 13, 1470–1477. [Google Scholar] [CrossRef]
- Fujiwara, T.; Matsuzawa, T.; Kubota, K.; Abe, Y.; Itoh, M.; Fukuda, H.; Hatazawa, J.; Yoshioka, S.; Yamaguchi, K.; Ito, K. Relationship between histologic type of primary lung cancer and carbon-11-L-methionine uptake with positron emission tomography. J. Nucl. Med. 1989, 30, 33–37. [Google Scholar]
- Hoffman, R.M.; Stern, P.H.; Coalson, D.W.; Wallace, C.D.; Erbe, R.W. Altered Methionine Metabolism in Cancer Cells. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer Nature: London, UK, 2019; Volume 1866, pp. 13–26. [Google Scholar]
- Cunningham, A.; Erdem, A.; Alshamleh, I.; Geugien, M.; Pruis, M.; Pereira-Martins, D.A.; van den Heuvel, F.A.J.; Wierenga, A.T.J.; Ten Berge, H.; Dennebos, R.; et al. Dietary methionine starvation impairs acute myeloid leukemia progression. Blood 2022, 140, 2037–2052. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Is DNA methylation the new guardian of the genome? Mol. Cytogenet. 2017, 10, 11. [Google Scholar] [CrossRef]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.-K.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef]
- Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells 2019, 8, 1322. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, L.; Lin, H.; Wang, J.; Fu, J.; Zhu, D.; Xu, W. Inhibition of MAT2A-Related Methionine Metabolism Enhances The Efficacy of Cisplatin on Cisplatin-Resistant Cells in Lung Cancer. Cell J. 2022, 24, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Inubushi, S.; Han, Q.; Tashiro, Y.; Sugisawa, N.; Hamada, K.; Aoki, Y.; Miyake, K.; Matsuyama, R.; Bouvet, M.; et al. Linkage of methionine addiction, histone lysine hypermethylation, and malignancy. iScience 2022, 25, 104162. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Birsoy, K. The Transsulfuration Pathway Makes, the Tumor Takes. Cell Metab. 2019, 30, 845–846. [Google Scholar] [CrossRef]
- Yamamoto, J.; Han, Q.; Inubushi, S.; Sugisawa, N.; Hamada, K.; Nishino, H.; Miyake, K.; Kumamoto, T.; Matsuyama, R.; Bouvet, M.; et al. Histone methylation status of H3K4me3 and H3K9me3 under methionine restriction is unstable in methionine-addicted cancer cells, but stable in normal cells. Biochem. Biophys. Res. Commun. 2020, 533, 1034–1038. [Google Scholar] [CrossRef]
- Garg, S.; Morehead, L.C.; Bird, J.T.; Graw, S.; Gies, A.; Storey, A.J.; Tackett, A.J.; Edmondson, R.D.; Mackintosh, S.G.; Byrum, S.D.; et al. Characterization of methionine dependence in melanoma cells. Mol. Omics 2024, 20, 37–47. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Liu, J.; Wang, X.; Teng, L.; Zhang, J.; Liu, Y.; Yao, Y.; Wang, J.; Qu, Y.; et al. IL1RN mediates the suppressive effect of methionine deprivation on glioma proliferation. Cancer Lett. 2019, 454, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.-W.; Chung, B.P.; Kaiser, P. S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J. Cell Sci. 2014, 127, 50–59. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 210. [Google Scholar] [CrossRef]
- Dai, X.; Ren, T.; Zhang, Y.; Nan, N. Methylation multiplicity and its clinical values in cancer. Expert. Rev. Mol. Med. 2021, 23, e2. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Murin, R.; Vidomanova, E.; Kowtharapu, B.S.; Hatok, J.; Dobrota, D. Role of S-adenosylmethionine cycle in carcinogenesis. Gen. Physiol. Biophys. 2017, 36, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Xu, Y.; Zheng, M.; Luo, C.; Lei, H.; Qu, H.; Shu, D. Methionine Attenuates Lipopolysaccharide-Induced Inflammatory Responses via DNA Methylation in Macrophages. Acs Omega 2019, 4, 2331–2336. [Google Scholar] [CrossRef]
- Tong, D.; Zhang, J.; Wang, X.; Li, Q.; Liu, L.; Lu, A.; Guo, B.; Yang, J.; Ni, L.; Qin, H.; et al. MiR-22, regulated by MeCP2, suppresses gastric cancer cell proliferation by inducing a deficiency in endogenous S-adenosylmethionine. Oncogenesis 2020, 9, 99. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Berglund, A.; Estrella, V.; Zhang, Y.; Liu, M.; Putney, R.M.; Yoder, S.J.; Johnson, J.; Brown, J.; Gatenby, R. Elevated Methionine Flux Drives Pyroptosis Evasion in Persister Cancer Cells. Cancer Res. 2023, 83, 720–734. [Google Scholar] [CrossRef]
- Aoki, Y.; Tome, Y.; Han, Q.; Yamamoto, J.; Hamada, K.; Masaki, N.; Bouvet, M.; Nishida, K.; Hoffman, R.M. Histone H3 lysine-trimethylation markers are decreased by recombinant methioninase and increased by methotrexate at concentrations which inhibit methionine-addicted osteosarcoma cell proliferation. Biochem. Biophys. Res. Commun. 2021, 28, 101177. [Google Scholar] [CrossRef]
- Hussain, S.; Tulsyan, S.; Dar, S.A.; Sisodiya, S.; Abiha, U.; Kumar, R.; Mishra, B.N.; Haque, S. Role of epigenetics in carcinogenesis: Recent advancements in anticancer therapy. Semin. Cancer Biol. 2022, 83, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. Int. J. Mol. Sci. 2023, 24, 2346. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11, 608. [Google Scholar] [CrossRef]
- Bhootra, S.; Jill, N.; Shanmugam, G.; Rakshit, S.; Sarkar, K. DNA methylation and cancer: Transcriptional regulation, prognostic, and therapeutic perspective. Med. Oncol. 2023, 40, 71. [Google Scholar] [CrossRef]
- Besselink, N.; Keijer, J.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E. The genome-wide mutational consequences of DNA hypomethylation. Sci. Rep. 2023, 13, 6874. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Long, M.D.; Smiraglia, D.J.; Campbell, M.J. The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer. Biomolecules 2017, 7, 15. [Google Scholar] [CrossRef]
- Hoffman, R.M. Is the Hoffman Effect for Methionine Overuse Analogous to the Warburg Effect for Glucose Overuse in Cancer? In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 273–278. [Google Scholar]
- Waterland, R.A. Assessing the effects of high methionine intake on DNA methylation. J. Nutr. 2006, 136, 1706s–1710s. [Google Scholar] [CrossRef]
- Hearnden, V.; Powers, H.J.; Elmogassabi, A.; Lowe, R.; Murdoch, C. Methyl-donor depletion of head and neck cancer cells in vitro establishes a less aggressive tumour cell phenotype. Eur. J. Nutr. 2018, 57, 1321–1332. [Google Scholar] [CrossRef]
- Xin, L.; Lu, H.; Liu, C.; Zeng, F.; Yuan, Y.-W.; Wu, Y.; Wang, J.-L.; Wu, D.-Z.; Zhou, L.-Q. Methionine deficiency promoted mitophagy via lncRNA PVT1-mediated promoter demethylation of BNIP3 in gastric cancer. Int. J. Biochem. Cell Biol. 2021, 141, 106100. [Google Scholar] [CrossRef]
- Menga, A.; Palmieri, E.M.; Cianciulli, A.; Infantino, V.; Mazzone, M.; Scilimati, A.; Palmieri, F.; Castegna, A.; Iacobazzi, V. SLC25A26 overexpression impairs cell function via mtDNA hypermethylation and rewiring of methyl metabolism. FEBS J. 2017, 284, 967–984. [Google Scholar] [CrossRef]
- Tang, Q.; Li, L.; Wang, Y.; Wu, P.; Hou, X.; Ouyang, J.; Fan, C.; Li, Z.; Wang, F.; Guo, C.; et al. RNA modifications in cancer. Br. J. Cancer 2023, 129, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, J.; Liu, B.; Xu, J.; Cai, B.; Yang, H.; Straube, J.; Yu, X.; Ma, T. Biological roles of RNA m(5)C modification and its implications in Cancer immunotherapy. Biomark. Res. 2022, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, M.; Zhao, Y.L.; Yang, Y.; Yang, Y.G. RNA methylations in human cancers. Semin. Cancer Biol. 2021, 75, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, M.; Zhu, X.; Xing, J.; Zhou, J.; Yin, X. The potential regulatory role of RNA methylation in ovarian cancer. RNA Biol. 2023, 20, 207–218. [Google Scholar] [CrossRef]
- Liang, W.; Yi, H.; Mao, C.; Meng, Q.; Wu, X.; Li, S.; Xue, J. Research Progress of RNA Methylation Modification in Colorectal Cancer. Front. Pharmacol. 2022, 13, 903699. [Google Scholar] [CrossRef]
- Li, T.; Tan, Y.-T.; Chen, Y.-X.; Zheng, X.-J.; Wang, W.; Liao, K.; Mo, H.-Y.; Lin, J.; Yang, W.; Piao, H.-L.; et al. Methionine deficiency facilitates antitumour immunity by altering m6A methylation of immune checkpoint transcripts. Gut 2023, 72, 501–511. [Google Scholar] [CrossRef]
- Zhang, C.; Du, Z.; Gao, Y.; Lim, K.S.; Zhou, W.; Huang, H.; He, H.; Xiao, J.; Xu, D.; Li, Q. Methionine secreted by tumor-associated pericytes supports cancer stem cells in clear cell renal carcinoma. Cell Metab. 2024, 36, 778–792.e10. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, B.; Deng, Y.; Deng, S.; Li, J.; Wei, W.; Wang, Y.; Wang, J.; Feng, Z.; Che, M.; et al. FBW7/GSK3β mediated degradation of IGF2BP2 inhibits IGF2BP2-SLC7A5 positive feedback loop and radioresistance in lung cancer. J. Exp. Clin. Cancer Res. 2024, 43, 34. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Mandumpala, J.J.; Baby, S.; Tom, A.A.; Godugu, C.; Shankaraiah, N. Role of histone demethylases and histone methyltransferases in triple-negative breast cancer: Epigenetic mnemonics. Life Sci. 2022, 292, 120321. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct. Target. Ther. 2020, 5, 143. [Google Scholar] [CrossRef] [PubMed]
- Sutopo, N.C.; Kim, J.H.; Cho, J.Y. Role of histone methylation in skin cancers: Histone methylation-modifying enzymes as a new class of targets for skin cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188865. [Google Scholar] [CrossRef]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. Rev. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.Q.; Yang, G.J.; Ma, D.L.; Wang, W.; Leung, C.H. The role and prospect of lysine-specific demethylases in cancer chemoresistance. Med. Res. Rev. 2023, 43, 1438–1469. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Liu, H.; Mi, R.; Huang, R.; Li, X.; Fan, F.; Xie, X.; Ding, J. The role of histone methylase and demethylase in antitumor immunity: A new direction for immunotherapy. Front. Immunol. 2022, 13, 1099892. [Google Scholar] [CrossRef]
- Aoki, Y.; Yamamoto, J.; Tome, Y.; Hamada, K.; Masaki, N.; Inubushi, S.; Tashiro, Y.; Bouvet, M.; Endo, I.; Nishida, K.; et al. Over-methylation of Histone H3 Lysines Is a Common Molecular Change Among the Three Major Types of Soft-tissue Sarcoma in Patient-derived Xenograft (PDX) Mouse Models. Cancer Genom. Proteom. 2021, 18, 715–721. [Google Scholar] [CrossRef]
- Bialopiotrowicz, E.; Noyszewska-Kania, M.; Kachamakova-Trojanowska, N.; Loboda, A.; Cybulska, M.; Grochowska, A.; Kopczynski, M.; Mikula, M.; Prochorec-Sobieszek, M.; Firczuk, M.; et al. Serine Biosynthesis Pathway Supports MYC-miR-494-EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells. Cancers 2020, 12, 580. [Google Scholar] [CrossRef]
- Yamamoto, J.; Aoki, Y.; Inubushi, S.; Han, Q.; Hamada, K.; Tashiro, Y.; Miyake, K.; Matsuyama, R.; Bouvet, M.; Clarke, S.G.; et al. Extent and Instability of Trimethylation of Histone H3 Lysine Increases with Degree of Malignancy and Methionine Addiction. Cancer Genom. Proteom. 2022, 19, 12–18. [Google Scholar] [CrossRef]
- Tang, X.; Keenan, M.M.; Wu, J.; Lin, C.-A.; Dubois, L.; Thompson, J.W.; Freedland, S.J.; Murphy, S.K.; Chi, J.-T. Comprehensive Profiling of Amino Acid Response Uncovers Unique Methionine-Deprived Response Dependent on Intact Creatine Biosynthesis. PLoS Genet. 2015, 11, e1005158. [Google Scholar] [CrossRef]
- Machover, D.; Rossi, L.; Hamelin, J.; Desterke, C.; Goldschmidt, E.; Chadefaux-Vekemans, B.; Bonnarme, P.; Briozzo, P.; Kopecny, D.; Pierige, F.; et al. Effects in Cancer Cells of the Recombinant L-Methionine Gamma-Lyase from Brevibacterium aurantiacum. Encapsulation in Human Erythrocytes for Sustained L-Methionine Eliminations. J. Pharmacol. Exp. Ther. 2019, 369, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Siblini, Y.; Namour, F.; Oussalah, A.; Gueant, J.-L.; Chery, C. Stemness of Normal and Cancer Cells: The Influence of Methionine Needs and SIRT1/PGC-1 alpha/PPAR-alpha Players. Cells 2022, 11, 3607. [Google Scholar] [CrossRef]
- Panatta, E.; Butera, A.; Mammarella, E.; Pitolli, C.; Mauriello, A.; Leist, M.; Knight, R.A.; Melino, G.; Amelio, I. Metabolic regulation by p53 prevents R-loop-associated genomic instability. Cell Rep. 2022, 41, 111568. [Google Scholar] [CrossRef]
- Raboni, S.; Montalbano, S.; Stransky, S.; Garcia, B.A.; Buschini, A.; Bettati, S.; Sidoli, S.; Mozzarelli, A. A Key Silencing Histone Mark on Chromatin Is Lost When Colorectal Adenocarcinoma Cells Are Depleted of Methionine by Methionine gamma-Lyase. Front. Mol. Biosci. 2021, 8, 735303. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Kong, P.; Huang, Y.; Wang, J.; Liu, X.; Hu, Y.; Chen, X.; Du, C.; Yang, H. Activation of MAT2A-ACSL3 pathway protects cells from ferroptosis in gastric cancer. Free Radic. Biol. Med. 2022, 181, 288–299. [Google Scholar] [CrossRef]
- Liu, C.C.; Chen, L.; Cai, Y.W.; Chen, Y.F.; Liu, Y.M.; Zhou, Y.J.; Shao, Z.M.; Yu, K.D. Targeting EMSY-mediated methionine metabolism is a potential therapeutic strategy for triple-negative breast cancer. Cell Rep. Med. 2024, 5, 101396. [Google Scholar] [CrossRef]
- Li, Y.; Liu, C.; Xin, L.; Liu, C.; Cao, J.; Yue, Z.; Sheng, J.; Yuan, Y.; Zhou, Q.; Liu, Z. Upregulation of E-cadherin by the combination of methionine restriction and HDAC2 intervention for inhibiting gastric carcinoma metastasis. Acta Biochim. Biophys. Sin. 2023, 56, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef]
- Pandit, M.; Kil, Y.S.; Ahn, J.H.; Pokhrel, R.H.; Gu, Y.; Mishra, S.; Han, Y.; Ouh, Y.T.; Kang, B.; Jeong, M.S.; et al. Methionine consumption by cancer cells drives a progressive upregulation of PD-1 expression in CD4 T cells. Nat. Commun. 2023, 14, 2593. [Google Scholar] [CrossRef]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 2020, 31, 250–266.e9. [Google Scholar] [CrossRef]
- Lee, J.M.; Hammarén, H.M.; Savitski, M.M.; Baek, S.H. Control of protein stability by post-translational modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef]
- Hong, X.L.; Huang, C.K.; Qian, H.; Ding, C.H.; Liu, F.; Hong, H.Y.; Liu, S.Q.; Wu, S.H.; Zhang, X.; Xie, W.F. Positive feedback between arginine methylation of YAP and methionine transporter SLC43A2 drives anticancer drug resistance. Nat. Commun. 2025, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhao, R.; Qu, Y.Y.; Mei, X.Y.; Zhang, X.; Zhou, Q.; Li, Y.; Yang, S.B.; Zuo, Z.G.; Chen, Y.M.; et al. Colonic Lysine Homocysteinylation Induced by High-Fat Diet Suppresses DNA Damage Repair. Cell Rep. 2018, 25, 398–412.e6. [Google Scholar] [CrossRef] [PubMed]
- Martínez, Y.; Li, X.; Liu, G.; Bin, P.; Yan, W.; Más, D.; Valdivié, M.; Hu, C.A.; Ren, W.; Yin, Y. The role of methionine on metabolism, oxidative stress, and diseases. Amino Acids 2017, 49, 2091–2098. [Google Scholar] [CrossRef]
- Bai, W.; Cheng, L.; Xiong, L.; Wang, M.; Liu, H.; Yu, K.; Wang, W. Protein succinylation associated with the progress of hepatocellular carcinoma. J. Cell Mol. Med. 2022, 26, 5702–5712. [Google Scholar] [CrossRef]
- Lu, K.; Han, D. A review of the mechanism of succinylation in cancer. Medicine 2022, 101, e31493. [Google Scholar] [CrossRef]
- Shi, L.; Duan, R.; Sun, Z.; Jia, Q.; Wu, W.; Wang, F.; Liu, J.; Zhang, H.; Xue, X. LncRNA GLTC targets LDHA for succinylation and enzymatic activity to promote progression and radioiodine resistance in papillary thyroid cancer. Cell Death Differ. 2023, 30, 1517–1532. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, R.; Wang, Y.; Duan, Y.; Zhan, H. Targeting succinylation-mediated metabolic reprogramming as a potential approach for cancer therapy. Biomed. Pharmacother. 2023, 168, 115713. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Upadhyayula, P.S.; Higgins, D.M.; Mela, A.; Banu, M.; Dovas, A.; Zandkarimi, F.; Patel, P.; Mahajan, A.; Humala, N.; Nguyen, T.T.T.; et al. Dietary restriction of cysteine and methionine sensitizes gliomas to ferroptosis and induces alterations in energetic metabolism. Nat. Commun. 2023, 14, 1187. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Xin, P.; Liu, G.; Wu, J. Biomedical applications of methionine-based systems. Biomater. Sci. 2021, 9, 1961–1973. [Google Scholar] [CrossRef]
- Jaune-Pons, E.; Vasseur, S. Role of amino acids in regulation of ROS balance in cancer. Arch. Biochem. Biophys. 2020, 689, 108438. [Google Scholar] [CrossRef]
- Malin, D.; Lee, Y.; Chepikova, O.; Strekalova, E.; Carlson, A.; Cryns, V.L. Methionine restriction exposes a targetable redox vulnerability of triple-negative breast cancer cells by inducing thioredoxin reductase. Breast Cancer Res. Treat. 2021, 190, 373–387. [Google Scholar] [CrossRef]
- Quere, M.; Alberto, J.-M.; Broly, F.; Hergalant, S.; Christov, C.; Gauchotte, G.; Gueant, J.-L.; Namour, F.; Battaglia-Hsu, S.-F. ALDH1L2 Knockout in U251 Glioblastoma Cells Reduces Tumor Sphere Formation by Increasing Oxidative Stress and Suppressing Methionine Dependency. Nutrients 2022, 14, 1887. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, W.; Wang, K.; Wang, X.; Yin, F.; Li, C.; Wang, C.; Zhao, B.; Zhong, C.; Zhang, J.; et al. Methionine and cystine double deprivation stress suppresses glioma proliferation via inducing ROS/autophagy. Toxicol. Lett. 2015, 232, 349–355. [Google Scholar] [CrossRef]
- Xue, Y.; Lu, F.; Chang, Z.; Li, J.; Gao, Y.; Zhou, J.; Luo, Y.; Lai, Y.; Cao, S.; Li, X.; et al. Intermittent dietary methionine deprivation facilitates tumoral ferroptosis and synergizes with checkpoint blockade. Nat. Commun. 2023, 14, 4758. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.; Poltorack, C.D.; Forcina, G.C.; Armenta, D.A.; Mallais, M.; Perez, M.A.; Wells, A.; Kahanu, A.; Magtanong, L.; Watts, J.L.; et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat. Chem. Biol. 2021, 17, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Bauer, C.; Newman, A.C.; Uribe, A.H.; Athineos, D.; Blyth, K.; Maddocks, O.D.K. Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat. Metab. 2020, 2, 1062–1076. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Peng, P.; Zhang, W.; Xing, X.; Jin, X.; Du, J.; Peng, W.; Hao, F.; Zhao, Z.; Dong, K.; et al. Methionine-SAM metabolism-dependent ubiquinone synthesis is crucial for ROS accumulation in ferroptosis induction. Nat. Commun. 2024, 15, 8971. [Google Scholar] [CrossRef]
- Kalev, P.; Hyer, M.L.; Gross, S.; Konteatis, Z.; Chen, C.C.; Fletcher, M.; Lein, M.; Aguado-Fraile, E.; Frank, V.; Barnett, A.; et al. MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent mRNA Splicing and Inducing DNA Damage. Cancer Cell 2021, 39, 209–224.e11. [Google Scholar] [CrossRef]
- Fan, N.; Zhang, Y.; Zou, S. Methylthioadenosine phosphorylase deficiency in tumors: A compelling therapeutic target. Front. Cell Dev. Biol. 2023, 11, 1173356. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; McDonald, E.R., 3rd; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef]
- Konteatis, Z.; Travins, J.; Gross, S.; Marjon, K.; Barnett, A.; Mandley, E.; Nicolay, B.; Nagaraja, R.; Chen, Y.; Sun, Y.; et al. Discovery of AG-270, a First-in-Class Oral MAT2A Inhibitor for the Treatment of Tumors with Homozygous MTAP Deletion. J. Med. Chem. 2021, 64, 4430–4449. [Google Scholar] [CrossRef]
- Atkinson, S.J.; Evans, L.; Scott, J.S. A patent review of MAT2a inhibitors (2018–2021). Expert. Opin. Ther. Pat. 2022, 32, 1043–1053. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, X.O.; Rozen, E.J.; Sun, X.; Sallis, B.; Verdejo-Torres, O.; Wigglesworth, K.; Moon, D.; Huang, T.; Cavaretta, J.P.; et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 2022, 13, 3955. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Dong, W.; Xie, R.; Wu, J.; Su, Q.; Li, W.; Yao, K.; Chen, Y.; Zhou, Q.; Zhang, Q.; et al. HSF1 facilitates the multistep process of lymphatic metastasis in bladder cancer via a novel PRMT5-WDR5-dependent transcriptional program. Cancer Commun. 2022, 42, 447–470. [Google Scholar] [CrossRef]
- Jiang, Y.; Yuan, Y.; Chen, M.; Li, S.; Bai, J.; Zhang, Y.; Sun, Y.; Wang, G.; Xu, H.; Wang, Z.; et al. PRMT5 disruption drives antitumor immunity in cervical cancer by reprogramming T cell-mediated response and regulating PD-L1 expression. Theranostics 2021, 11, 9162–9176. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Yu, C.; Qiu, C.; Wu, Q.; Huang, C.; Li, X.; She, X.; Wan, K.; Liu, L.; Li, M.; et al. PRMT5 methylating SMAD4 activates TGF-β signaling and promotes colorectal cancer metastasis. Oncogene 2023, 42, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Sloan, S.L.; Brown, F.; Long, M.E.; Weigel, C.; Koirala, S.; Chung, J.H.; Pray, B.; Villagomez, L.; Hinterschied, C.; Sircar, A.; et al. PRMT5 supports multiple oncogenic pathways in mantle cell lymphoma. Blood 2023, 142, 887–902. [Google Scholar] [CrossRef]
- Szewczyk, M.M.; Luciani, G.M.; Vu, V.; Murison, A.; Dilworth, D.; Barghout, S.H.; Lupien, M.; Arrowsmith, C.H.; Minden, M.D.; Barsyte-Lovejoy, D. PRMT5 regulates ATF4 transcript splicing and oxidative stress response. Redox Biol. 2022, 51, 102282. [Google Scholar] [CrossRef]
- Wang, Z.; Li, R.; Hou, N.; Zhang, J.; Wang, T.; Fan, P.; Ji, C.; Zhang, B.; Liu, L.; Wang, Y.; et al. PRMT5 reduces immunotherapy efficacy in triple-negative breast cancer by methylating KEAP1 and inhibiting ferroptosis. J. Immunother. Cancer 2023, 11, e006890. [Google Scholar] [CrossRef]
- Hwang, J.W.; Kim, S.-N.; Myung, N.; Song, D.; Han, G.; Bae, G.-U.; Bedford, M.T.; Kim, Y.K. PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun. Biol. 2020, 3, 428. [Google Scholar] [CrossRef]
- Fu, S.; Zheng, Q.; Zhang, D.; Lin, C.; Ouyang, L.; Zhang, J.; Chen, L. Medicinal chemistry strategies targeting PRMT5 for cancer therapy. Eur. J. Med. Chem. 2022, 244, 114842. [Google Scholar] [CrossRef]
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal 2019, 17, 30. [Google Scholar] [CrossRef]
- Aoki, Y.; Tome, Y.; Han, Q.; Yamamoto, J.; Hamada, K.; Masaki, N.; Kubota, Y.; Bouvet, M.; Nishida, K.; Hoffman, R.M. Deletion of MTAP Highly Sensitizes Osteosarcoma Cells to Methionine Restriction with Recombinant Methioninase. Cancer Genom. Proteom. 2022, 19, 299–304. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Ji, M.; Xu, X.; Xu, Q.; Hsiao, Y.C.; Martin, C.; Ukraintseva, S.; Popov, V.; Arbeev, K.G.; Randall, T.A.; Wu, X.; et al. Methionine restriction-induced sulfur deficiency impairs antitumour immunity partially through gut microbiota. Nat. Metab. 2023, 5, 1526–1543. [Google Scholar] [CrossRef]
- Liao, Y.; Weng, J.; Chen, L.; Hu, N.; Yuan, X.; Wang, J.; He, F.; Cai, Y.; Huang, Q.; Wang, J.; et al. Comprehensive analysis of SLC43A2 on the tumor immune microenvironment and prognosis of liver hepatocellular carcinoma. Front. Genet. 2022, 13, 911378. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chu, Z.; Liu, M.; Zou, Q.; Li, J.; Liu, Q.; Wang, Y.; Wang, T.; Xiang, J.; Wang, B. Amino acid metabolism in immune cells: Essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J. Hematol. Oncol. 2023, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Qiu, Y.; Xu, Y.; Chen, L.; Ma, K.; Tao, M.; Frankiw, L.; Yin, H.; Xie, E.; Pan, X.; et al. Extracellular acidosis restricts one-carbon metabolism and preserves T cell stemness. Nat. Metab. 2023, 5, 314–330. [Google Scholar] [CrossRef]
- Hung, M.H.; Lee, J.S.; Ma, C.; Diggs, L.P.; Heinrich, S.; Chang, C.W.; Ma, L.; Forgues, M.; Budhu, A.; Chaisaingmongkol, J.; et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat. Commun. 2021, 12, 1455. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Hao, Y.; Yu, H.; Gu, X.; Peng, Q.; Zhuo, H.; Li, Y.; Liu, Z.; Wang, J.; Chen, Y.; et al. Methionine restriction promotes cGAS activation and chromatin untethering through demethylation to enhance antitumor immunity. Cancer Cell 2023, 41, 1118–1133.e12. [Google Scholar] [CrossRef]
- Morehead, L.C.; Garg, S.; Wallis, K.F.; Simoes, C.C.; Siegel, E.R.; Tackett, A.J.; Miousse, I.R. Increased Response to Immune Checkpoint Inhibitors with Dietary Methionine Restriction in a Colorectal Cancer Model. Cancers 2023, 15, 4467. [Google Scholar] [CrossRef]
- Binz, R.L.; Sadhukhan, R.; Miousse, I.R.; Garg, S.; Koturbash, I.; Zhou, D.; Hauer-Jensen, M.; Pathak, R. Dietary Methionine Deficiency Enhances Genetic Instability in Murine Immune Cells. Int. J. Mol. Sci. 2021, 22, 2378. [Google Scholar] [CrossRef]
- Yang, C.; Ou, Y.; Zhou, Q.; Liang, Y.; Li, W.; Chen, Y.; Chen, W.; Wu, S.; Chen, Y.; Dai, X.; et al. Methionine orchestrates the metabolism vulnerability in cisplatin resistant bladder cancer microenvironment. Cell Death Dis. 2023, 14, 525. [Google Scholar] [CrossRef] [PubMed]
- Joulia, E.; Metallo, C.M. Methionine and H2S alter cancer-immune dialogue. Nat. Metab. 2023, 5, 1456–1458. [Google Scholar] [CrossRef]
- Yu, T.; Nie, F.Q.; Zhang, Q.; Yu, S.K.; Zhang, M.L.; Wang, Q.; Wang, E.X.; Lu, K.H.; Sun, M. Effects of methionine deficiency on B7H3-DAP12-CAR-T cells in the treatment of lung squamous cell carcinoma. Cell Death Dis. 2024, 15, 12. [Google Scholar] [CrossRef]
- Vega, A.A.; Marshall, E.A.; Noonan, A.J.C.; Filho, F.S.L.; Yang, J.; Stewart, G.L.; Johnson, F.D.; Vucic, E.A.; Pewarchuk, M.E.; Shah, P.P.; et al. Methionine-producing tumor micro(be) environment fuels growth of solid tumors. Cell. Oncol. 2023, 46, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Nan, D.; Yao, W.; Huang, L.; Liu, R.; Chen, X.; Xia, W.; Sheng, H.; Zhang, H.; Liang, X.; Lu, Y. Glutamine and cancer: Metabolism, immune microenvironment, and therapeutic targets. Cell Commun. Signal. 2025, 23, 45. [Google Scholar] [CrossRef]
- Bjelosevic, S.; Gruber, E.; Newbold, A.; Shembrey, C.; Devlin, J.R.; Hogg, S.J.; Kats, L.; Todorovski, I.; Fan, Z.; Abrehart, T.C.; et al. Serine Biosynthesis Is a Metabolic Vulnerability in FLT3-ITD-Driven Acute Myeloid Leukemia. Cancer Discov. 2021, 11, 1582–1599. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef]
- Do, L.K.; Lee, H.M.; Ha, Y.S.; Lee, C.H.; Kim, J. Amino acids in cancer: Understanding metabolic plasticity and divergence for better therapeutic approaches. Cell Rep. 2025, 44, 115529. [Google Scholar] [CrossRef]
- Dong, C.; Zhao, Y.; Han, Y.; Li, M.; Wang, G. Targeting glutamine metabolism crosstalk with tumor immune response. Biochim. Biophys. Acta Rev. Cancer 2025, 1880, 189257. [Google Scholar] [CrossRef]
- Kim, M.; Hwang, S.; Kim, B.; Shin, S.; Yang, S.; Gwak, J.; Jeong, S.M. YAP governs cellular adaptation to perturbation of glutamine metabolism by regulating ATF4-mediated stress response. Oncogene 2023, 42, 2828–2840. [Google Scholar] [CrossRef]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Hoffman, R.M.; Bertino, J.R. Exploiting methionine restriction for cancer treatment. Biochem. Pharmacol. 2018, 154, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Bertino, J.R. Methods to Study the Role of Methionine-Restricted Diet and Methioninase in Cancer Growth Control. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 1–12. [Google Scholar]
- Orentreich, N.; Matias, J.R.; DeFelice, A.; Zimmerman, J.A. Low methionine ingestion by rats extends life span. J. Nutr. 1993, 123, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Han, Q.; Aoki, Y.; Masaki, N.; Obara, K.; Hamada, K.; Hozumi, C.; Wong, A.C.W.; Bouvet, M.; Tsunoda, T.; et al. Synergy of Combining Methionine Restriction and Chemotherapy: The Disruptive Next Generation of Cancer Treatment. Cancer Diagn. Progn. 2023, 3, 272–281. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Stern, P.H. Dietary Methionine Restriction-Based Cancer Chemotherapy in Rodents. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 83–94. [Google Scholar]
- Hoffman, R.M. Clinical Studies of Methionine-Restricted Diets for Cancer Patients. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 95–105. [Google Scholar]
- Hoffman, R.M.; Tan, Y.; Li, S.; Han, Q.; Zavala, J., Sr.; Zavala, J., Jr. Pilot Phase I Clinical Trial of Methioninase on High-Stage Cancer Patients: Rapid Depletion of Circulating Methionine. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 231–242. [Google Scholar]
- Lu, W.C.; Saha, A.; Yan, W.; Garrison, K.; Lamb, C.; Pandey, R.; Irani, S.; Lodi, A.; Lu, X.; Tiziani, S.; et al. Enzyme-mediated depletion of serum l-Met abrogates prostate cancer growth via multiple mechanisms without evidence of systemic toxicity. Proc. Natl. Acad. Sci. USA 2020, 117, 13000–13011. [Google Scholar] [CrossRef]
- Bondarev, N.; Ivanenko, K.; Khabusheva, E.; Lebedev, T.; Manukhov, I.; Prassolov, V. MGL S3 Chimeric Enzyme Drives Apoptotic Death of EGFR-Dependent Cancer Cells through ERK Downregulation. Int. J. Mol. Sci. 2022, 23, 12807. [Google Scholar] [CrossRef]
- Bondarev, N.A.; Bagaeva, D.F.; Bazhenov, S.V.; Buben, M.M.; Bulushova, N.V.; Ryzhykau, Y.L.; Okhrimenko, I.S.; Zagryadskaya, Y.A.; Maslov, I.V.; Anisimova, N.Y.; et al. Methionine gamma lyase fused with S3 domain VGF forms octamers and adheres to tumor cells via binding to EGFR. Biochem. Biophys. Res. Commun. 2024, 691, 149319. [Google Scholar] [CrossRef]
- Gay, F.; Aguera, K.; Senechal, K.; Tainturier, A.; Berlier, W.; Maucort-Boulch, D.; Honnorat, J.; Horand, F.; Godfrin, Y.; Bourgeaux, V. Methionine tumor starvation by erythrocyte-encapsulated methionine gamma-lyase activity controlled with per os vitamin B6. Cancer Med. 2017, 6, 1437–1452. [Google Scholar] [CrossRef]
- Perreault, M.; Means, J.; Gerson, E.; James, M.; Cotton, S.; Bergeron, C.G.; Simon, M.; Carlin, D.A.; Schmidt, N.; Moore, T.C.; et al. The live biotherapeutic SYNB1353 decreases plasma methionine via directed degradation in animal models and healthy volunteers. Cell Host Microbe 2024, 32, 382–395.e10. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, S.; Zheng, K.; Li, Z.; Hu, E.; Mu, Y.; Mai, J.; Zhao, A.; Zhao, Z.; Li, F. Salmonella-mediated methionine deprivation drives immune activation and enhances immune checkpoint blockade therapy in melanoma. J. Immunother. Cancer 2024, 12, e008238. [Google Scholar] [CrossRef] [PubMed]
- Lombardini, J.B.; Sufrin, J.R. Chemotherapeutic potential of methionine analogue inhibitors of tumor-derived methionine adenosyltransferases. Biochem. Pharmacol. 1983, 32, 489–495. [Google Scholar] [CrossRef]
- Abels, J.; Kroes, A.C.; Ermens, A.A.; van Kapel, J.; Schoester, M.; Spijkers, L.J.; Lindemans, J. Anti-leukemic potential of methyl-cobalamin inactivation by nitrous oxide. Am. J. Hematol. 1990, 34, 128–131. [Google Scholar] [CrossRef]
- Secker, K.A.; Bloechl, B.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Schneidawind, C. MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis. Cancers 2020, 12, 1342. [Google Scholar] [CrossRef]
- Zhang, W.; Sviripa, V.; Chen, X.; Shi, J.; Yu, T.; Hamza, A.; Ward, N.D.; Kril, L.M.; Vander Kooi, C.W.; Zhan, C.G.; et al. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem. Biol. 2013, 8, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Muylaert, C.; Wyns, A.; Vlummens, P.; De Veirman, K.; Vanderkerken, K.; Zaal, E.; Berkers, C.; Moreaux, J.; De Bruyne, E.; et al. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability in multiple myeloma. Haematologica 2024, 109, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Johnson, M.; Heist, R.S.; Shapiro, G.I.; Postel-Vinay, S.; Wilson, F.H.; Garralda, E.; Wulf, G.; Almon, C.; Nabhan, S.; et al. MAT2A inhibitor AG-270/S095033 in patients with advanced malignancies: A phase I trial. Nat. Commun. 2025, 16, 423. [Google Scholar] [CrossRef]
- Li, M.; Konteatis, Z.; Nagaraja, N.; Chen, Y.; Zhou, S.; Ma, G.; Gross, S.; Marjon, K.; Hyer, M.L.; Mandley, E.; et al. Leveraging Structure-Based Drug Design to Identify Next-Generation MAT2A Inhibitors, Including Brain-Penetrant and Peripherally Efficacious Leads. J. Med. Chem. 2022, 65, 4600–4615. [Google Scholar] [CrossRef]
- Li, C.; Gui, G.; Zhang, L.; Qin, A.; Zhou, C.; Zha, X. Overview of Methionine Adenosyltransferase 2A (MAT2A) as an Anticancer Target: Structure, Function, and Inhibitors. J. Med. Chem. 2022, 65, 9531–9547. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Y.; Buettner, R.; Rosen, S.T. Targeting the methionine-methionine adenosyl transferase 2A-S-adenosyl methionine axis for cancer therapy. Curr. Opin. Oncol. 2022, 34, 546–551. [Google Scholar] [CrossRef]
- Hutchinson, K.; Silva, D.B.; Bohlke, J.; Clausen, C.; Thomas, A.A.; Bonomi, M.; Schlessinger, A. Describing inhibitor specificity for the amino acid transporter LAT1 from metainference simulations. Biophys. J. 2022, 121, 4476–4491. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Gynther, M.; Huttunen, J.; Puris, E.; Spicer, J.A.; Denny, W.A. A Selective and Slowly Reversible Inhibitor of l-Type Amino Acid Transporter 1 (LAT1) Potentiates Antiproliferative Drug Efficacy in Cancer Cells. J. Med. Chem. 2016, 59, 5740–5751. [Google Scholar] [CrossRef] [PubMed]
- Häfliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, H. Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef]
- Hens, J.R.; Sinha, I.; Perodin, F.; Cooper, T.; Sinha, R.; Plummer, J.; Perrone, C.E.; Orentreich, D. Methionine-restricted diet inhibits growth of MCF10AT1-derived mammary tumors by increasing cell cycle inhibitors in athymic nude mice. BMC Cancer 2016, 16, 349. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Xu, Q.; Li, Y.; Gao, X.; Kang, K.; Williams, J.G.; Tong, L.; Liu, J.; Ji, M.; Deterding, L.J.; Tong, X.; et al. HNF4α regulates sulfur amino acid metabolism and confers sensitivity to methionine restriction in liver cancer. Nat. Commun. 2020, 11, 3978. [Google Scholar] [CrossRef]
- Strekalova, E.; Malin, D.; Good, D.M.; Cryns, V.L. Methionine Deprivation Induces a Targetable Vulnerability in Triple-Negative Breast Cancer Cells by Enhancing TRAIL Receptor-2 Expression. Clin. Cancer Res. 2015, 21, 2780–2791. [Google Scholar] [CrossRef]
- Forney, L.A.; Wanders, D.; Stone, K.P.; Pierse, A.; Gettys, T.W. Concentration-dependent linkage of dietary methionine restriction to the components of its metabolic phenotype. Obesity 2017, 25, 730–738. [Google Scholar] [CrossRef]
- Kreis, W.; Hession, C. Isolation and purification of L-methionine-alpha-deamino-gamma-mercaptomethane-lyase (L-methioninase) from Clostridium sporogenes. Cancer Res. 1973, 33, 1862–1865. [Google Scholar] [PubMed]
- Kreis, W.; Hession, C. Biological effects of enzymatic deprivation of L-methionine in cell culture and an experimental tumor. Cancer Res. 1973, 33, 1866–1869. [Google Scholar] [PubMed]
- Tokoro, M.; Asai, T.; Kobayashi, S.; Takeuchi, T.; Nozaki, T. Identification and characterization of two isoenzymes of methionine gamma-lyase from Entamoeba histolytica: A key enzyme of sulfur-amino acid degradation in an anaerobic parasitic protist that lacks forward and reverse trans-sulfuration pathways. J. Biol. Chem. 2003, 278, 42717–42727. [Google Scholar] [CrossRef] [PubMed]
- Amarita, F.; Yvon, M.; Nardi, M.; Chambellon, E.; Delettre, J.; Bonnarme, P. Identification and functional analysis of the gene encoding methionine-gamma-lyase in Brevibacterium linens. Appl. Environ. Microbiol. 2004, 70, 7348–7354. [Google Scholar] [CrossRef]
- Mamaeva, D.V.; Morozova, E.A.; Nikulin, A.D.; Revtovich, S.V.; Nikonov, S.V.; Garber, M.B.; Demidkina, T.V. Structure of Citrobacter freundii L-methionine gamma-lyase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 546–549. [Google Scholar] [CrossRef]
- El-Sayed, A.S. Microbial L-methioninase: Production, molecular characterization, and therapeutic applications. Appl. Microbiol. Biotechnol. 2010, 86, 445–467. [Google Scholar] [CrossRef]
- Kulikova, V.V.; Morozova, E.A.; Revtovich, S.V.; Kotlov, M.I.; Anufrieva, N.V.; Bazhulina, N.P.; Raboni, S.; Faggiano, S.; Gabellieri, E.; Cioni, P.; et al. Gene cloning, characterization, and cytotoxic activity of methionine γ-lyase from Clostridium novyi. IUBMB Life 2017, 69, 668–676. [Google Scholar] [CrossRef]
- Tanaka, H.; Esaki, N.; Soda, K. A versatile bacterial enzyme: L-methionine gamma-lyase. Enzym. Microb. 1985, 7, 530–531. [Google Scholar] [CrossRef]
- Takakura, T.; Takimoto, A.; Notsu, Y.; Yoshida, H.; Ito, T.; Nagatome, H.; Ohno, M.; Kobayashi, Y.; Yoshioka, T.; Inagaki, K.; et al. Physicochemical and pharmacokinetic characterization of highly potent recombinant L-methionine gamma-lyase conjugated with polyethylene glycol as an antitumor agent. Cancer Res. 2006, 66, 2807–2814. [Google Scholar] [CrossRef]
- Tan, Y.; Xu, M.; Tan, X.; Tan, X.; Wang, X.; Saikawa, Y.; Nagahama, T.; Sun, X.; Lenz, M.; Hoffman, R.M. Overexpression and large-scale production of recombinant L-methionine-alpha-deamino-gamma-mercaptomethane-lyase for novel anticancer therapy. Protein Expr. Purif. 1997, 9, 233–245. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Miyake, K.; Kiyuna, T.; Miyake, M.; Chemielwski, B.; Nelson, S.D.; et al. Intra-tumor L-methionine level highly correlates with tumor size in both pancreatic cancer and melanoma patient-derived orthotopic xenograft (PDOX) nude-mouse models. Oncotarget 2018, 9, 11119–11125. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Yano, S. Tumor-Specific S/G2-Phase Cell Cycle Arrest of Cancer Cells by Methionine Restriction. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 49–60. [Google Scholar]
- Hoffman, R.M.; Yano, S.; Igarashi, K. Methioninase Cell-Cycle Trap Cancer Chemotherapy. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 133–148. [Google Scholar]
- Hoffman, R.M.; Murakami, T.; Kawaguchi, K.; Igarashi, K.; Tan, Y.; Li, S.; Han, Q. High Efficacy of Recombinant Methioninase on Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Cancer. In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 149–161. [Google Scholar]
- Kawaguchi, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Murakami, T.; Unno, M.; Hoffman, R.M. Efficacy of Recombinant Methioninase (rMETase) on Recalcitrant Cancer Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models: A Review. Cells 2019, 8, 410. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Miyake, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Kiyuna, T.; Miyake, M.; Higuchi, T.; Oshiro, H.; et al. Oral recombinant methioninase (o-rMETase) is superior to injectable rMETase and overcomes acquired gemcitabine resistance in pancreatic cancer. Cancer Lett. 2018, 432, 251–259. [Google Scholar] [CrossRef]
- Igarashi, K.; Kawaguchi, K.; Li, S.; Han, Q.; Tan, Y.; Murakami, T.; Kiyuna, T.; Miyake, K.; Miyake, M.; Singh, A.S.; et al. Recombinant methioninase in combination with doxorubicin (DOX) overcomes first-line DOX resistance in a patient-derived orthotopic xenograft nude-mouse model of undifferentiated spindle-cell sarcoma. Cancer Lett. 2018, 417, 168–173. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Han, Q.; Kawaguchi, K.; Li, S.; Tan, Y. Afterword: Oral Methioninase-Answer to Cancer and Fountain of Youth? In Methionine Dependence of Cancer and Aging: Methods and Protocols; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1866, pp. 311–322. [Google Scholar]
- Kawaguchi, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Kiyuna, T.; Miyake, K.; Miyake, M.; Chmielowski, B.; Nelson, S.D.; et al. Targeting methionine with oral recombinant methioninase (o-rMETase) arrests a patient-derived orthotopic xenograft (PDOX) model of BRAF-V600E mutant melanoma: Implications for chronic clinical cancer therapy and prevention. Cell Cycle 2018, 17, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Han, Q.; Bouvet, M.; Hoffman, R.M.; Park, J.H. Recombinant Oral Methioninase (o-rMETase) Combined with Oxaliplatinum Plus 5-Fluorouracil Improves Survival of Mice with Massive Colon-Cancer Peritoneal Carcinomatosis. Anticancer Res. 2023, 43, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.I.; Yamamoto, J.; Han, Q.; Sun, Y.U.; Nishino, H.; Tashiro, Y.; Sugisawa, N.; Tan, Y.; Choi, H.J.; Nam, S.J.; et al. Response of Triple-negative Breast Cancer Liver Metastasis to Oral Recombinant Methioninase in a Patient-Derived Orthotopic Xenograft (PDOX) Model. In Vivo 2020, 34, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Miyake, K.; Han, Q.; Igarashi, K.; Kawaguchi, K.; Barangi, M.; Kiyuna, T.; Sugisawa, N.; Higuchi, T.; Oshiro, H.; et al. Synergy of oral recombinant methioninase (rMETase) and 5-fluorouracil on poorly differentiated gastric cancer. Biochem. Biophys. Res. Commun. 2023, 643, 48–54. [Google Scholar] [CrossRef]
- Sugisawa, N.; Higuchi, T.; Han, Q.; Hozumi, C.; Yamamoto, J.; Tashiro, Y.; Nishino, H.; Kawaguchi, K.; Bouvet, M.; Murata, T.; et al. Oral recombinant methioninase combined with paclitaxel arrests recalcitrant ovarian clear cell carcinoma growth in a patient-derived orthotopic xenograft (PDOX) nude-mouse model. Cancer Chemother. Pharmacol. 2021, 88, 61–67. [Google Scholar] [CrossRef]
- Sugisawa, N.; Yamamoto, J.; Han, Q.; Tan, Y.; Tashiro, Y.; Nishino, H.; Inubushi, S.; Hamada, K.; Kawaguchi, K.; Unno, M.; et al. Triple-Methyl Blockade with Recombinant Methioninase, Cycloleucine, and Azacitidine Arrests a Pancreatic Cancer Patient-Derived Orthotopic Xenograft Model. Pancreas 2021, 50, 93–98. [Google Scholar] [CrossRef]
- Han, Q.; Hoffman, R.M. Chronic Treatment of an Advanced Prostate-cancer Patient with Oral Methioninase Resulted in Long-term Stabilization of Rapidly Rising PSA Levels. In Vivo 2021, 35, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Paley, O.; Hu, J.; Ekerdt, B.; Cheung, N.K.; Georgiou, G. De novo engineering of a human cystathionine-γ-lyase for systemic (L)-Methionine depletion cancer therapy. ACS Chem. Biol. 2012, 7, 1822–1829. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Lin, Y.; Zhao, Z.; Lai, Y.; Lu, M.; Shao, Z.; Mo, X.; Mu, Y.; Liang, Z.; Wang, X.; et al. Targeted deprivation of methionine with engineered Salmonella leads to oncolysis and suppression of metastasis in broad types of animal tumor models. Cell Rep. Med. 2023, 4, 101070. [Google Scholar] [CrossRef]
- Sviripa, V.M.; Zhang, W.; Balia, A.G.; Tsodikov, O.V.; Nickell, J.R.; Gizard, F.; Yu, T.; Lee, E.Y.; Dwoskin, L.P.; Liu, C.; et al. 2′,6′-Dihalostyrylanilines, pyridines, and pyrimidines for the inhibition of the catalytic subunit of methionine S-adenosyltransferase-2. J. Med. Chem. 2014, 57, 6083–6091. [Google Scholar] [CrossRef]
- Lien, E.C.; Ghisolfi, L.; Geck, R.C.; Asara, J.M.; Toker, A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci. Signal 2017, 10, eaao6604. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Y.; Liu, Y.; Huang, Y.; Wang, W. Amino Acid-Starved Cancer Cells Utilize Macropinocytosis and Ubiquitin-Proteasome System for Nutrient Acquisition. Adv. Sci. 2024, 11, e2304791. [Google Scholar] [CrossRef] [PubMed]
- Ampomah, P.B.; Cai, B.; Sukka, S.R.; Gerlach, B.D.; Yurdagul, A., Jr.; Wang, X.; Kuriakose, G.; Darville, L.N.F.; Sun, Y.; Sidoli, S.; et al. Macrophages use apoptotic cell-derived methionine and DNMT3A during efferocytosis to promote tissue resolution. Nat. Metab. 2022, 4, 444–457. [Google Scholar] [CrossRef]
- Puris, E.; Fricker, G.; Gynther, M. The Role of Solute Carrier Transporters in Efficient Anticancer Drug Delivery and Therapy. Pharmaceutics 2023, 15, 364. [Google Scholar] [CrossRef]
- Wu, G.; Xu, J.; Wang, Q.; Fang, Z.; Fang, Y.; Jiang, Y.; Zhang, X.; Cheng, X.; Sun, J.; Le, G. Methionine-Restricted Diet: A Feasible Strategy Against Chronic or Aging-Related Diseases. J. Agric. Food Chem. 2023, 71, 5–19. [Google Scholar] [CrossRef]
- Zhang, Y.; Jelleschitz, J.; Grune, T.; Chen, W.; Zhao, Y.; Jia, M.; Wang, Y.; Liu, Z.; Höhn, A. Methionine restriction—Association with redox homeostasis and implications on aging and diseases. Redox Biol. 2022, 57, 102464. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, C.; Xu, A.; Zuo, L.; Li, Q.; Fan, F.; Hu, Y.; Sun, C. Methionine Dependency and Restriction in Cancer: Exploring the Pathogenic Function and Therapeutic Potential. Pharmaceuticals 2025, 18, 640. https://doi.org/10.3390/ph18050640
Ma C, Xu A, Zuo L, Li Q, Fan F, Hu Y, Sun C. Methionine Dependency and Restriction in Cancer: Exploring the Pathogenic Function and Therapeutic Potential. Pharmaceuticals. 2025; 18(5):640. https://doi.org/10.3390/ph18050640
Chicago/Turabian StyleMa, Chi, Aoshuang Xu, Liping Zuo, Qun Li, Fengjuan Fan, Yu Hu, and Chunyan Sun. 2025. "Methionine Dependency and Restriction in Cancer: Exploring the Pathogenic Function and Therapeutic Potential" Pharmaceuticals 18, no. 5: 640. https://doi.org/10.3390/ph18050640
APA StyleMa, C., Xu, A., Zuo, L., Li, Q., Fan, F., Hu, Y., & Sun, C. (2025). Methionine Dependency and Restriction in Cancer: Exploring the Pathogenic Function and Therapeutic Potential. Pharmaceuticals, 18(5), 640. https://doi.org/10.3390/ph18050640