Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase

Abstract
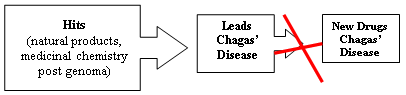
{kind=link}
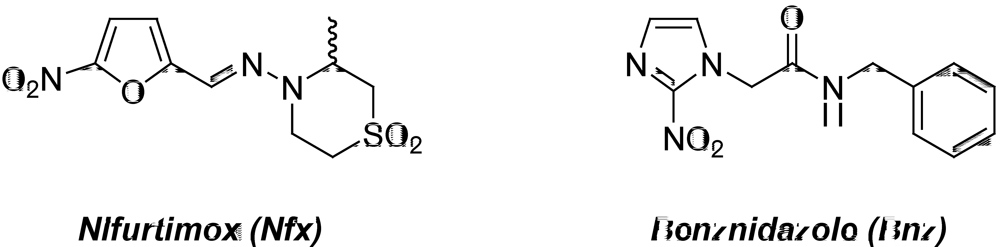{kind=link}
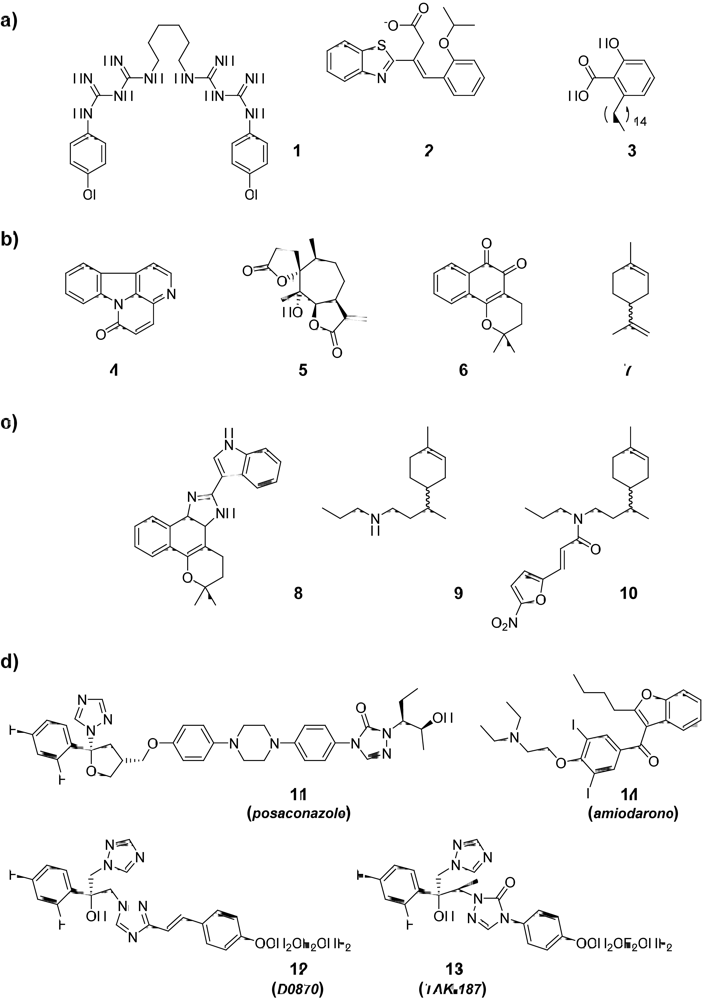{kind=link}
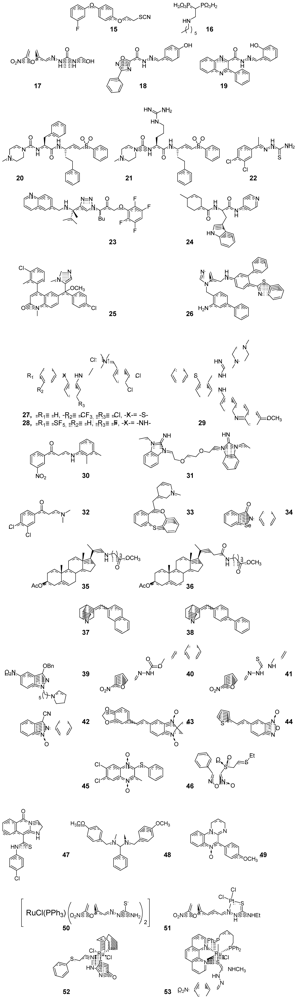{kind=link}
{kind=link}
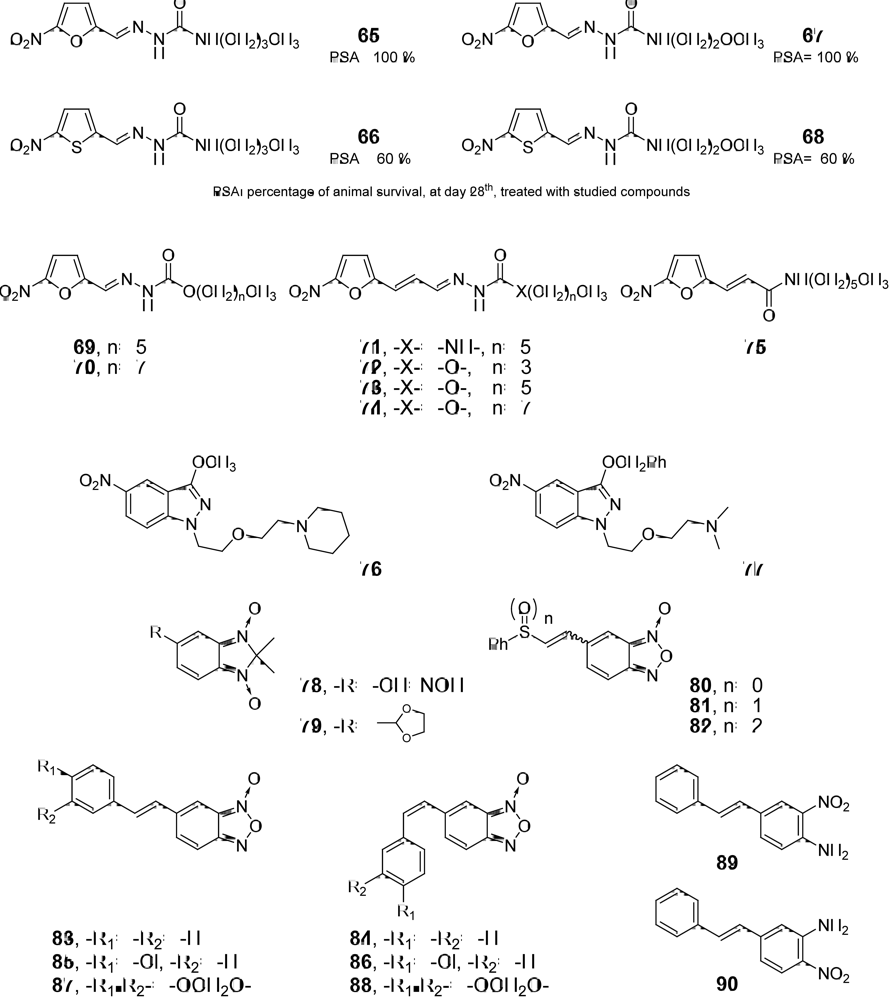{kind=link}
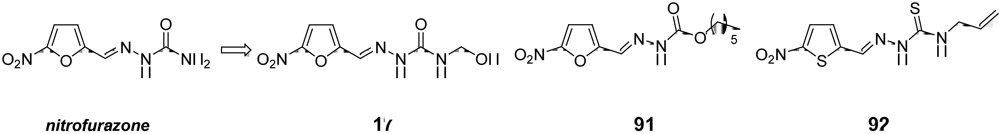{kind=link}
Share and Cite
Cerecetto, H.; González, M. Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase. Pharmaceuticals 2010, 3, 810-838. https://doi.org/10.3390/ph3040810
Cerecetto H, González M. Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase. Pharmaceuticals. 2010; 3(4):810-838. https://doi.org/10.3390/ph3040810
Chicago/Turabian StyleCerecetto, Hugo, and Mercedes González. 2010. "Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase" Pharmaceuticals 3, no. 4: 810-838. https://doi.org/10.3390/ph3040810
APA StyleCerecetto, H., & González, M. (2010). Synthetic Medicinal Chemistry in Chagas’ Disease: Compounds at The Final Stage of “Hit-To-Lead” Phase. Pharmaceuticals, 3(4), 810-838. https://doi.org/10.3390/ph3040810