Anti-Inflammatory Drug Design Using a Molecular Hybridization Approach


Abstract
: The design of new drugs with better physiochemical properties, adequate absorption, distribution, metabolism, and excretion, effective pharmacologic potency and lacking toxicity remains is a challenge. Inflammation is the initial trigger of several different diseases, such as Alzheimer's disease, asthma, atherosclerosis, colitis, rheumatoid arthritis, depression, cancer; and disorders such as obesity and sexual dysfunction. Although inflammation is not the direct cause of these disorders, inflammatory processes often increase related pain and suffering. New anti-inflammatory drugs developed using molecular hybridization techniques to obtain multiple-ligand drugs can act at one or multiple targets, allowing for synergic action and minimizing toxicity. This work is a review of new anti-inflammatory drugs developed using the molecular modification approach.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
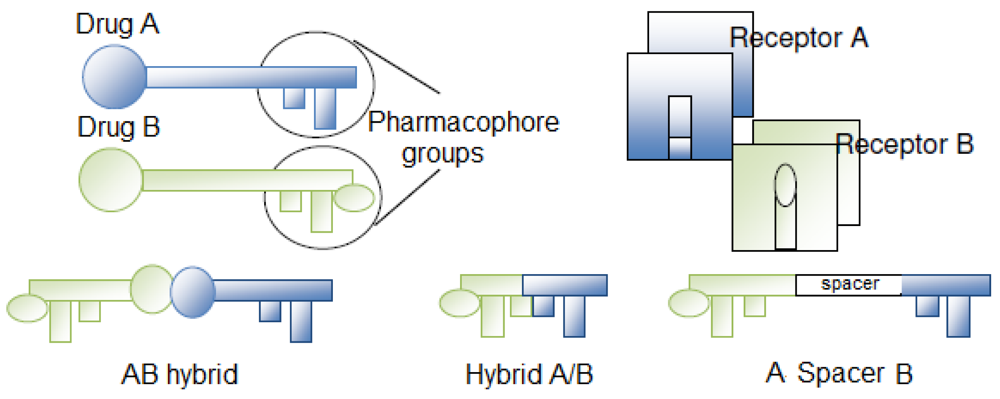
Molecular hybridization is a molecular modification approach to obtain multiple-ligands/compounds with pharmacokinetic advantages over concomitant administration of two different drugs [1]. The term “multiple ligands”, proposed by Morphy et al., applies to drugs that recognize more than one receptor. The advantages of multiple ligands are their ability: (1) to activate different targets by a single molecule, thereby increasing therapeutic efficacy and (2) to change the bioavailability profile in the cell and be effectively eliminated after exerting their effects [2].
The hybridization process is closely related to the strategy of obtaining a mutual prodrug, with the main difference being that the prodrug action is dependent on its in vivo cleavage while hybrid compounds can also act “per se” at their specific receptors or targets (Figure 1). Hybrid compounds can be constructed by linking pharmacophore subunits directly or with spacer agents. The simple association of two distinct active principles can also be considered a hybrid compound [3].
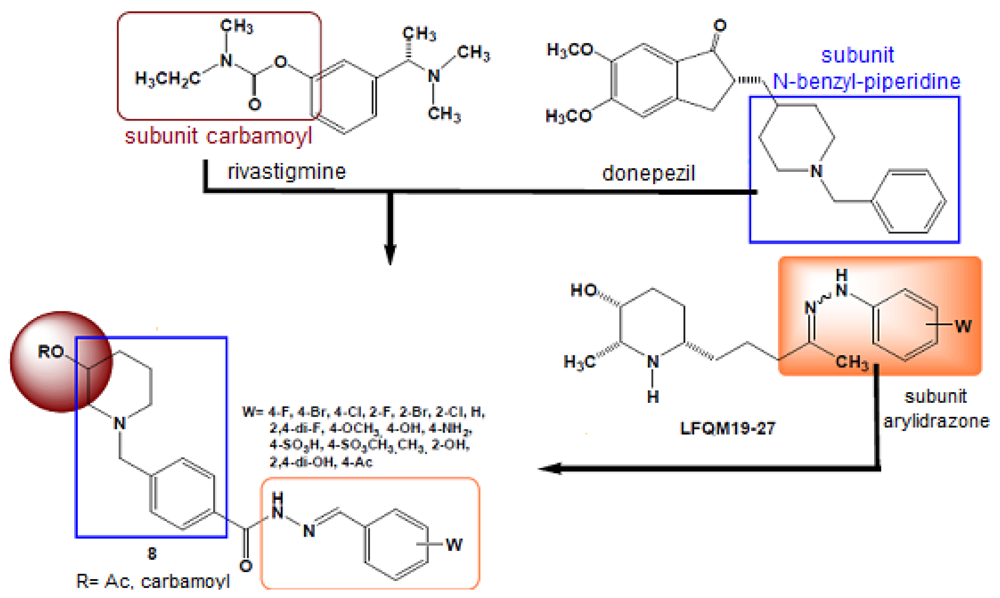
Hybridization techniques (of new entities or mutual prodrugs) to develop new anti-inflammatory drugs have been used by several researchers to obtain new compounds with better actions than those of classical nonsteroidal anti-inflammatory drugs (NSAIDs). For example, Rocha and Viegas-Jr reported using this strategy to obtain anti-inflammatory compounds useful in treating chronic degenerative diseases, such as Alzheimer's disease. The authors designed new molecules by linking together three different subunits: the carbamoyl from rivastigmine, the N-benzylpiperidine subunit of donezepil, and the arylhydrazone subunit of the anti-inflammatory compound LFQM 19-27 (Figure 2) [4].
New anti-inflammatory compounds were designed by the molecular hybridization of thalidomide. This drug was introduced in the pharmaceutical market in 1956 by the German Pharmaceutical Industry Chemie Grunenthal, which explored the sedative activity of the drug. In 1958, the use of thalidomide was expanded to combat nausea during pregnancy [5,6]. A few years later, however, the drug was withdrawn from the market due to its teratogenic effects [5]. In 1965, an Israeli dermatologist used thalidomide to treat his leprosy patients suffering from insomnia. To his surprise, he found that in addition to its hypnotic effects, the drug also alleviated the wounds (erythema nodosum) caused by Mycobacterium leprae [6-10].
The beneficial effects of thalidomide can be attributed to its anti-inflammatory, immunomodulatory, and angiogenic activities [11]. Although the mechanism of action is not yet fully understood, it is known that thalidomide inhibits chemotaxis of lymphocytes and neutrophils, and decreases the levels of cytokines such as tumor necrosis factor (TNF)-α and interferon (IFN)-γ.
In addition, thalidomide is involved in the regulation of T lymphocytes (TH1 and TH2), increases the production of TH2 and cytokines such as interleukin (IL)-4 and IL-5, and inhibits the production of inflammatory lymphocytes (TH1) and the cytokine IFN-γ in peripheral blood cells stimulated by antigens and mitogens [5,11]. In July 1998, the Food and Drug Administration approved the use of thalidomide for the treatment of erythema nodosum lepromatous [12].
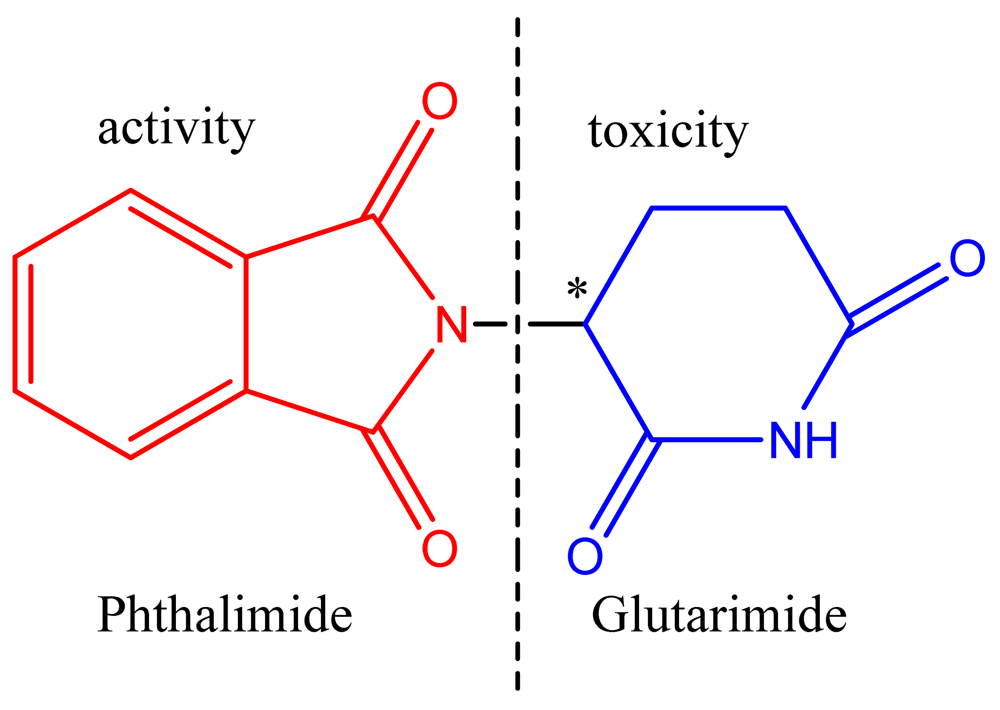
Several research groups have now developed thalidomide analogues for the treatment of chronic inflammation using hybridization techniques to improve the pharmacodynamic and pharmacokinetic properties, and thereby reducing the teratogenic effects. The design of new thalidomide analogues devoid of teratogenicity resulted from studies demonstrating that the teratogenic effects are due to the toxicophore glutarimide subunit (Figure 3) [9].
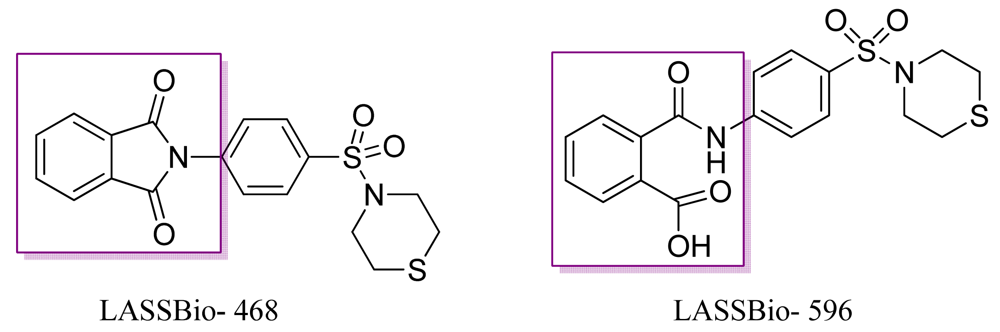
Patients with chronic inflammatory diseases have elevated levels of the pro-inflammatory cytokine TNF-α, which triggers a series of detrimental changes that promote the development of inflammatory, immunopathologic, and autoimmune diseases [13-19]. One such disease, asthma, affects the airways involving many cells and cellular elements, particularly mast cells, eosinophils, T lymphocytes, macrophages, and neutrophils of epithelial cells. The inflammatory process also causes an associated increase in the existing exacerbated bronchial response to a variety of stimuli. Based on this knowledge, Lima and coworkers obtained a series of new N-substituted phthalimide derivatives, structurally designed as hybrids of thalidomide (1) and aryl sulfonamides (2), to selectively inhibit phosphodiesterase, which is involved in the asthma process (Figure 4) [20].
These authors showed that the best compound, LASSBio 468 (Figure 5), inhibited the neutrophil infiltration induced by lipopolysaccharides (LPS) with an ED50 of 2.5 mg kg−1 as well as the increased TNF-α in the bronchoalveolar lavage fluid from mice treated with LPS. The preliminary structure activity relationship of this compound revealed the importance of the sulfonyl group, the nature of the N-terminal piperazine ring, and the role of the phthalimide ring in the anti-inflammatory activity of this compound (468) [20]. LASSBio-596 is the metabolic compound of LASSBio-468, with activity similar to that of the parent drug.
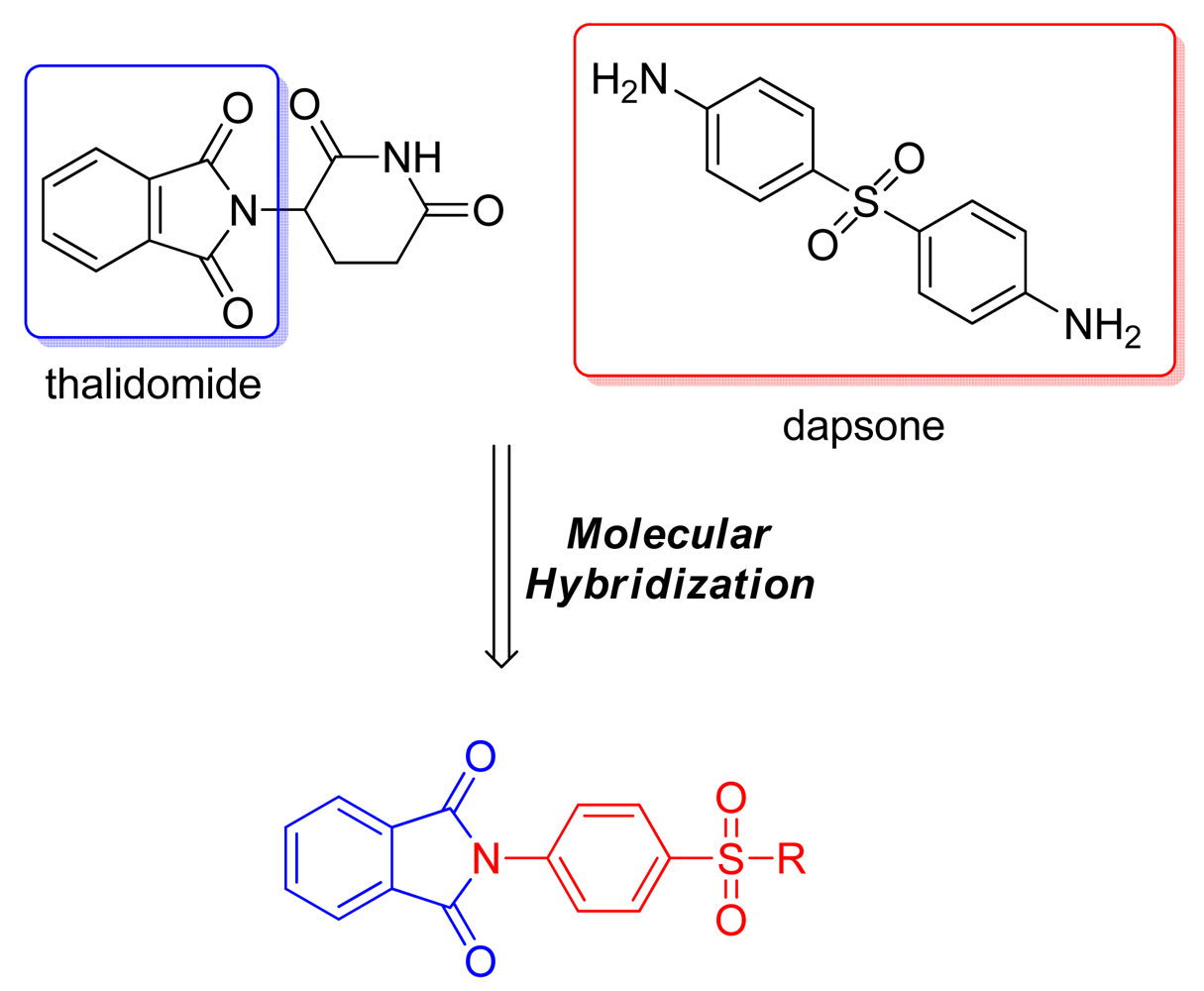
Using the same concept of the phthalimide moiety as a TNF-α inhibitor, Santos and coworkers used molecular hybridization to develop a series of derivatives of thalidomide and dapsone to obtain new anti-inflammatory compounds to treat erythema nodosum lepromatous (Figure 6). All the compounds demonstrated analgesic/anti-inflammatory activity and reduced in vitro TNF-α levels. Furthermore, the compounds reduced the number of bacillus/paw in nude mice 12 months after inoculation of 1.0 × 104 bacilli/mL in comparison with controls (unpublished results). Curiously, when the same compounds were evaluated against Mycobacterium tuberculosis, three of them had minimum inhibitory concentrations of 3.9, 5.0, and 7.8 (μg/mL). These compounds demonstrated an excellent selectivity ratio, decreasing the number of M. tuberculosis without macrophage toxicity [21].
Santos and coworkers also used a phthalimide moiety to construct new hybrid derivative drug candidates useful for treating the symptoms of sickle cell disease. This disease is characterized by a point mutation that changes glutamic acid (Glu6) to valine (Val6) in the β chain of hemoglobin. In the deoxy state, this modification leads to polymerizations that modify the cytoskeletal structure to form a sickle morphology. Patients with sickle cell disease present with a well-known chronic inflammatory condition. The only drug approved by the Food and Drug Administration to treat sickle cell disease is the antineoplasic hydroxyurea, which controversially increases the expression of pro-inflammatory cytokines.
Based on the inflammatory aspects of sickle cell disease, Santos and coworkers developed new compounds from thalidomide and hydroxyurea (Figure 7). The synthesized compounds had higher analgesic and anti-inflammatory activities than controls and reduced the TNF-α levels of transgenic sickle animals. Furthermore, the important property of hydroxyurea to induce gammaglobulin expression was maintained and, surprisingly, the potency of the compounds was increased compared with that of hydroxyurea. Thus, these results showed the successful application of this molecular modification tool to improve the pharmacodynamic properties of new drug candidates to treat sickle cell disease [22].
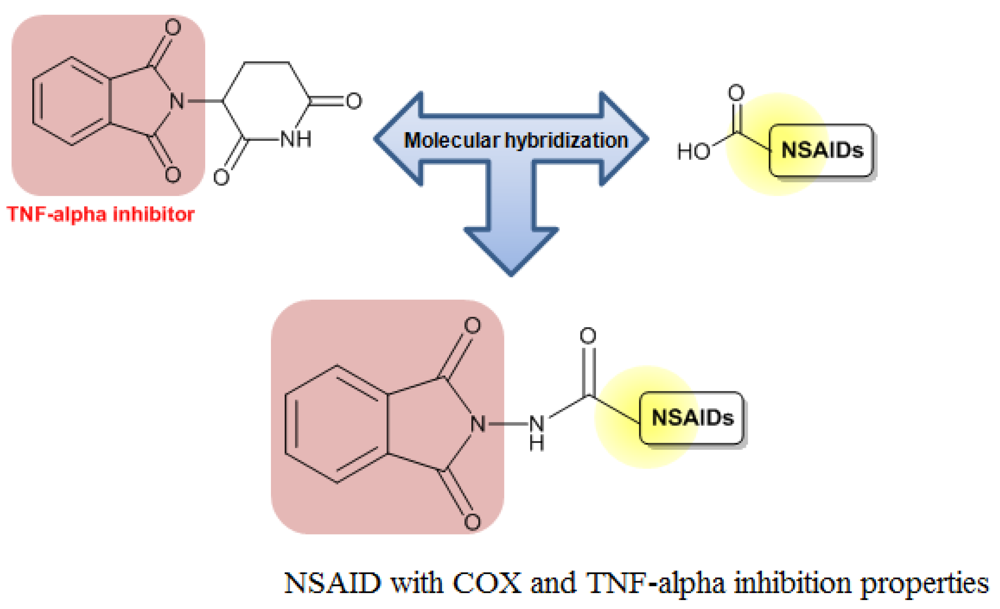
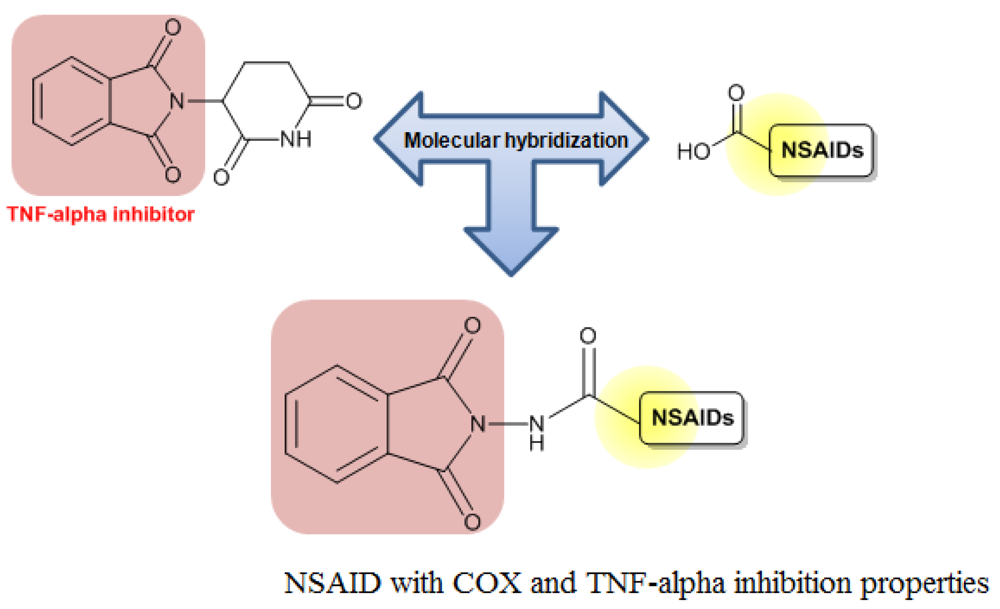
In addition, Santos and coworkers obtained a series of hybrid derivatives of classical NSAIDs with a phthalimide group (Figure 8). These hybrid compounds may act synergistically, inhibiting cyclooxygenase (COX)-2 and TNF-α with potential activity against chronic inflammation [23]. Indeed, Vizioli (2009) showed that these hybrid NSAIDs were effective against acute anti-inflammatory and ulcerative colitis and showed no gastrotoxicity [3].
Jung and coworkers obtained a prodrug of mesalasine and taurine to selectively target the colon. Extensive mechanistic studies, however, showed that chlorinated taurine (Tau-Cl) has additive anti-inflammatory activity by inhibiting IL-1b mediated nuclear factor-κB activation [24,25].
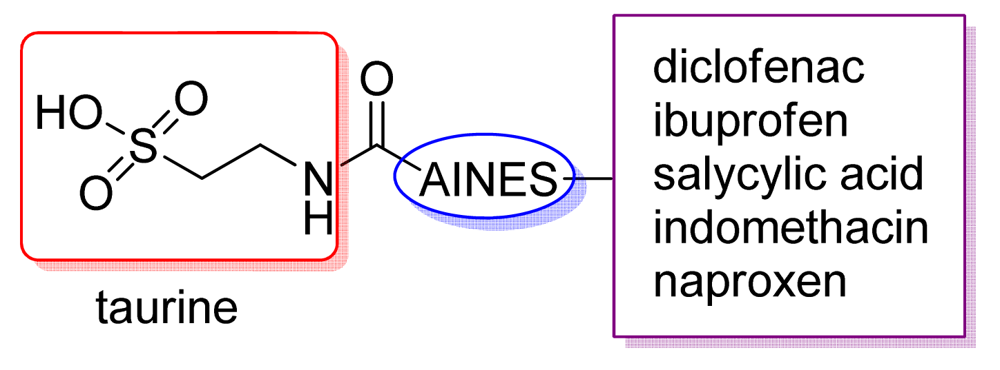
Vizioli reported the synthesis and biologic activity of mutual prodrugs of classical NSAIDs and taurine (Figure 9). These compounds showed no gastric toxicity and abolished ulcerative colitis induced by acetic acid in rats, decreasing mortality in comparison with NSAIDs [26,27].
Excessive production of reactive oxygen species is associated with inflammation and leads to the condition of oxidative stress, which can contribute to the high mortality rates associated with several diseases [28].
Natural and synthetic antioxidants possess anti-inflammatory properties [29,30]. Lipoic acid (1,2-dithiolane-3-valeric acid) is a naturally occurring compound present in all kinds of prokaryotic and eukaryotic cells and synthesized by animals. It exists as both the reduced dithiol form (dihydrolipoic acid) and the oxidized disulfide form [31,32] and has been described as a universal antioxidant that reacts with RO2•, ascorbyl radicals HO• and NO•, and tocopherol radicals O2•− and hypochlorous acid [33-35].
Melagraki and coworkers developed hybrid compounds of lipoic acid and coumarins [36]. Coumarins exhibit anti-inflammatory and antioxidant activities [37,38]. The inhibition of inflammation ranged from 41% to 73%, while the reference drug indomethacin showed 47% inhibition at an equivalent dose. All compounds showed activity, but conjugates 3 and 4 (Figure 10) were more potent [36].
The increase in reactive oxygen species is associated with death caused by malaria due to erythrocyte hemolysis. Based on this concept, Raji and coworkers obtained a NSAID (ibuprofen, ketoprofen, fenoprofen, ketoprofen hydroxy) and methylene analogue (diclofenac or indomethacin) hybrid of primaquine (PQ) to decrease the inflammatory activity of reactive oxygen species in malaria. Compound 5 comprises one reduced ketoprofen moiety and two PQ units (one bound by an amide and the other by the carbamate bond) (Figure 11). All the tested PQ-NSAID conjugates showed antioxidant activity and moderate antiradical activity, with an EC50 between 269.5 ± 10.7 and 379.3 ± 59.1 mg L−1. Based on the β-carotene-linoleic acid assay, the PQ-ketoprofen hybrid (compound 5) possessed the strongest antioxidant activity. The most active conjugate, according both to the antioxidant activity relative to water control value (69.4 ± 0.9%) or the absolute changes in absorbance at t = 60 (58.4 ± 3.1%) and t = 120 min (59.3 ± 1.8%), was compound 5 with two PQ units [39].
Nitric oxide (NO), which is naturally generated from L-arginine by NO synthase, is a key signaling molecule involved in the regulation of many physiologic processes, including vascular relaxation, neurotransmission, and immune system events.
Most recent studies of hybrid anti-inflammatory agents report using NO donors as a moiety to improve activity in treating several diseases, including atherosclerosis, which is a disease related to endothelial dysfunction resulting from an increase in plasma lipids, peroxidation of low-density lipoproteins, and impaired endothelial-derived relaxing factor (NO, NO•)-mediated bioactions. The oxidative stress of low-density lipoproteins leads to the formation of foam cells, the precursors of atherosclerotic plaques. In an atherosclerotic blood vessel, NO• bioactions are impaired by a number of processes, including a possible decrease in NO• production, an increase in NO• inactivation, and a decrease in the responsiveness of the target cells to NO•.
Cena and coworkers obtained a series of hybrids of ascorbic acid (antioxidant derivatives)/NO donor (furoxans, nitrate), as shown in Figure 12. All compounds inhibited ferrous salt/ascorbate-induced lipid peroxidation of membrane lipids of rat hepatocytes and showed potent in vitro vasodilation activity. These hybrids promoted dose-dependent dilation of rat aorta strips pre-contracted with phenylephrine [40].
Several researchers have synthesized derivatives of NSAIDs with NO donor moieties. In 2001, a series of NSAIDs obtained by linking ibuprofen to selected furoxan moieties and related furazans were synthesized and tested for their anti-inflammatory, antiplatelet, and anti-ulcerogenic properties (Figure 13). All the derivatives showed anti-inflammatory activities comparable to that of ibuprofen and, unlike ibuprofen, they showed reduced acute gastrotoxicity [41].
Ibuprofen propyl ester 9 significantly reduced edema after 4 and 6 hours (37.6% and 49.6%, respectively). All the other products of the series also showed significant anti-inflammatory activity, with comparable efficacy, less irritation, and less platelet aggregation than ibuprofen. With regard to platelet aggregation, the NO-donor 6 was more potent than ibuprofen, while compounds 7 and 8 were approximately equipotent with the parent drug [41].
Cena and coworkers reported in 2003 a series of hybrids of aspirin/NO donors linked to furoxan moieties, with different NO-releasing abilities (Figure 14) [42]. All the compounds showed anti-inflammatory activity in a carrageenan rat paw edema model. Derivatives 11, 13, and 14 showed significant anti-inflammatory and antiplatelet activities, without gastrotoxicity. The most potent antiplatelet compounds of the series were the benzenesulfonyl and cyano-substituted furoxans 10 and 14, respectively [42]. Turnbull and coworkers tested the TNF-α inhibition properties of these aspirin/NO donor hybrid compounds. Compound 14 (R = CN) had a significant inhibitory effect on TNF-α release in human monocyte-derived macrophages treated with LPS (36 ± 10% of LPS control, P < 0.01; n = 5–10). The effect was equivalent in magnitude to that of dexamethasone, but was not shared by DEA/NO (2-(N,N-diethylamino)-diazenolate-2-oxide), furazan, aspirin, or NCX4016 (3-(nitroxymethyl)phenyl 2-(acetoxy)benzoate).
None of the treatments studied caused significant cell death compared with untreated macrophages and monocytes. Levels of lactate dehydrogenase released following the treatments were comparable with those released from untreated control samples (∼0.5 × 105 cells/treatment) [43].
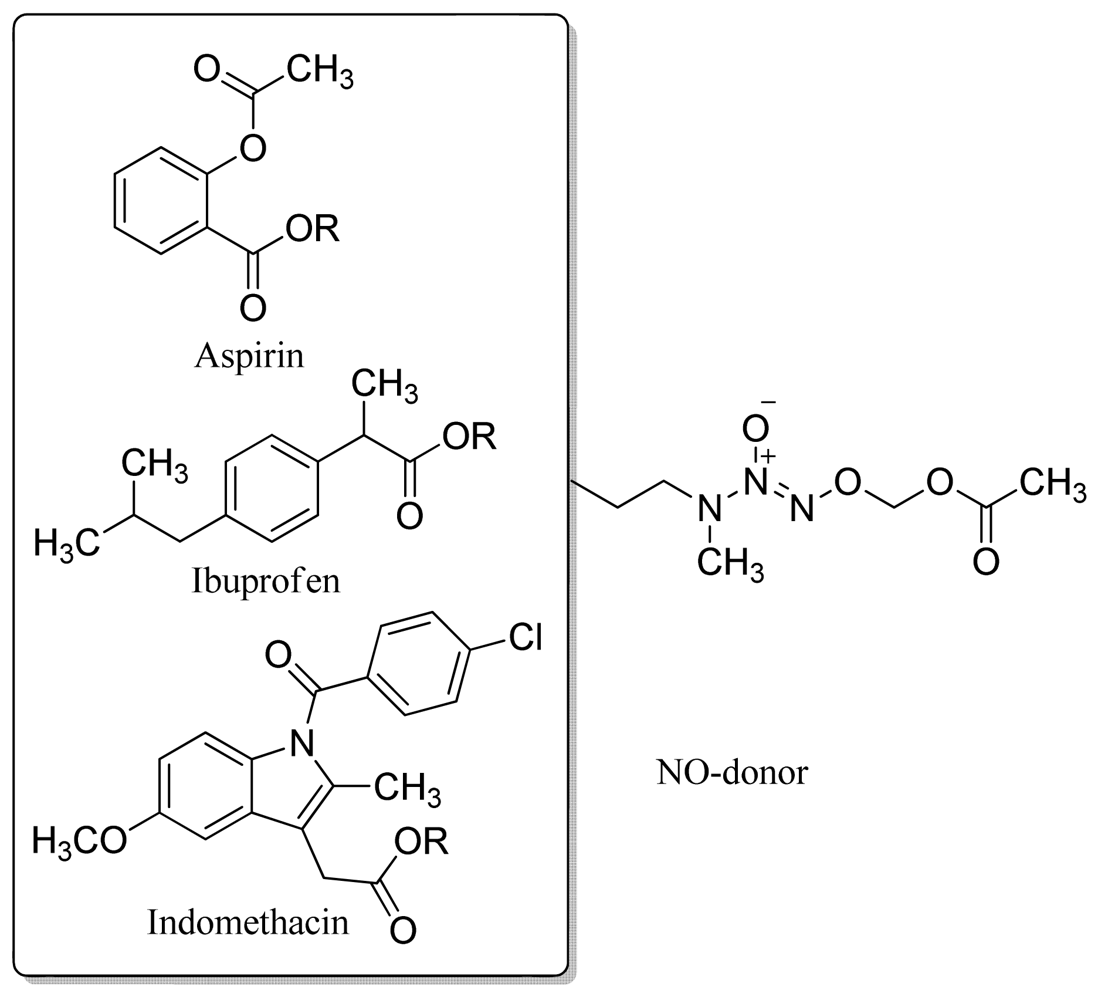
Based on the same concept, Gasco and coworkers designed a class of NO-donor aspirin-like drugs. These hybrid compounds were derived from aspirin by linking acyl moieties possessing NO-donor O-nitro groups to the −OH function of salicylic acid (Figure 15) [44].
Oral administration of mono O-nitro derivatives 15-20 and di (O-nitro) derivatives 21-25 to rats at a dose equimolar with aspirin (120 mg/Kg i.g.) showed less gastric damage after 3 h compared with aspirin. These derivatives also inhibit platelet aggregation induced by collagen in human platelet-rich plasma. Compounds 15 and 21 were the best antiplatelet compounds and due to the presence of O-nitro NO-donor moieties, all the compounds promoted relaxation in rat aorta strips pre-contracted with phenylephrine [44].
In 2009, a series of nitrooxyacyloxy methyl esters of aspirin were synthesized and evaluated as new NO-donor/aspirin hybrids by Lazzarato and coworkers using nitrates as the NO donor moieties. All the compounds showed in vitro vasodilator activity, but no antiplatelet activity. The best results were obtained using aromatic nitrooxyacyloyl moieties.
In the case of the benzoyl derivatives, the most active compounds presented NO-donor chains at the p- or at m-position (Figure 16). All the compounds, including the nitrooxy-substituted acid metabolites displayed in vitro vasodilator activity. The aromatic derivative series showed higher activity than the aliphatic derivative series [45].
In 2011, the same authors obtained an aspirin hybrid with H2S donor properties (Figure 17) and all compounds were stable in acid and physiologic pH and promoted fast aspirin release when incubated in human serum. The compounds inhibited collagen-induced platelet aggregation in human plasma. This effect was observed only with hybrid compounds while the control NO-donor and H2S-donor moieties did not show anti-platelet activity. These new compounds produce the expected NO-dependent and H2S-dependent vasodilator activities, respectively, and the authors suggested the use of these compounds as safer derivatives of aspirin for antithrombotic and anti-inflammatory therapies [46].
In addition, NSAIDs such as aspirin have shown promising effects in colorectal cancer clinical trials. Exploration of the use of NO with NSAIDs (NO/NSAIDs) in chemotherapy represented a logical progression, because NO/NSAIDs were originally designed to deliver the biologic activity of NO to reduce the gastrointestinal damage caused by NSAIDs [47].
Glucocorticoids have been widely used since the 1950s for their potent anti-inflammatory and immunosuppressive actions [48]. Fang and coworkers obtained a series of hybrid compound derivatives of glucocorticoids with NO donors (Figure 18) [49].
The results of Fang and coworkers showed that compound 26 was effective against acute and chronic inflammation (paw edema and rheumatoid arthritis model in rats). The most important effect in chronic inflammation is the higher activity observed than when using hydrocortisone alone in the later phase of the disease (days 21 and 24) [49].
Two triple-hybrid compounds containing a cysteine subunit (known to enhance the activity of organic nitrates and reduce nitrate tolerance), naproxen, and an organic nitrate (nitrooxypivaloic acid) were synthesized. Derivatives 28 and 29 (Figure 19), comprising naproxen, cysteine ethyl ester, and nitrooxypivaloic acid, showed good anti-inflammatory activity and decreased gastrointestinal tract damage. This new hybrid compound showed a reduced induction of tolerance and an attenuated decrease in blood pressure with nitrates (data not published) [50]. The authors, however, showed only the in vitro results.
Intermediate 27 and compounds 28 and 29 were tested for their COX inhibitory properties relative to naproxen. Naproxen inhibits COX-1 with an IC50 of 0.1 μM and COX-2 with an IC50 of 9.0 μM. Compound 27 showed no inhibition of COX-2 and weakly inhibition of COX-1 (IC50 = 6.0 μM). The N-3-nitrooxypivaoyl-S-(+)-2-(6-methoxy-2-naphthyl)-propanoyl-l-cysteine ethyl ester (28) was inactive against both COX-1 and COX-2 at a concentration range of 0.1 to 10 μM, while N-(+)-2-(6-methoxy-2-naphthyl)propanoyl-S-3-nitrooxypivaloyl-l-cysteine ethyl ester (29) exhibited no COX-2 inhibition activity at the same concentration range and low COX-1 inhibition activity with an IC50 of 5.6 μM [50].
Knaus and coworkers obtained a series of selective COX-2 inhibitor/NO donor hybrid agents (Figure 20). 3,4-Diphenyl-1,2,5-oxadiazole-2-oxides (3,4-diphenylfuroxans) and the corresponding N-desoxy 3,4-diphenyl-1,2,5-oxadiazoles (3,4-diphenylfurazans) analogs were evaluated and showed COX-2 selectivity. The methanesulfonyl regioisomers 30a,b [COX-1 IC50 = 11.6 μM; COX-2 IC50 = 0.12 μM; COX-2 selectivity index (SI) = 97] and aminosulfonyl regioisomers 31 (COX-1 IC50 = 9.8 μM; COX-2 IC50 = 0.78 μM; COX-2 SI = 12), reference drug celecoxib (COX-1 IC50 = 33.1 μM; COX-2 IC50 = 0.07 μM; COX-2 SI = 472), were potent in vitro COX-2 inhibitors with a good COX-2 selectivity index.
The authors suggested that the 1,2,5-oxadiazole-2-oxide (furoxan) ring system possesses beneficial features desirable for the design of hybrid COX-2 inhibitor/NO donor anti-arthritic agents with a low ulcerogenicity profile and minimal potential to induce adverse cardiovascular events such as heart attacks and strokes [51].
The same authors obtained a series of hybrid NO-releasing NSAIDs possessing a 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate or 1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate moiety attached via a one-carbon methylene spacer to the carboxylic acid group of the classical NSAIDs aspirin, ibuprofen, and indomethacin (Figure 21) [52].
In vitro studies of COX showed that none of these compounds inhibited either the COX-1 or COX-2 isoenzyme at the highest test compound concentration used (100 μM). When administered orally to rats, however, the carrageenan-induced rat paw edema assay indicated ID50 values similar to those obtained for the reference drugs [52].
In 2007, Knaus and coworkers tried other NO donor groups as NSAID linkers. They obtained NO-releasing nonsteroidal anti-inflammatory mutual prodrugs possessing an O-2-acetoxymethyl-1-[N-(2-hydroxyethyl)-N-methylamino]diazen-1-ium-1,2-diolate moiety (NONO-NSAIDs) (Figure 22) [53].
These derivatives did not inhibit the catalytic activity of COX-1/COX-2 isozymes in vitro, but showed equipotent anti-inflammatory properties compared to their parent NSAID in a carrageenan-induced rat paw edema model in vivo, without significant gastric toxicity, when administered orally. The authors report the benefits of using mutual NONO-aspirin prodrugs for the prevention of thrombus formation and adverse cardiovascular events such as stroke and myocardial infarction [53].
In 2008, Knaus and coworkers obtained a nonselective COX hybrid of NO donor anti-inflammatory compounds wherein an O-2-acetoxymethyl-1-(N-ethyl-N-methylamino)diazen-1-ium-1,2-diolate (32a-d), or 2-nitrooxyethyl (33a-d) NO-donor moiety is attached directly to the carboxylic acid group of (E)-3-(4-methanesulfonylphenyl)-2-(phenyl)acrylic acids (Figure 23). The O2-acetoxymethyl-1-(N-ethyl-N-methylamino)diazen-1-ium-1,2-diolate (32a-d) derivatives undergo extensive ester cleavage by esterase (in rats serum), followed by a significant release of NO (∼76.2%−83.0%) [54].
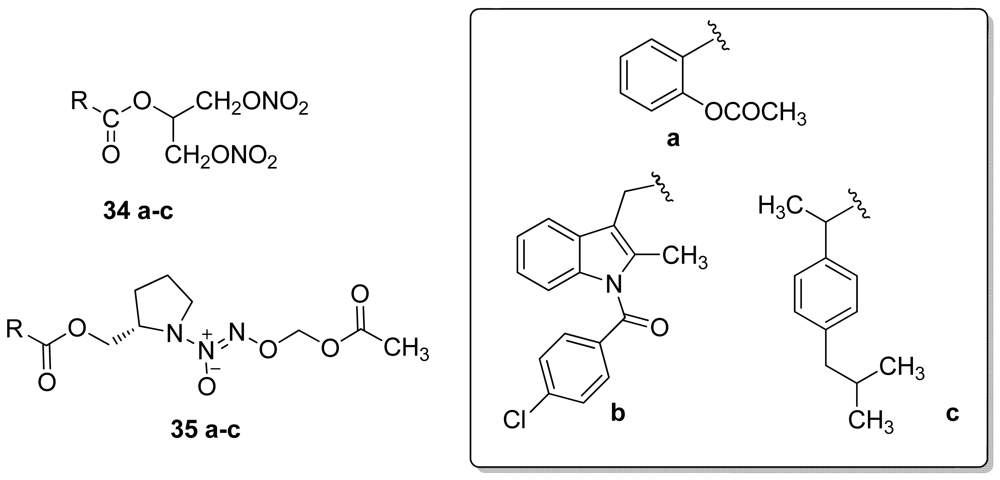
In 2009, the same authors developed NONO/NSAIDs wherein a 1,3-dinitrooxy-2-propyl (34a–c), or O2-acetoxymethyl-1-[2-(methyl)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate (35a–c) NO-donor moiety was directly linked to the carboxylic acid group of aspirin, indomethacin, or ibuprofen (Figure 24). All compounds showed COX-2 activity without COX-1 enzyme inhibition at the highest concentration used (100 μM) [55].
In the same year, Shoman and coworkers obtained a series of 3,5-diaryl-2-pyrazoline derivatives via the reaction of chalcones with hydrazine hydrate in ethanol. A group of NO-donating-2-pyrazoline derivatives was synthesized by carrying a nitrate ester group or an oxime group onto the prepared pyrazoline derivatives through different spacers (Figure 25) [56].
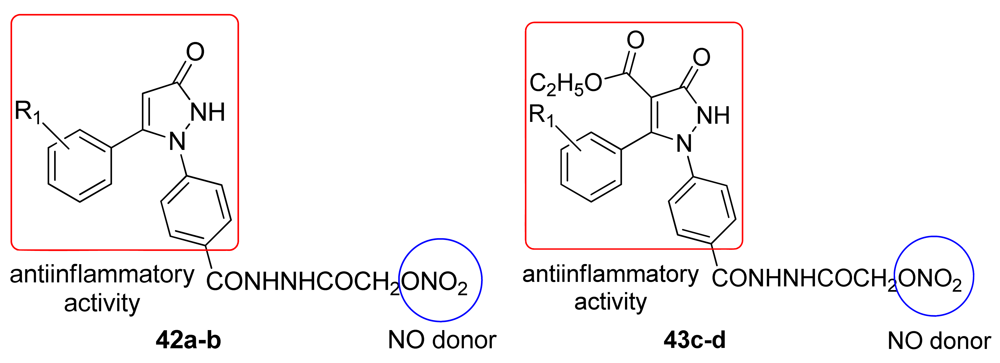
Most of the prepared compounds showed significant anti-inflammatory activity at 100 mg/kg and were safer than indomethacin with respect to gastric toxicity. Incorporation of the NO-donating group into the parent pyrazoline derivatives caused a nonsignificant reduction in the anti-inflammatory activity, but there was also a marked decrease in gastric ulcerations induced by their parent drug [56]. Bhandari and coworkers obtained several substituted 1,5-diarylpyrazol-3-one derivatives 42a-d and evaluated their analgesic, anti-inflammatory, and ulcerogenic activities, and their ability to release NO (Figure 26).
Some of these compounds exhibited analgesic and anti-inflammatory activities similar to indomethacin and all of the compounds showed analgesic activity and an absence of gastric damage compared with diclofenac [57].
In 2004, a series of novel pyrazoles containing a nitrate (ONO2) moiety functionality were synthesized by Ranatunge and coworkers. Modifications of the pyrazole moiety increased the COX-2 inhibitory potency compared to celecoxib and improved gastric tolerance (compound 44, Figure 27). Compound 45a proved most potent, with 78% inhibition of prostaglandin E2 formation and 43% inhibition of white cell infiltration at an oral dose of 45 μmol/kg. This compound 45a, however, showed poor selectivity (IC50 for COX-1 and COX-2 was 1.5 μM and 2.5 μM, respectively). Among selective pyrazole carbonyl derivatives, only compounds 45b and 45c showed good anti-inflammatory activity. Both inhibited prostaglandin E2 production by 45% at 45 μmol/kg [58].
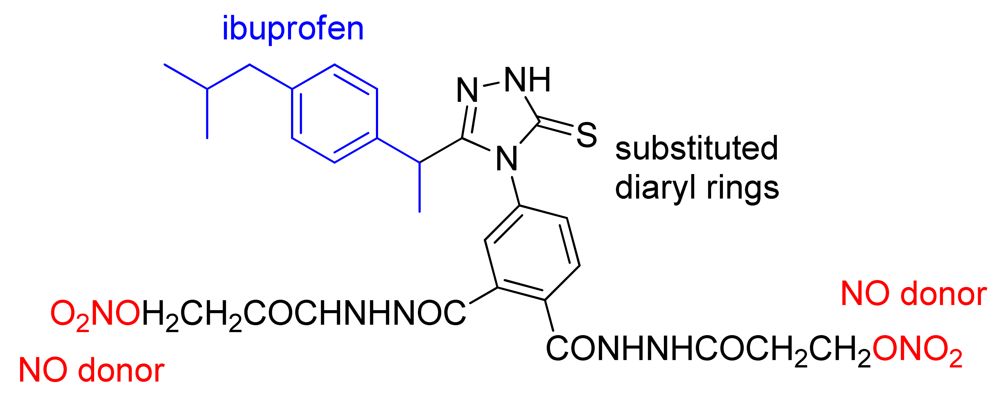
More recently, a series of hybrid molecules containing the pharmacophore moiety of ibuprofen and substituted diaryl rings on a 5-membered heterocycle similar to coxibs and an NO releasing moiety were obtained by Sarkate and coworkers (Figure 28). The compounds obtained showed anti-inflammatory activity similar to that of ibuprofen and decreased gastrotoxicity compared with diclofenac [59].
Therapy with selective COX-2 inhibitory anti-inflammatory drugs promotes an imbalance in the natural prostacyclin (PGI2) and thromboxane A2 biochemical pathway [60,61]. This process is responsible for myocardial infarction, a serious adverse reaction to selective COX-2 inhibitors that caused the market withdrawal of rofecoxib from the market. Hybridization of selective COX-2 inhibitors and NO donors can increase the safety of COX-2 inhibitors by increasing vasodilation, and inhibiting the platelet aggregation and adhesion effects. In fact, several researchers have obtained different COX-2 inhibitors linked with NO donors.
In 2007, Knaus and coworkers synthesized NO-releasing anti-inflammatory drug hybrids (49-51) possessing a 1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (a), or 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate (b), an NO donor moiety attached via a one-carbon methylene spacer to the carboxylic acid group of (E)-3-(4-methanesulfonylphenyl)-2-phenylacrylic acids (Figure 29). The most potent COX-2 inhibitor compound, however, was less potent than celecoxib [62].
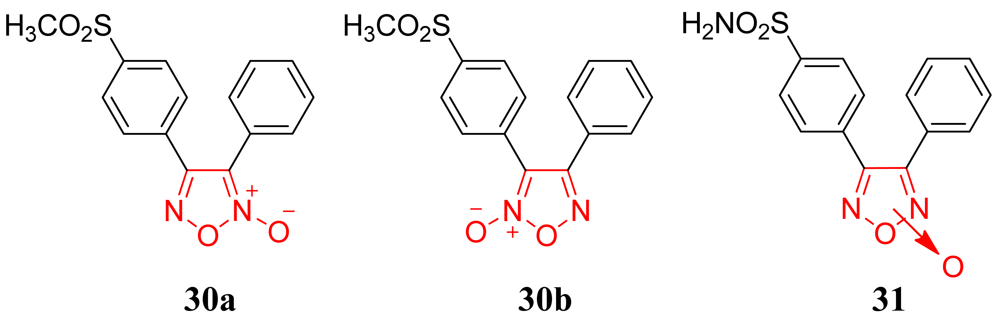
Knaus and coworkers prepared a series of bioisosteres of celecoxib linked to NO donor moieties (Figure 30).
The compounds were 4-[2-(4-methyl(amino)sulfonylphenyl)-5-trifluoromethyl-2H-pyrazol-3-yl]-1,2,3,6-tetrahydropyridines 52-55 possessing a variety of substituents (CH3, CO2CH2CH3, H, N = O) attached to the 1,2,3,6-tetrahydropyridyl N1-nitrogen atom. The anti-inflammatory activity of the two most potent compounds, 52a (SO2CH3; ED50 = 95.3 mg/kg p.o) and 52b (SO2NH2; ED50 = 61.2 mg/kg having an N-methyl-1,2,3,6-tetrahydropyridyl moiety, exhibited anti-inflammatory activities with potencies between those of the reference drugs celecoxib (ED50 = 10.8 mg/kg p.o) and aspirin (ED50 = 128.7 mg/kg p.o) [63]. In 2009 and 2010, other research groups tried to obtain a better COX-2/NO donor hybrid. Although none of the compounds demonstrated better anti-inflammatory activity than celecoxib, they showed fewer adverse effects [64,65]. The data indicated that product 57, obtained by Boschi and coworkers, and derived from substituting the nitrooxymethyl function for the methyl group of celecoxib (IC50 = 1.3 ± 0.4 μM), was a weak COX-2 inhibitor, being ca. 50 times less potent than the lead drug, but it retained a good degree of COX-2 selectivity (IC50 = 67 ± 19 μM): it also displayed negligible COX-1 activity when tested at 100 μM concentration (Figure 31) [64].
Hybrid nitric oxide donor prodrugs such as the N-(4-nitrooxybutyl) piperidines 62a–b and the corresponding 1,2,3,6-tetrahydropyridine analogs 63a–b constitute a potential class of selective COX-2 inhibitor agents that are devoid of adverse cardiovascular properties (Figure 32).
The COX-2 selectivity indices shown by compounds 62a–b and 63a–b are higher than those of the reference drug aspirin (selectivity index = 0.13) and comparable with those of ibuprofen (selectivity index = 2.64) [65]. The most potent COX-2 inhibitor, 1,2,3,6-tetrahydropyridyl compound 63b having a H2NSO2 moiety, is approximately equipotent with aspirin, but less potent than celecoxib and ibuprofen [65].
2. Conclusions
The current treatment of inflammation is limited by several adverse effects. Combinations of adequate subunits through the molecular hybridization of anti-inflammatory drugs can create new entities with superior therapeutic activity and better safety profiles.
























Acknowledgments
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP Process: 10/12495-6 and 2010/50330-9) and Conselho Nacional de Pesquisa (CNPq process 308157/2010-9).
Conflict of Interest
The authors declare no conflict of interest.
References
- Barreiro, E.J.; Fraga, C.A.M.; Miranda, A.L.P.; Rodrigues, C.R. A química medicinal de N-acilidrazonas: Novos compostos-protótipos de fármacos analgésicos, antiinflamatórios e anti-trombóticos. Quim. Nova 2002, 25, 129–148. [Google Scholar]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar]
- Vizioli, E.O. Ensaios Pré-clínicos de Híbridos Ftalimídicos e Pró-fármacos Taurínicos Derivados de Antiinflamatórios não Esteróides. Thesis, Faculdade de Ciências Farmacêuticas, UNESP, Araraquara, São Paulo, Brazil, 2009. [Google Scholar]
- Rocha, M.D.; Viegas-Jr, C. Síntese de Novos Candidatos a Fármacos Híbridos Carbamoilacilidrazônicos Planejados Como Antiinflamatórios e Anticolinesterásicos. Proceedings of 32a Reunião Anual da Sociedade Brasileira de Química, São Paulo, Brazil, 26–29 May 2008; Centro de Convenções do Hotel Monte Real Resort, Águas de Lindóia.
- Tseng, S.; Pak, G.; Washenik, K.; Pomeranz, M.K.; Shupack, J.L. Rediscovering thalidomide: A review of its mechanism of action, side effects, and potential uses. J. Am. Acad. Dermatol. 1996, 35, 969–979. [Google Scholar]
- Grosshans, E.; Illy, G. Thalidomide therapy for inflammatory dermatoses. Int. J. Dermatol. 1984, 23, 598–602. [Google Scholar]
- Stirling, D.I. Thalidomide and its impact in dermatology. Semin. Cutan. Med. Surg. 1988, 17, 231–242. [Google Scholar]
- Ordi-ros, J.; Cortes, F.; Cucurull, E.; Mauri, M.; Bujan, S.; Vilardell, M. Thalidomide in the treatment of cutaneous lupus refractory to conventional therapy. J. Rheumatol. 2000, 27, 1429–1433. [Google Scholar]
- Lima, L.M.; Fraga, C.A.M.; Barreiro, E.J. O renascimento de um fármaco: Talidomida. Quim. Nova 2001, 24, 683–688. [Google Scholar]
- Wannmacher, L. Inibidores seletivos de cicloxigenase-2 revisitados um ano depois. Uso racional de medicamentos. Temas Selecionados OPAS-OMS 2005, 2, 1–6. [Google Scholar]
- Borges, L.G.; Froehlich, P.E. Talidomida: Novas perspectivas para utilização como antiinflamatório, imunossupressor e antiangiogênico. Rev. Assoc. Med. Bras. 2003, 49, 96–102. [Google Scholar]
- Marriot, J.B.; Müller, G.; Dalgleish, A.G. Thalidomide as an emerging immunotherapeutic agent. Immunol. Today 1999, 20, 538–540. [Google Scholar]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar]
- Suryaprasad, A.G.; Prindiville, T. The biology of TNF blockade. Autoimmun. Rev. 2003, 2, 346–357. [Google Scholar]
- Kodama, S.; Davis, M.; Faustman, D.L. The therapeutic potential of tumor necrosis factor for autoimmune disease: A mechanistically based hypothesis. Cell. Mol. Life Sci. 2005, 62, 1850–1862. [Google Scholar]
- Popa, C.; Netea, M.G.; van Riel, P.L.; van der Meer, J.W.; Stalenhoef, A.F. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J. Lipid Res. 2007, 48, 751–762. [Google Scholar]
- Clark, P.R.; Pober, J.S.; Kluger, M.S. Knockdown of TNFR1 by the sense strand of an ICAM-1 siRNA: Dissection of an off-target effect. Nucleic Acids Res. 2008, 36, 1081–1097. [Google Scholar]
- Koch, A.; Zacharowski, K.; Boehm, O.; Stevens, M.; Lipfert, P.; von Giesen, H.J.; Wolf, A.; Freynhagen, R. Nitric oxide and pro-inflammatory cytokines correlate with pain intensity in chronic pain patients. Inflamm. Res. 2007, 56, 32–37. [Google Scholar]
- Schenk, M.; Bouchon, A.; Seibold, F.; Mueller, C. TREM-1--expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J. Clin. Invest. 2007, 117, 3097–3106. [Google Scholar]
- Lima, L.M.; Castro, P.; Machado, A.L.; Fraga, C.A.M.; Lugnier, C.; Moraes, V.L.G.; Barreiro, E.J. Synthesis and anti-Inflammatory activity of phthalimide derivatives, designed as new thalidomide analogues. Bioorg. Med. Chem. 2002, 10, 3067–3073. [Google Scholar]
- Santos, J.L.; Yamasaki, P.R.; Chung, M.C.; Takashi, C.H.; Pavan, F.R.; Leite, C.Q. Synthesis and in vitro anti Mycobacterium tuberculosis activity of a series of phthalimide derivatives. Bioorg. Med. Chem. 2009, 17, 3795–3799. [Google Scholar]
- Dos Santos, J.L.; Lanaro, C.; Lima, L.; Gambero, S.; Franco-Penteado, C.F.; Alexandre-Moreira, M.S.; Wade, M.; Yerigenahally, S.; Kutlar, A.; Meiler, S.E.; et al. Design, synthesis, and pharmacological evaluation of novel hybrid compounds to treat sickle cell disease symptoms. J. Med. Chem. 2011, 54, 5811–5819. [Google Scholar]
- Santos, J.L.; Oliveira, E.V.; Chung, M.C.; Menegon, R.F.; Blau, L. Derivados Ftalimídicos de Compostos Antiinflamatórios Não-Esteróide e/ou Moduladores de TNF-, Processo de sua Obtenção, Composições Farmacêuticas Contendo os Mesmos e Seus Usos no Tratamento de Doenças Inflamatórias”; Depositada em 06 de Abril de 2009, INPI 020090033479.
- Jung, Y.J.; Kim, H.H.; Kong, H.S.; Kim, Y.M. Synthesis and properties of 5-aminosalicyl-taurine as a colon-specific prodrug of 5-aminosalicylic acid. Arch. Pharm. Res. 2003, 26, 264–269. [Google Scholar]
- Joo, K.; Lee, Y.; Choi, D.; Han, J.; Hong, S.; Kim, Y.M.; Jung, Y. An anti-inflammatory mechanism of taurine conjugated 5aminosalicylic acid against experimental colitis: Taurine chloramines potentiatesinhibitory effect of 5-aminosalicylic acid on IL-1beta-mediated NFkappaB activation. Eur. J. Pharmacol. 2009, 618, 91–97. [Google Scholar]
- Vizioli, E.O.; Chung, M.C.; Menegon, R.F.; Blau, L.; Santos, J.L.; Longo, M.C. Novel compounds derived from taurine, process of their preparation and pharmaceutical compositions containing these. PCT/BR 2009/0409, WO/2009/124371.
- Vizioli, E.O.; Chung, M.C.; Menegon, R.F.; Blau, L.; Santos, J.L.; Longo, M.C. Novel compounds derived from taurine, process of their preparation and pharmaceutical compositions containing these. United States Patent Application 20110118303.
- Brawek, B.; Löffler, M.; Wagner, K.; Huppertz, H.J.; Wendling, A.S.; Weyerbrock, A.; Jackisch, R.; Feuerstein, T.J. Reactive oxygen species (ROS) in the human neocortex: Role of aging and cognition. Brain Res. Bull. 2010, 81, 484–490. [Google Scholar]
- Halliwell, R.B.; Aeschbach, J.L.; Löliger, J.; Aruoma, O.I. The characterization of antioxidants. Food Chem. Toxicol. 1995, 33, 601–617. [Google Scholar]
- Garrido, G.; GonzáLez, D.; Delporte, C.; Backhouse, N.; Quintero, G.; Núnez-Sélles, A.J.; Morales, M.A. Analgesic and anti-inflammatory effects of Mangifera indica L. extract (Vimang). Phytother. Res. 2001, 15, 18–21. [Google Scholar]
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 1997, 29, 315–331. [Google Scholar]
- Packer, L.; Witt, E.H.; Tritschler, H.J. alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar]
- Moini, H.; Packer, L.; Saris, N.-E.L. Antioxidant and prooxidant activities of alpha-lipoic acid and dihydrolipoic acid. Toxicol. Appl. Pharmacol. 2002, 182, 84–90. [Google Scholar]
- Gurer, H.; Ozgunes, H.; Oztezcan, S.; Ercal, N. Antioxidant role of alpha-lipoic acid in lead toxicity. Free Radic. Biol. Med. 1999, 27, 75–81. [Google Scholar]
- Alleva, R.; Nasole, E.; di Donato, F.; Borghi, B.; Neuzil, J.; Tomasetti, M. alpha-Lipoic acid supplementation inhibits oxidative damage, accelerating chronic wound healing in patients undergoing hyperbaric oxygen therapy. Biochem. Biophys. Res. Commun. 2005, 333, 404–410. [Google Scholar]
- Melagraki, G.; Afantitis, A.; Igglessi-Markopoulou, O.; Detsi, A.; Koufaki, M.; Kontogiorgis, C.; Hadjipavlou-Litina, D.J. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur. J. Med. Chem. 2009, 44, 3020–3026. [Google Scholar]
- Kontogiorgis, C.A.; Hadjipavlou-Litina, D. Synthesis and antiinflammatory activity of coumarin derivatives. J. Med. Chem. 2005, 48, 6400–6408. [Google Scholar]
- Ghate, M.; Manohar, D.; Kulkarni, V.; Shobha, R.; Kattimani, S.Y. Synthesis of vanillin ethers from 4-(bromomethyl) coumarins as anti-inflammatory agents. Eur. J. Med. Chem. 2003, 38, 297–302. [Google Scholar]
- Raji, Z.; Koncik, M.Z.; Miloloza, K.; Perkovic, I.; Butula, I.; Bucar, F.; Zorc, B. Primaquine-NSAID twin drugs: Synthesis, radical scavenging, antioxidant and Fe2+ chelating activity. Acta Pharm. 2010, 60, 325–337. [Google Scholar]
- Cena, C.; Boschi, D.; Tron, G.C.; Chegaev, K.; Lazzarato, L.; di Stilo, A.; Aragno, M.; Fruttero, R.; Gasco, A. Development of a new class of potential antiatherosclerosis agents: NO-donor antioxidants. Bioorg. Med. Chem. Lett. 2004, 14, 5971–5974. [Google Scholar]
- Lolli, M.L.; Cena, C.; Medana, C.; Lazzarato, L.; Morini, G.; Manarini, G.S.; Fruttero, R.; Gasco, A. A new class of ibuprofen derivatives with reduced gastrotoxicity. J. Med. Chem. 2001, 44, 3463–3468. [Google Scholar]
- Cena, C.; Lolli, M.L.; Lazzarato, L.; Guaita, E.; Morini, G.; Coruzzi, G.; McElroy, S.P.; Megson, I.L.; Fruttero, R.; Gasco, A. Antiinflammatory, gastrosparing, and antiplatelet properties of new NO-donor esters of aspirin. J. Med. Chem. 2003, 46, 747–754. [Google Scholar]
- Turnbull, C.M.; Marcarino, P.; Sheldrake, T.A.; Lazzarato, L.; Cena, C.; Fruttero, R.; Gasco, A.; Fox, S.; Megson, I.L.; Rossi, A.G. A novel hybrid aspirin-NO-releasing compound inhibits TNF-alpha release from LPS activated human monocytes and macrophages. J. Inflamm. (Lond) 2008, 5, 1–10. [Google Scholar]
- Gasco, A.; Boschi, D.; Chegaev, K.; Cena, C.; di Stilo, A.; Fruttero, R.; Lazzarato, L.; Rolando, B.; Tosco, P. Multitarget drugs: Focus on the NO-donor hybrid drugs. Pure Appl. Chem. 2008, 80, 1693–1701. [Google Scholar]
- Lazzarato, L.; Donnola, M.; Rolando, B.; Chegaev, K.; Marini, E.; Cena, C.; di Stilo, A.; Fruttero, R.; Biondi, S.; Ongini, E.; et al. (Nitrooxyacyloxy) methyl esters of aspirin as novel nitric oxide releasing aspirins. J. Med. Chem. 2009, 52, 5058–5068. [Google Scholar]
- Lazzarato, L.; Chegaev, K.; Marini, E.; Rolando, B.; Borretto, E.; Guglielmo, S.; Joseph, S.; di Stilo, A.; Fruttero, R.; Gasco, A. New nitric oxide or hydrogen sulfide releasing aspirins. J. Med. Chem. 2011, 11, 5478–5484. [Google Scholar]
- Hulsman, N.; Medema, J.P.; Bos, C.; Jongejan, A.; Leurs, R.; Smit, M.J.; Esch, I.J.P.; Richel, D.; Wijtmans, M. Chemical insights in the concept of hybrid drugs: The antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem. 2007, 50, 2424–2431. [Google Scholar]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar]
- Fang, L.; Zhang, Y.; Lehmann, J.; Wang, Y.; Ji, H.; Ding, D. Design and synthesis of furoxan-based nitric oxide-releasing glucocorticoid derivatives with potent anti-inflammatory activity and improved safety. Bioorg. Med. Chem. Lett. 2007, 17, 1062–1066. [Google Scholar]
- Kartasasmita, R.E.; Laufer, S.; Lehmann, J. NO-donors (VII [I]): Synthesis and cyclooxygenase inhibitory properties of N- and S-nitrooxypivaloyl-cysteine derivatives of naproxen – a novel type of NO-NSAID. Arch. Pharm. (Weinheim) 2002, 335, 363–366. [Google Scholar]
- Velazquez, C.; Rao, P.N.P.; McDonald, R.; Knaus, E.E. Synthesis and biological evaluation of 3,4-diphenyl-1,2,5-oxadiazole-2-oxides and 3,4-diphenyl-1,2,5-oxadiazoles as potential hybrid COX-2 inhibitor/nitric oxide donor agents. Bioorg. Med. Chem. 2005, 13, 2749–2757. [Google Scholar]
- Velázquez, C.; Rao, P.N.P.; Knaus, E.E. Novel nonsteroidal antiinflammatory drugs possessing a nitric oxide donor diazen-1-ium-1,2-diolate moiety: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem. 2005, 48, 4061–4067. [Google Scholar]
- Velazquez, C.A.; Rao, P.N.P.; Citro, M.L.; Keefer, L.K.; Knaus, E.E. O2-Acetoxymethyl-protected diazeniumdialate-based NSAIDs (NONO-NSAIDs): Synthesis, nitric oxide release, and biological evaluation studies. Bioorg. Med. Chem. 2007, 15, 4767–4774. [Google Scholar]
- Abdellatif, K.R.A.; Chowdhury, M.A.; Dong, Y.; Chen, Q.H.; Knaus, E.E. Diazen-1-ium-1,2-diolated and nitrooxyethyl nitric oxide donor ester prodrugs of anti-inflammatory (E)-2-(aryl)-3-(4-methanesulfonylphenyl)acrylic acids: Synthesis, cyclooxygenase inhibition, and nitric oxide release studies. Bioorg. Med. Chem. 2008, 16, 3302–3308. [Google Scholar]
- Abdellatif, K.R.A.; Chowdhury, M.A.; Dong, Y.; Das, D.; Yu, G.; Velázquez, C.A.; Suresh, M.R.; Knaus, E.E. Dinitroglyceryl and diazen-1-ium-1,2-diolated nitric oxide donor ester prodrugs of aspirin, indomethacin and ibuprofen: Synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett. 2009, 19, 3014–3018. [Google Scholar]
- Shoman, M.E.; Abdel-Aziz, M.; Aly, O.M.; Farag, H.H.; Morsy, M.A. Synthesis and investigation of anti-inflammatory activity and gastric ulcerogenicity of novel nitric oxide-donating pyrazoline derivatives. Eur. J. Med. Chem. 2009, 44, 3068–3076. [Google Scholar]
- Bhandari, S.V.; Dangre, S.C.; Bothara, K.G.; Patil, A.A.; Sarkate, A.P.; Lokwani, D.K.; Gore, S.T.; Deshmane, B.J.; Raparti, V.T.; Khachane, C.V. Design, synthesis and pharmacological screening of novel nitric oxide donors containing 1,5-diarylpyrazolin-3-one as nontoxic NSAIDs. Eur. J. Med. Chem. 2009, 44, 4622–4636. [Google Scholar]
- Ranatunge, R.R.; Augustyniak, M.; Bandarage, U.K.; Earl, R.A.; Ellis, J.L.; Garvey, D.S.; Janero, D.R.; Letts, L.G.; Martino, A.M.; Murty, D.R.; et al. Synthesis and selective cyclooxygenase-2 inhibitory activity of a series of novel, nitric oxide donorcontaining pyrazoles. J. Med. Chem. 2004, 47, 2180–2193. [Google Scholar]
- Sarkate, A.P.; Lokwani, D.K.; Patil, A.A.; Bhandari, S.V.; Bothara, K.G. Synthesis and evaluation of anti-inflammatory, analgesic, ulcerogenicity and nitric oxide-releasing studies of novel ibuprofen analogs as nonulcerogenic derivatives. Med. Chem. Res. 2010, 20, 795–808. [Google Scholar]
- Hinz, B.; Brune, K.J. Cyclooxygenase-2—10 years later. J. Pharmacol. Exp. Ther. 2002, 300, 367–375. [Google Scholar]
- Mukherjee, D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem. Pharmacol. 2002, 63, 817–821. [Google Scholar]
- Khaled, R.A.; Abdellatif, Y.D.; Chen, Q.H.; Chowdhury, M.A.; Knaus, E.E. Novel (E)-2-(aryl)-3-(4-methanesulfonylphenyl) acrylic ester prodrugs possessing a diazen-1-ium-1,2-diolate moiety: Design, synthesis, cyclooxygenase inhibition, and nitric oxide release studies. Bioorg. Med. Chem. 2007, 15, 6796–6801. [Google Scholar]
- Chowdhury, M.A.; Abdellatif, K.R.A.; Dong, Y.; Knaus, E.E. Synthesis of new 4-[2-(4-methyl(amino)sulfonylphenyl)-5-trifluoromethyl-2H-pyrazol-3-yl]-1,2,3,6-tetrahydropyridines: A search for novel nitric oxide donor anti-inflammatory agents. Bioorg. Med. Chem. 2008, 16, 8882–8888. [Google Scholar]
- Boschi, D.; Lazzarato, L.; Rolando, B.; Filieri, A.; Cena, C.; di Stilo, A.; Fruttero, R.; Gasco, A. Nitrooxymethyl substituted analogues of celecoxib: Synthesis and pharmacological characterization. Chem. Biodivers. 2009, 6, 369–379. [Google Scholar]
- Chowdhury, M.A.; Abdellatif, K.R.A.; Dong, Y.; Yu, G.; Huang, Z.; Rahman, M.; Das, D.; Velázquez, C.A.; Suresh, M.R.; Knaus, E.E. Celecoxib analogs possessing a N-(4-nitrooxybutyl)piperidin-4-yl or N-(4-nitrooxybutyl)-1,2,3,6-tetrahydropyridin-4-yl nitric oxide donor moiety: Synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett. 2010, 20, 1324–1329. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bosquesi, P.L.; Melo, T.R.F.; Vizioli, E.O.; Santos, J.L.d.; Chung, M.C. Anti-Inflammatory Drug Design Using a Molecular Hybridization Approach. Pharmaceuticals 2011, 4, 1450-1474. https://doi.org/10.3390/ph4111450
Bosquesi PL, Melo TRF, Vizioli EO, Santos JLd, Chung MC. Anti-Inflammatory Drug Design Using a Molecular Hybridization Approach. Pharmaceuticals. 2011; 4(11):1450-1474. https://doi.org/10.3390/ph4111450
Chicago/Turabian StyleBosquesi, Priscila Longhin, Thais Regina Ferreira Melo, Ednir Oliveira Vizioli, Jean Leandro dos Santos, and Man Chin Chung. 2011. "Anti-Inflammatory Drug Design Using a Molecular Hybridization Approach" Pharmaceuticals 4, no. 11: 1450-1474. https://doi.org/10.3390/ph4111450
APA StyleBosquesi, P. L., Melo, T. R. F., Vizioli, E. O., Santos, J. L. d., & Chung, M. C. (2011). Anti-Inflammatory Drug Design Using a Molecular Hybridization Approach. Pharmaceuticals, 4(11), 1450-1474. https://doi.org/10.3390/ph4111450