Improved Histone Deacetylase Inhibitors as Therapeutics for the Neurodegenerative Disease Friedreich’s Ataxia: A New Synthetic Route

Abstract
: Friedreich's ataxia (FRDA) is caused by transcriptional repression of the nuclear FXN gene encoding the essential mitochondrial protein frataxin. Based on the hypothesis that the acetylation state of the histone proteins is responsible for gene silencing in FRDA, previous work in our lab identified a first generation of HDAC inhibitors (pimelic o-aminobenzamides), which increase FXN mRNA in lymphocytes from FRDA patients. Importantly, these compounds also function in a FRDA mouse model to increase FXN mRNA levels in the brain and heart. While the first generation of HDAC inhibitors hold promise as potential therapeutics for FRDA, they have two potential problems: less than optimal brain penetration and metabolic instability in acidic conditions. Extensive optimization focusing on modifying the left benzene ring, linker and the right benzene ring lead to a novel class of HDAC inhibitors that have optimized pharmacological properties (increased brain penetration and acid stability) compared to the previous HDAC inhibitors. This article will describe the chemical synthesis and pharmacological properties of these new HDAC inhibitors.1. Introduction
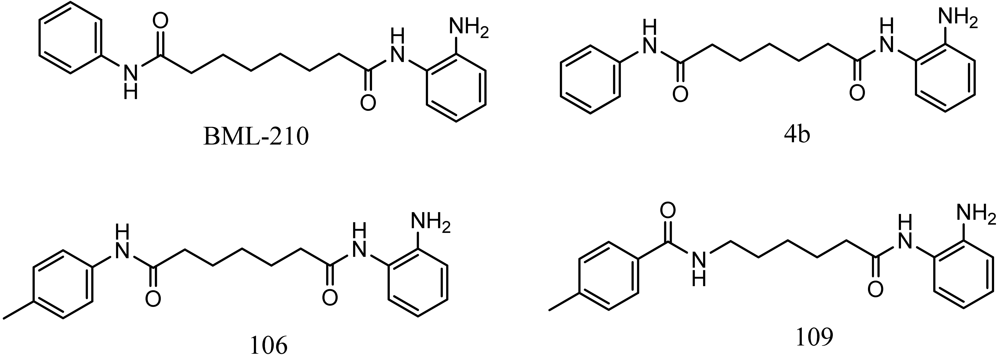
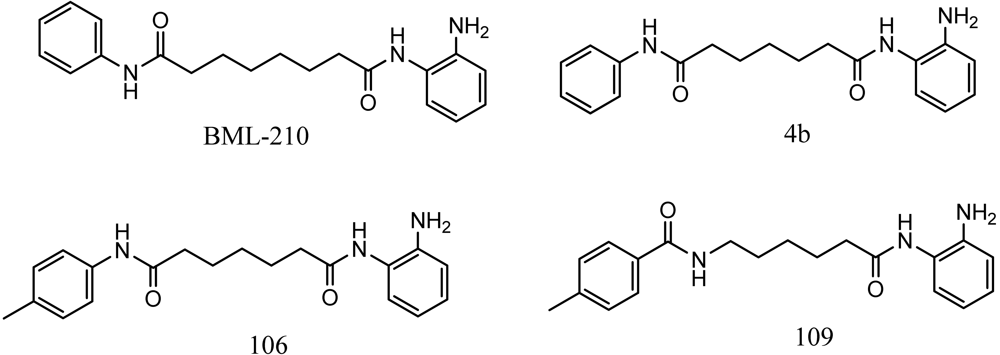
Friedreich's ataxia (FRDA) is caused by transcriptional repression of the nuclear FXN gene encoding the essential mitochondrial protein frataxin [1-3]. Currently there is no effective therapy for FRDA that addresses the cause of the disease. Unlike many triplet-repeat diseases (e.g., the polyglutamine expansion diseases), expanded GAA•TTC triplets in FXN are in an intron and do not alter the amino acid sequence of frataxin protein; thus, gene activation would be of therapeutic benefit. Based on the hypothesis that chromatin structure and the histone modification state of the FXN gene are responsible for gene repression on alleles containing expanded GAA•TTC repeats, our lab found that a commercially available histone deacetylase inhibitor (BML-210, Figure 1) and derivatives 4b, 106 and 109 that we synthesized (Figure 1) relieve repression of the FXN gene in lymphoid cell lines derived from FRDA patients, in primary lymphocytes from donor FRDA patient blood [4-6], and in mouse models [7-9]. Recently, these compounds have also shown efficacy in FRDA patient iPSC-derived neuronal cells [10].
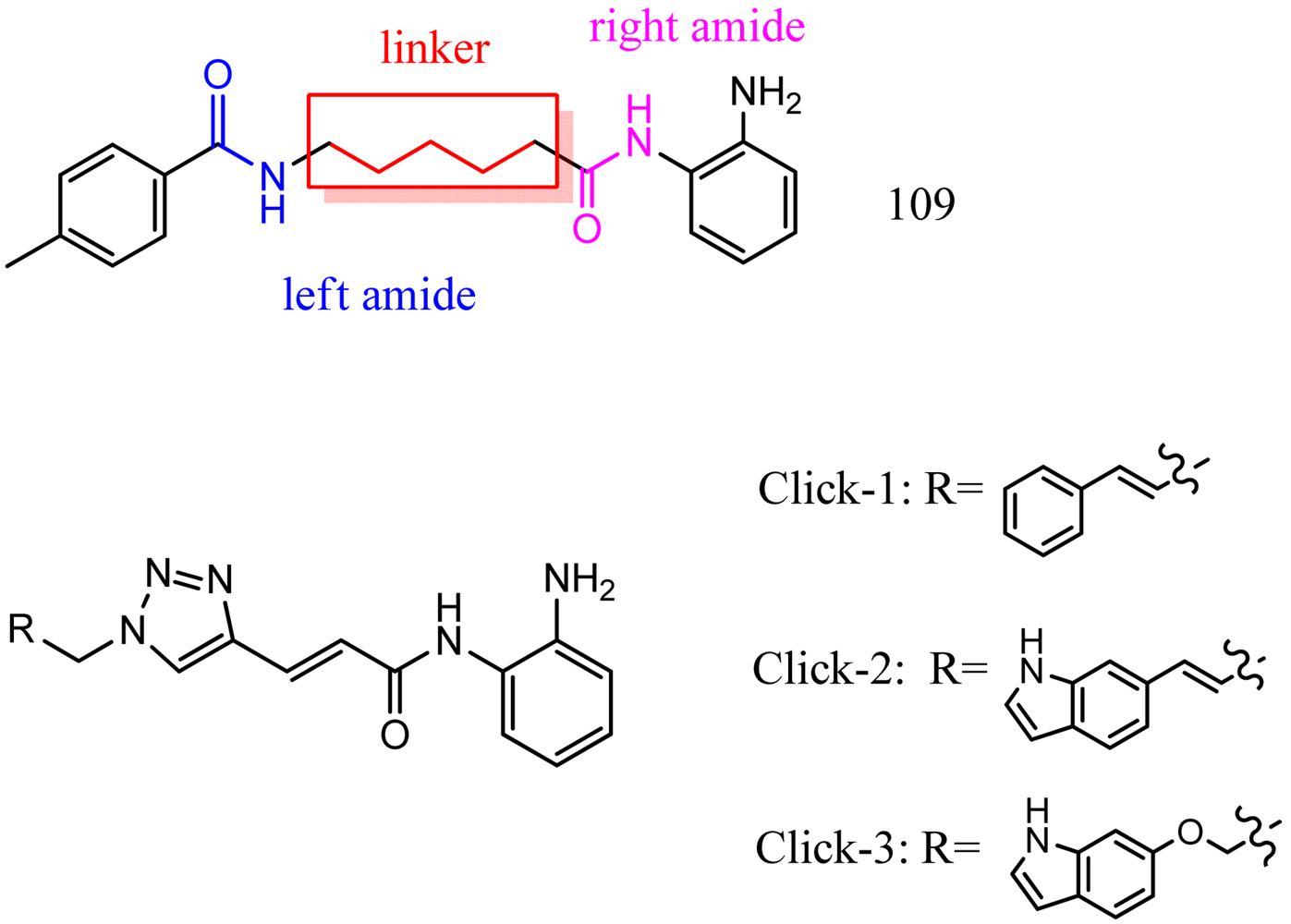
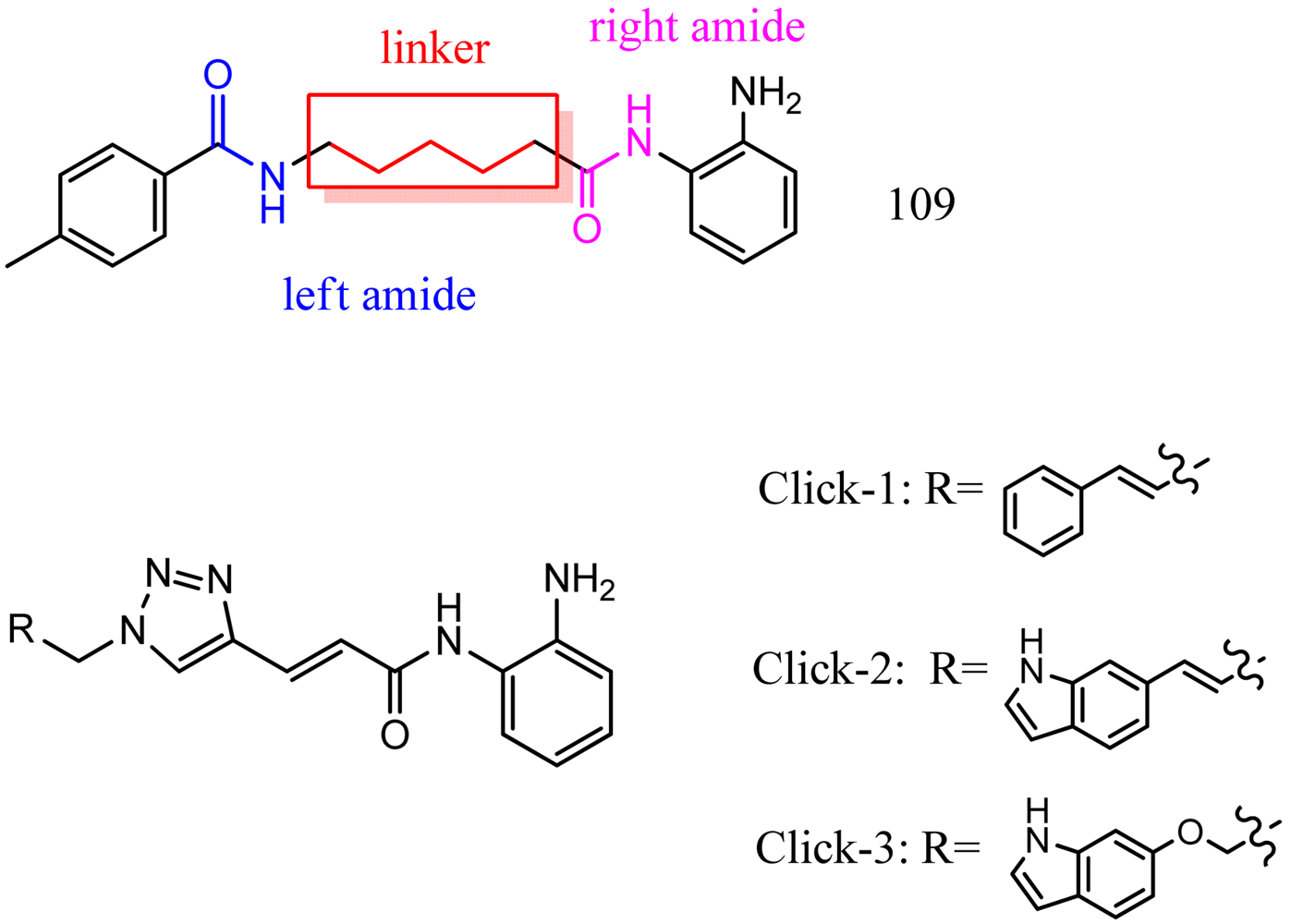
Optimization of our original series of compounds, exemplified by 4b [4] and 106 [7] lead to the development of HDACi 109 [8,9]. This compound has been subjected to full preclinical development and will soon enter human clinical trials for FRDA. While 109 holds promise as a FRDA therapeutic, this molecule has two potential problems: less than optimal brain penetration [11] and cyclization to a benzimidazole in vivo. We have identified two structural features that solve these problems: replacement of the “left” amide with an ether, olefin or ketone to improve brain penetration and introduction of an unsaturated linkage adjacent to the “right” amide to prevent formation of a benzimidazole metabolic byproduct. Based on these results, we devised and synthesized a new lead compound click-1 (Figure 2) using Cu(I)-catalyzed click chemistry [12]. Structure-activity relationship at the left side (cap) of the molecule was evaluated by replacement of the left benzene ring with an indole ring, which generated derivative click-2. The left double bond on click-2 was replaced with an ether to give another derivative click-3 (Figure 2). Activity against recombinant HDACs (HDAC1, 2 and 3), activity in restoring FXN transcription in patient cell lines, blood brain barrier penetration and acid stability have been assayed for each of these derivatives.
2. Results and Discussion
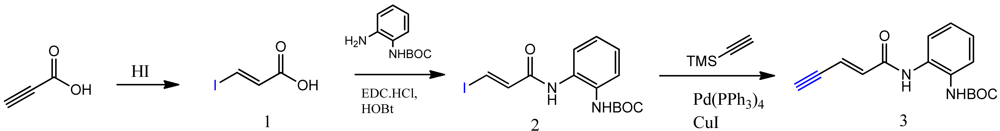
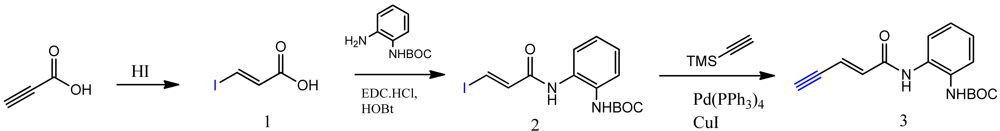
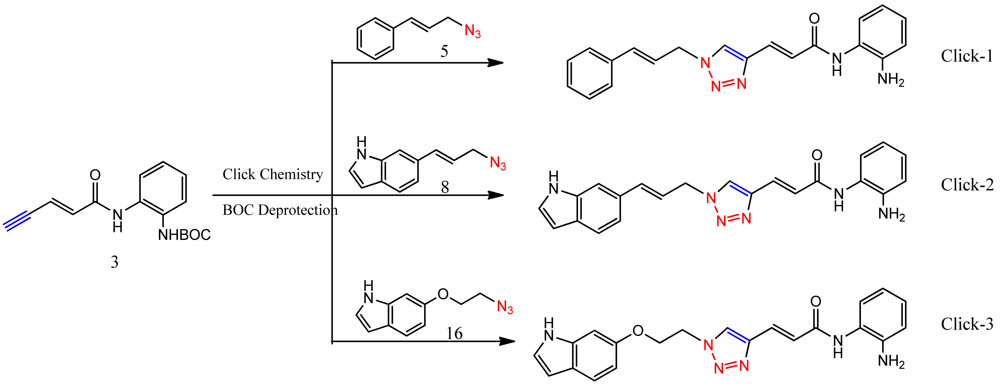
The synthesis of compounds click-1, -2, and -3 relies on the formation of a triazole ring by coupling of a common enyne intermediate with various azides using a Cu(I) catalyst. Hydrogen iodide addition to propiolic acid generated acrylic iodide 1, which was subjected to standard amide coupling with N-Boc-1,2-phenylenediamine to give compound 2. Sonogashira coupling of vinyl iodide 2 with trimethylsilyl acetylene lead to the common intermediate enyne 3 for the following click chemistry (Scheme 1).
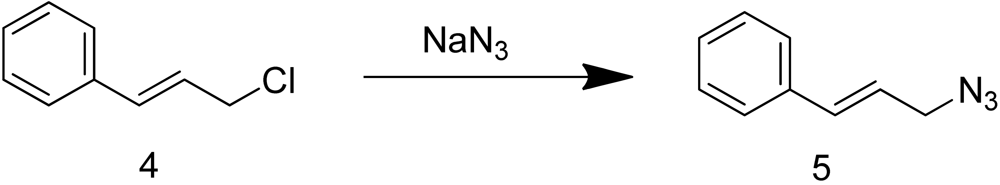
Cinnamyl azide 5 was prepared by treating commercially available cinnamyl chloride 4 with sodium azide (Scheme 2).
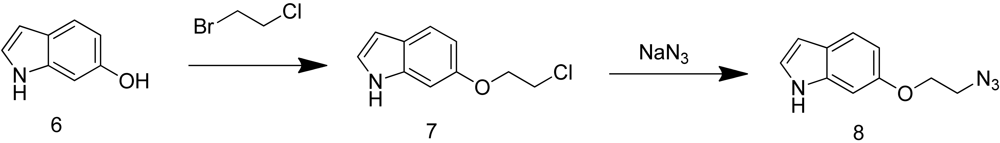
Both 6-(2-azidoethoxy)-1H-indole (8) and (E)-N-(t-butoxycarbonyl)-6-(3-azidoprop-1-en-1-yl)-1H-indole (16) were synthesized by Aberjona Laboratories (Beverly, MA, USA) through the following synthetic routes.
6-(2-azidoethoxy)-1H-indole (8) was obtained through reaction of sodium azide with the corresponding chloride 7, which was prepared by substitution reaction of 6-hydroxy-1H-indole with 1-bromo-2-chloroethane (Scheme 3).
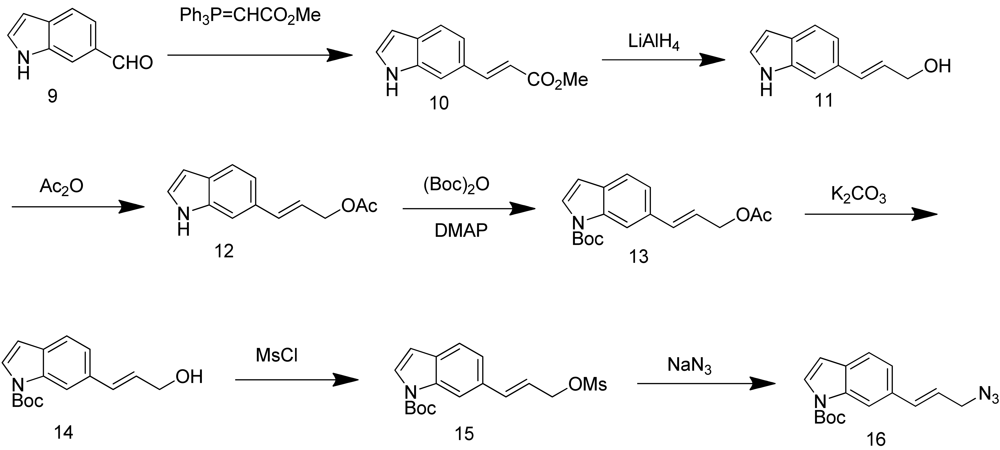
Synthesis of (E)-N-(t-butoxycarbonyl)-6-(3-azidoprop-1-en-1-yl)-1H-indole (16) started with commercially available compound 1H-indole-6-carbaldehyde (9). Wittig olefination of aldehyde 9 afforded compound 10. Reduction of the ester 10, followed by acetylation of the alcohol 11 lead to compound 12. Boc protection of the indole amine, removal of acetyl group with potassium carbonate, followed by mesylation of the free alcohol 14 generated compound 15, which was reacted with sodium azide to give azide 16 (Scheme 4).
All three azides 5, 8 and 16 were subjected to click chemistry with common intermediate 3 to give the corresponding triazole products click-1, click-2 and click-3 (Scheme 5). Each compound was characterized by 1H-, 13C-NMR and mass spectrometry. In vitro HDAC assays, in-cell deacetylase (DAC) assays, and cellular assays for effects on FXN transcription were carried out for all three click compounds, and the results compared to those for the original lead compound 109. Compound click-3 displayed poor stability; hence, studies with this compound were not pursued further.
HDAC assays: Previous studies have established that the pimelic o-aminobenzamides are potent inhibitors of class I HDAC enzymes [6]. Therefore, the inhibitory activity of compounds click-1 and click-2 against the recombinant class I HDACs 1-3 was determined (Table 1). By measuring IC50 values, we find that these compounds retain full activity as inhibitors of these enzymes, compared to HDACi 109. In fact, both click compounds exhibit lower IC50 values against these three enzymes than 109. Notably, the click compounds are more potent against HDACs 1 and 2 than 109. This increased activity against HDACs 1 and 2 reduces the selectivity of the click compounds for HDAC3, where compound 109 has a 4.3-fold selectivity for HDAC3 over HDAC1, while click-1 and -2 are only 2.4- and 1.2-fold selective for HDAC3 over HDAC1, respectively. In-cell efficacy of these compounds, as measured in the DAC assay (see Experimental section), is also not significantly different from 109. It should be also pointed out that both click-1 and click-2 have comparable inhibitory activity against recombinant HDACs compared to the well-studied benzamide MS-275 (IC50s: HDAC1 = 0.036 μM; HDAC2 = 0.219 μM; HDAC3 = 0.114 μM and DAC = 1.9 μM); however, MS-275 has cytotoxicity issue and does not penetrate the central nervous system [15].
Cytotoxicity assays: Cytotoxicity was determined using an antiproliferation assay with Hct116 cells. MS-275, being cytotoxic, gave an IC50 of 1 μM, while click-1 and click-2 had IC50s = 23 and 27 μM respectively. Comparing these IC50 values for blocking cell proliferation to the in-cell DAC assay values (Table 1) yields ratios of 0.53 for MS-275 (cytotoxicity/DAC), 4.1 for click-1 and 5.3 for click-2. Thus, our click compounds demonstrate a more favorable therapeutic index for a chronic disease compared to MS-275.
Frataxin mRNA assays: Through target identification, our lab has recently demonstrated that HDAC3 plays a key role in FXN gene transcription [5]. In general, the activity of a compound against HDAC3 is related to its ability to increase FXN gene transcription [4]. Compounds click-1 and -2 were assayed for their ability to increase transcription of the FXN gene in primary lymphocytes from a FRDA patient, as previously described [4,5].
Quantitative RT-PCR was used to estimate FXN mRNA levels after treatment with 10 μM compound for 48 h, and values were normalized to the level of FXN mRNA observed for a DMSO vehicle control. Importantly, compounds click-1 and click-2 are highly active in promoting transcription of the silenced FXN gene in primary lymphocytes isolated from Friedreich's ataxia patient donor blood, pointing to the therapeutic potential of these compounds (Figure 3).
Brain penetration and acid stability: Since HDACi 109 suffers from poor brain penetration (∼10–15% of plasma levels) and from formation of a benzimidazole under acid conditions, we monitored these two properties for click-1 and click-2, and compared the results with those obtained for HDACi 109. The half-life of each compound in acidic conditions was used to quantify their relative stability. At pH = 2 and 50 °C, t1/2 = 6 h for 109, t1/2 = 33 h for click-1 and t1/2 = 24 h for click-2, representing improvements of 5.5- and 4-fold respectively relative to HDACi 109. These results demonstrate that placing a double bond next to the right amide bond improves stability in acidic conditions. Blood brain barrier penetration was measured in the rat for compound 109 and click-1. Brain penetration (as measured by brain/plasma ratio at tmax) was determined to be 0.15 for 109 and 0.33 for click-1 (Cmax: 800 ng/g in the brain at Tmax = 5 min post-dose (5 mg/kg i.v. rat)), representing a significant increase in brain penetration for click-1. Click-2 was tested in mouse only, where brain penetration was limited with a brain/plasma of 0.03, as can be expected because of the indole cap (relatively hydrophilic and capable of intermolecular H-bonding).
3. Experimental
3.1. General
Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. NMR spectra were recorded on Bruker DRX-500, Varian Inova-400 and Varian Mercury-300 instruments and calibrated using residual undeuterated solvent as an internal reference (CHCl3 @ 7.24 ppm 1H-NMR, 77.0 ppm 13C-NMR, CD3OD @ 3.35 ppm 1H-NMR, 49.3 ppm 13C-NMR). High resolution mass spectra (HRMS) were recorded at the Center of Mass Spectrometry, the Scripps Research Institute.
3.2. (E)-Tert-butyl (2-(3-Iodoacrylamido)phenyl)carbamate (2)

(E)-3-Iodo-2-propenoic acid (0.40 g, 2.02 mmol) was dissolved in anhydrous dichloromethane (8 mL) under argon protection. Oxalyl chloride (0.34 mL, 3.96 mmol) was added dropwise to the above solution, one drop of DMF was added to catalyze the reaction, and the reaction was stirred at room temperature for 2 h. After removal of the solvent, the residue was used directly in the next step. Anhydrous dichloromethane (10 mL) was added to the above acid chloride, and N-Boc phenylenediamine (0.35 g, 1.68 mmol) was added to the solution, followed by diisopropylethylamine (0.42 mL, 2.42 mmol). The reaction was kept at room temperature for 1 h. Removal of the solvent followed by column chromatography gives 0.51 g of product 2. Yield: 78%; 1H-NMR (300 MHz, CD3OD) (ppm): 8.53 (1H), 8.06 (d, 1H, 3J = 15Hz), 6.96 (d, 1H, 3 J = 15Hz), 6.92–7.50 (4H), 1.52 (s, 9H); 13C-NMR (75 MHz, CD3OD) (ppm): 165.7, 137.2, 135.0, 129.7, 128.2, 127.4, 126.6, 125.7, 99.3, 77.6, 27.6.
3.3. (E)-Tert-butyl (2-(Pent-2-en-4-ynamido)phenyl)carbamate (3)

Iodo propenamide 2 (0.19 g, 0.49 mmol) was dissolved in DMF/Et3N (4:1, 2 mL), Pd(PPh3)2Cl2 (17.3 mg, 0.0245 mmol) and copper iodide (9.4 mg, 0.049 mmol) were added to the solution and the mixture was degassed for 5 minutes. Trimethylsilylacetylene (0.13 mL, 0.91 mmol) was added to the above solution under argon protection. The whole reaction was kept at room temperature for 1 h. The mixture was then poured into sat. (NH4)2SO4 (50 mL) and extracted with ethyl acetate (50 mL × 3). The combined organic layers were washed with brine, dried with sodium sulfate, filtered, and concentrated. The residue was subjected to flash column chromatography to yield 0.15 g trimethylsilyl enyne product, yield: 86%. The trimethylsilyl enyne product (0.15 g, 0.42 mmol) was redissolved in THF/Methanol (1:1, 2 mL), cesium fluoride (76 mg, 0.50 mmol, 1.2 eq.) was then added and the reaction was monitored by TLC analysis. The product is slightly more polar. Upon the disappearance of the reactant, the reaction mixture was concentrated and purified by column chromatography to yield the enyne product 3 (0.11 g, 95%); 1H-NMR (300 MHz, CDCl3) (ppm): 8.59 (1H), 6.97–7.47 (4H), 6.78 (dd, 1H, 3J= 15.3 Hz, 4 J= 2.4 Hz), 6.44 (d, 1H, 3J = 15.3 Hz), 3.32 (s, 1H), 1.52 (s, 9H); 13C-NMR (75 MHz, CDCl3) (ppm): 166.2, 135.0, 129.7, 126.6, 125.7, 124.6, 121.8, 81.5, 80.8, 77.6, 28.5.
3.4. 6-(2-Azidoethoxy)-1H-indole (8)

Sodium azide (416 mg, 2 eq.) was added to 6-(2-chloroethoxy)-1H-indole (626 mg, 3.2 mmol) in DMSO (10 mL) at room temperature. The mixture was heated for 8 h at 80 °C. After the mixture was cooled to room temperature it was diluted with CH2Cl2 and washed twice with water. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by reversed phase preparative LC to afford compound 8, (425 mg, 66%); 1H-NMR (300 MHz, CDCl3) (ppm): 8.04 (br s, 1H), 7.54 (d, 3J = 8.7 Hz), 7.12 (m, 1H), 6.91 (br s, 1H), 6.83 (dd, 3J = 8.7 Hz and 4J = 2.1 Hz, 1H), 6.50 (m, 1H), 4.19 (t, 3J = 4.8 Hz, 2H), 3.61 (t, 3J = 4.8 Hz, 2H).
3.5. (E)-Tert-butyl 6-(3-((Methylsulfonyl)oxy)-prop-1-en-1-yl)-1H-indole-1-carboxylate (15)

The mesylate 15 was prepared by addition of mesyl chloride (320 L, 1.5 eq.) and triethylamine (770 L, 2 eq.) to a solution of (E)-tert-butyl 6-(3-hydroxyprop-1-en-1-yl)-1H-indole-1-carboxylate (800 mg, 2.9 mmol) in dichloromethane (30 mL). The mixture was stirred at room temperature for 1 h. It was then washed with water and brine, dried over Na2SO4, and concentrated to give crude 15, which was used for the next step without further purification.
3.6. (E)-N-(t-Butoxycarbonyl)-6-(3-azidoprop-1-en-1-yl)-1H-indole (16)

Crude (E)-tert-butyl 6-(3-((methylsulfonyl)oxy)prop-1-en-1-yl)-1H-indole-1-carboxylate (15) was dissolved in DMSO (10 mL). Sodium azide (355 mg, 5.46 mmol) was added at room temperature and the mixture was heated for 8 h at 80 °C. The mixture was cooled to room temperature and diluted with dichloromethane. It was then washed twice with water. The organic layer was dried over Na2SO4, and concentrated. The residue was purified by reversed phase preparative LC to give compound 16 (261 mg, 30%); 1H-NMR (300 MHz, CDCl3) (ppm): 8.25 (br s, 1H), 7.58 (d, 4J = 3.6 Hz, 1H), 7.51 (d, 3J = 8.1 Hz, 1H), 7.31 (dd, 3J = 8.1 and 4J = 1.5 Hz), 6.76 (d, 3J = 15.9 Hz, 1H), 6.54 (d, 4J = 3.6 Hz, 1H), 6.30 (dt, 3J = 15.9 and 3J = 6.6 Hz, 1H), 3.96 (d, 3J = 6.6 Hz, 2H), 1.7 (s, 9H).
3.7. (E)-N-(2-Aminophenyl)-3-(1-cinnamyl-1H-1,2,3-triazol-4-yl)acrylamide (click 1)

A mixture of cinnamyl azide 5 (0.18 g, 1.15 mmol), enyne intermediate 3 (0.27 g, 0.96 mmol), sodium ascorbate (38 mg, 0.192 mmol, 20 mol%) and CuSO4 (12 mg, 0.048 mmol, 5 mol%) in t-BuOH-H2O (2:1, 3.9 mL) was stirred at room temperature for 16 h. Water (20 mL) was added, and the product was extracted with ethyl acetate (30 mL, 3×). The combined organic layers were washed with brine (60 mL), dried with sodium sulfate, filtered, and concentrated. The resulting residue was subjected to column chromatography to yield the desired product (0.36 g, yield: 84%). The Boc-protected intermediate (0.36 g, 0.81 mmol) from the above reaction was dissolved in 20% TFA in dichloromethane (3.5 mL). The solution was stirred at room temperature for 1 h. The solvent was removed, and the reaction mixture was redissolved in 10 mL dichloromethane and concentrated again. The resulting residue was purified by column chromatography to give the desired product (0.25 g, yield: 90%).1H-NMR (500 MHz, CD3OD) (ppm): 8.31 (s, 2H), 7.76 (d, 1H, 3J = 15.5 Hz), 7.26–7.46 (10H), 7.04 (d, 1H, 3J = 16 Hz), 6.75 (d, 1H, 3J = 15.5 Hz), 6.47 (dd, 1H, 3J = 6.5 Hz, 16 Hz), 5.23 (dd, 2H, 3J = 6.75 Hz and 4J = 1.5 Hz); 13C-NMR (125 MHz, CD3OD) (ppm): 166.1, 143.7, 136.2, 135.6, 131.1, 128.7, 128.6, 128.4, 127.6, 126.8, 125.6, 125.0, 123.7, 122.2, 121.0, 52.4; ESI-TOF: Expected: 346.1662, Observed: 346.1661.
3.8. (E)-3-(1-((E)-3-(1H-Indol-6-yl)allyl)-1H-1,2,3-triazol-4-yl)-N-(2-aminophenyl)acrylamide (click 2)
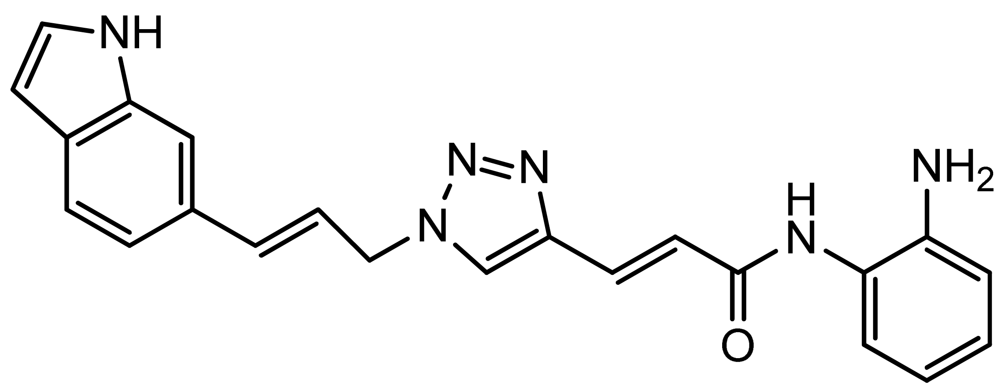
A mixture of (E)-N-(t-butoxycarbonyl)-6-(3-azidoprop-1-en-1-yl)-1H-indole (16, 0.125 g, 0.42 mmol), enyne intermediate 3 (0.100 g, 0.35 mmol), sodium ascorbate (14 mg, 0.071 mmol, 20 mol%) and CuSO4 (5 mg, 0.002 mmol, 5 mol%) in t-BuOH-H2O (2:1, 1.5 mL) was stirred at room temperature for 16 h. Water (10 mL) was added, and the product was extracted with ethyl acetate (15 mL, 3×). The combined organic layers were washed with brine (30 mL), dried with sodium sulfate, filtered, and concentrated. The resulting residue was subjected to column chromatography to yield the desired Boc-protected product (0.20 g, yield: 81%). The Boc-protected product (0.20 g, 0.34 mmol) from the above reaction was dissolved in 20% TFA in dichloromethane (2 mL) and the solution was stirred at room temperature for 1 h. The solvents were removed, and the reaction product was redissolved in 10 mL dichloromethane and concentrated again. The residue was purified by column chromatography to give click-2 (0.12 g, yield: 92%); 1H-NMR (500 MHz, CD3OD) (ppm): 8.30 (1H, s), 8.04 (1H, s), 7.71 (1H, d, 3J = 15.5 Hz), 7.57 (1H, d, 3J = 8.0 Hz), 7.51 (1H, s), 7.32 (1H, d, 4J= 3.0 Hz), 7.27 (2H, td, 3J= 8.5 and 4J = 1.5 Hz), 7.06–7.13 (2H), 6.90–6.95 (2H), 6.79–6.83 (1H), 6.44–6.50 (2H), 5.27 (2H, dd, 3J= 4.0 and 4J = 1.5 Hz); 13C-NMR (125 MHz, CD3OD) (ppm): 165.5, 144.0, 142.1, 137.4, 136.8, 129.6, 129.4, 128.9, 127.3, 125.9, 125.8, 124.5, 124.0, 122.3, 120.4, 119.2, 118.5, 117.9, 117.6, 110.3, 101.5, 52.7; ESI-TOF: Expected: 385.1771, Observed: 385.1781.
3.9. (E)-3-(1-(2-((1H-Indol-6-yl)oxy)ethyl)-1H-1,2,3-triazol-4-yl)-N-(2-aminophenyl)acrylamide (click 3)

A mixture of 6-(2-azidoethoxy)-1 H-indole (8, 0.085 g, 0.42 mmol), enyne intermediate 3 (0.100 g, 0.35 mmol), sodium ascorbate (14 mg, 0.071 mmol, 20 mol%) and CuSO4 (5 mg, 0.002 mmol, 5 mol%) in t-BuOH-H2O (2:1, 1.5 mL) was stirred at room temperature for 16 h. Water (10 mL) was added, and the product was extracted with ethyl acetate (15 mL, 3×). The combined organic layers were washed with brine (30 mL), dried with sodium sulfate, filtered, and concentrated, and the resulting residue was subjected to column chromatography to yield the desired product (0.13 g, yield: 78%). The bis-Boc protected product (0.16 g, 0.33 mmol) from the above reaction was dissolved in 2 mL 20% TFA in dichloromethane and the mixture was stirred at room temperature for 1 h. Solvent was removed, and the reaction product was redissolved in 10 mL dichloromethane and concentrated again. The resulting residue was purified by column chromatography to give pure click-3 (0.12 g, yield: 95%). 1H-NMR (500 MHz, CD3OD) (ppm): 8.47 (1H, s), 8.37 (1H, s), 7.50-7.61 (5H), 7.42 (2H, d, 4J = 1.5 Hz), 7.15-7.25 (5H), 7.029 (1H, d, 3J = 15.5 Hz), 6.97 (1H, d, 4J = 1.5 Hz), 6.65 (1H, dd, 3J = 8.5 and 4J = 2 Hz), 6.40 (1H, m), 4.83 (2H, t, 3J= 4.5 Hz), 4.43 (2H, t, 3J = 4.5 Hz); 13C-NMR (125 MHz, CD3OD) (ppm): 165.2, 154.5, 154.2, 143.6, 137.1, 130.1, 126.6, 125.3, 125.1, 123.3, 122.9, 121.6, 110.2, 101.9, 96.8, 80.8, 67.4, 50.4; ESI-TOF: Expected: 389.1720, Observed: 389.1714.
3.10. HDAC Enzyme Inhibition Assay
Serial dilutions of HDAC inhibitors were prepared in HDAC assay buffer (25 mM Tris/HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8) from DMSO stock solutions in 96-well assay plates (Fisher scientific, #07-200-309) and were pre-incubated for 2 h at ambient temperature in the presence of 125 μg/mL BSA and purified HDAC1 (#50051, BPS Bioscience, San Diego, CA, USA), HDAC2 (#50053, BPS Bioscience), or HDAC3/NcoR2 (#50003, BPS Bioscience) at concentrations of 1.25, 1.32, and 0.167 μg/mL, respectively. Following pre-incubation, Fluor-de-LysTM substrate (Enzo Life Sciences, Farmingdale, NY, USA) was added to a final concentration of 10 μM and plates were further incubated for 30 minutes at room temperature. The enzymatic reaction was stopped by addition of trichostatin A (Sigma-Aldrich, St. Louis, MO, USA, #T8552, final concentration: 100 nM) and trypsin (#02101179, MP Biomedicals, Solon, OH, USA) was added to reach a final concentration of 100 μg/mL. After a 15-minute incubation at room temperature, fluorescence was recorded using a Spectramax M2 fluorometer (Molecular Devices, Sunnyvale, CA, USA) with excitation at 365 nm and emission at 460 nm. IC50 values were calculated by using a sigmoidal dose-response (variable slope) equation in GraphPad Prism® 5 for Windows (GraphPad Software, La Jolla, CA, USA).
3.11. In-Cell Deacetylase Inhibition Assay (DAC Assay)
GM15850 cells (an FRDA lymphoblastoid cell line from the Coriell Institute NIGMS collection) were seeded in 96-well plates at an appropriate density (100,000 cells/well) in 90 μL RPMI1640 medium containing 10% v/v fetal bovine serum (FBS), 1% v/v penicillin/streptomycin, and 1% v/v L-glutamine. Compound dilutions were made in 100% DMSO followed by parallel dilution in media with 2% DMSO. 10 μL of the compound dilutions were added to the cells to achieve the desired concentrations. The final concentration of DMSO in each well was 0.2%. Cells were incubated for 4 h at 37 °C with 5% CO2. After incubation, cells were centrifuged and the supernatant was removed. Cell pellets were washed with 100 μL phosphate-buffered saline (PBS) and then lysed with 45 μL lysis buffer (HDAC assay buffer at pH 8.0 (25 mM Tris/HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2 plus 1%, v/v, Igepal CA-630). To initiate the reaction, HDAC substrate KI-104 (Enzo Life Sciences) was added at a final concentration of 50 μM. The reaction was stopped after 30 min incubation by addition of 50 μL developer solution (6 mg/mL trypsin in HDAC assay buffer). The reaction was allowed to develop for 30 min at room temperature and the fluorescence signal was detected using a fluorometer (Spectramax M2, Molecular Devices, Sunnyvale, CA, USA) with excitation and emission wavelengths of 360 nm and 470 nm respectively. Data was fitted to a sigmoidal dose response equation with variable slope in GraphPad Prism 5.0 to determine IC50 values. Minimum and maximum values were fixed to the average fluorescence response of control wells with no cells and cells treated with 0.2% DMSO only respectively.
3.12. Brain Penetration Studies
Compound was dosed as a solution containing 5% DMSO by volume, 30% hydroxypropyl-β-cyclodextrin by weight in 100 mM acetate buffer at pH = 5.4. Dose volume was 1 mL/kg for iv dosing and 10 mL/kg for oral gavage (po dosing). Blood samples were taken at 0.083, 0.25, 0.5, 1, 2, 4, and 8 h post-dose. Serial sampling from the same animals (N = 3) was used for the p.o. dosed group. Terminal samples were obtained from animals dosed iv. Brain was collected at sacrifice to determine drug levels. Blood samples were collected in EDTA containing tubes and plasma was isolated. Plasma samples were analyzed by LC/MS/MS on an API4000 triple quadrupole (AB Sciex, Foster city, CA, USA) after protein precipitation from plasma or homogenized brain samples using acetonitrile containing an internal standard as the precipitant. Standards were prepared by serial dilution of analyte stock solution in corresponding matrix (rat plasma or brain tissue). Quantitation was generated automatically by the AB Sciex Analyst software. Data analysis was performed using Phoenix Winnonlin 6.1 (Pharsight Corporation, St. Louis, MO, USA) using the non-compartmental approach and the iv bolus or extravascular models provided in the software.
3.13. Acid Stability Determination
A 100 M solution of test compound was prepared by dilution of a 10 mM DMSO stock solution in a 0.01 M solution of HCl in deionized water. Immediately after mixing, an aliquot (100 μL) was sampled and analyzed by HPLC/UV. The area under the compound peak was determined and used as the time zero reference point. The remainder of the acid sample was incubated at 50 °C and samples were taken after 2, 4, and 24 h of incubation. These were analyzed by the same HPLC/UV method and the area of the peak corresponding to the test compound was measured. Percent remaining at a given time point was then calculated as the ratio of the area under the peak after incubation to that at time zero times 100.
4. Conclusions
In this report, we describe a new synthetic route to a class of benzamide HDAC inhibitors that show efficacy in restoring transcription to the silenced FXN gene in primary lymphocytes from Friedreich's ataxia patients. Our use of Cu(I)-catalyzed click chemistry enabled the facile synthesis of a series of derivatives of our current clinical candidate, HDACi 109. Compound 109, which has been shown to be effective in restoring FXN gene transcription both in primary patient cells and in animal models [8,9], suffers from two liabilities, namely, less than optimal brain penetration and poor stability under acidic conditions. Previous medicinal chemistry efforts (V.J. and J.R.R., unpublished) lead to the identification of two chemical features that can be incorporated into the pimelic o-aminobenzamide scaffold to overcome these limitations. These are replacement of the “left” amide with an ether, olefin or ketone to increase brain penetration and introduction of an unsaturated bond adjacent to the “right” amide to prevent formation of the benzimidazole product. Using click chemistry, we find that these two features can now be incorporated into a single molecule that improves on both properties while retaining the ability to increase FXN gene expression in patient cells. Importantly, compound click-1 has limited cytotoxicity issues compared to other benzamide HDAC inhibitors (such as MS-275), but retains its ability to inhibit recombinant HDACs (Table 1) and increase FXN gene expression in FRDA patient cells (Figure 3). These features suggest that this new class of compounds have improved efficacy as therapeutics for this neurodegenerative disease.
* For consistency with previous publications [4,5,7,8] we use here the compound numbers employed in those previous reports.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | HDAC1 IC50(μM) | HDAC2 IC50(μM) | HDAC3 IC50(μM) | DAC1 IC50(μM) |
|---|---|---|---|---|---|
| 109 |  | 0.322 | 1.98 | 0.075 | 3.5 |
| Click-1 |  | 0.066 | 0.477 | 0.028 | 5.6 |
| Click-2 |  | 0.046 | 0.466 | 0.039 | 5.1 |
| Click-3 |  | ND | ND | ND | 29.4 |
1In-cell deacetylase assay.
Acknowledgments
This work was supported by grants from The Friedreich's Ataxia Research Alliance and GoFAR.
Conflict of Interest
V.J. and J.R.R. are employees of Repligen Corporation and J.M.G. serves as a consultant to Repligen Corporation.
References
- Pandolfo, M. Friedreich ataxia. Semin. Pediatr. Neurol. 2003, 10, 163–172. [Google Scholar]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich's ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar]
- Bidichandani, S.I.; Ashizawa, T.; Patel, P.I. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet. 1998, 62, 111–121. [Google Scholar]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar]
- Xu, C.; Soragni, E.; Chou, C.J.; Herman, D.; Plasterer, H.L.; Rusche, J.R.; Gottesfeld, J.M. Chemical probes identify a role for histone deacetylase 3 in Friedreich's ataxia gene silencing. Chem. Biol. 2009, 16, 980–989. [Google Scholar]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar]
- Rai, M.; Soragni, E.; Jenssen, K.; Burnett, R.; Herman, D.; Gottesfeld, J.M.; Pandolfo, M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One 2008, 3, e1958. [Google Scholar]
- Rai, M.; Soragni, E.; Chou, C.J.; Barnes, G.; Jones, S.; Rusche, J.R.; Gottesfeld, J.M.; Pandolfo, M. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PLoS One 2010, 5, e8825. [Google Scholar]
- Sandi, C.; Pinto, R.M.; Al-Mahdawi, S.; Ezzatizadeh, V.; Barnes, G.; Jones, S.; Rusche, J.R.; Gottesfeld, J.M.; Pook, M.A. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol. Dis. 2011, 42, 496–505. [Google Scholar]
- Ku, S.; Soragni, E.; Campau, E.; Thomas, E.A.; Altun, G.; Laurent, L.C.; Loring, J.F.; Napierala, M.; Gottesfeld, J.M. Friedreich's ataxia induced pluripotent stem cells model intergenerational GAA-TTC triplet repeat instability. Cell Stem Cell 2010, 7, 631–637. [Google Scholar]
- de Boer, A.G.; Gaillard, P.J. Strategies to improve drug delivery across the blood-brain barrier. Clin. Pharmacokinet. 2007, 46, 553–576. [Google Scholar]
- Finn, M.G.; Fokin, V.V. Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC). In Catalysis without Precious Metals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; Chapter 10; pp. 235–260. [Google Scholar]
- Shen, J.; Woodward, R.; Kedenburg, J.P.; Liu, X.; Chen, M.; Fang, L.; Sun, D.; Wang, P.G. histone deacetylase inhibitors through click chemistry. J. Med. Chem. 2008, 51, 7417–7427. [Google Scholar]
- Garrais, S.; Turkington, J.; Goldring, W.P.D. Synthesis of isomeric polyacetylenes based on natural hydroxy matricaria esters. Tetrahedron 2009, 65, 8418–8427. [Google Scholar]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone deacetylase inhibitor MS-275 exhibits poor brain penetration: Pharmacokinetic studies of [11C]MS-275 using positron emission tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/)
Share and Cite
Xu, C.; Soragni, E.; Jacques, V.; Rusche, J.R.; Gottesfeld, J.M. Improved Histone Deacetylase Inhibitors as Therapeutics for the Neurodegenerative Disease Friedreich’s Ataxia: A New Synthetic Route. Pharmaceuticals 2011, 4, 1578-1590. https://doi.org/10.3390/ph4121578
Xu C, Soragni E, Jacques V, Rusche JR, Gottesfeld JM. Improved Histone Deacetylase Inhibitors as Therapeutics for the Neurodegenerative Disease Friedreich’s Ataxia: A New Synthetic Route. Pharmaceuticals. 2011; 4(12):1578-1590. https://doi.org/10.3390/ph4121578
Chicago/Turabian StyleXu, Chunping, Elisabetta Soragni, Vincent Jacques, James R. Rusche, and Joel M. Gottesfeld. 2011. "Improved Histone Deacetylase Inhibitors as Therapeutics for the Neurodegenerative Disease Friedreich’s Ataxia: A New Synthetic Route" Pharmaceuticals 4, no. 12: 1578-1590. https://doi.org/10.3390/ph4121578
APA StyleXu, C., Soragni, E., Jacques, V., Rusche, J. R., & Gottesfeld, J. M. (2011). Improved Histone Deacetylase Inhibitors as Therapeutics for the Neurodegenerative Disease Friedreich’s Ataxia: A New Synthetic Route. Pharmaceuticals, 4(12), 1578-1590. https://doi.org/10.3390/ph4121578