Myocardial Opioid Receptors in Conditioning and Cytoprotection

{kind=link}
Abstract
: Opioid compounds and G-protein coupled opioid receptors (ORs) have been studied widely in terms of central nervous system (CNS) actions relating to pain management and drug abuse. Opioids are also linked to induction of mammalian hibernation, a natural state of tolerance involving prolonged and orchestrated shifts in cellular metabolism, growth and stress resistance. It is not surprising then that OR agonism induces acute or delayed cytoprotective states in myocardium, rendering ORs an attractive target for protection of cardiac tissue from the potentially fatal consequences of ischemic heart disease. Cardiac ORs are implicated in triggering/mediating so-called ‘conditioning’ responses, in which powerful cytoprotection arises following transient receptor ligation prior to or immediately following ischemic insult. These responses involve one or more OR sub-types engaging pro-survival kinase cascades to ultimately modulate cell stress and mitochondrial end-effectors. However, important questions remain regarding the role of endogenous opioids, OR signalling, and the transduction and mediation of these protective responses. We briefly review opioid-mediated cardioprotection, focussing on recent developments in signal transduction, the role of receptor ‘cross-talk’, and the effects of sustained OR ligand activation.1. Introduction
Opioids modulate cellular function via Gi/o coupled members of the G-protein protein coupled receptor (GPCR) superfamily—the μ-, κ-, and δ-ORs. They are activated by opioid peptides derived from the endorphin, dynorphin and enkephalin families, share ∼50% homology, and differ in binding properties, tissue distribution, and cell signalling. The μ-ORs appear sensitive to endorphins, κ-ORs preferentially bind dynorphins, while δ-ORs have a higher affinity for enkephalins. Pharmacological evidence suggests that the κ-, and δ-OR sub-families may include κ1, κ2, δ1, and δ2 sub-types. For a comprehensive review of OR pharmacology specifically within the cardiovascular system, readers are directed to comprehensive review [1].
While initially considered to act indirectly via modulation of nervous/sympathetic control, it is known that myocardial cells are sites of opioid peptide synthesis, storage and release [2]. Stressors such as ischemia elevate myocardial peptide turnover [3], and κ- and δ-ORs are expressed in myocardium, whereas binding and gene expression studies excluded μ-ORs from adult myocardium [4,5]. However, immunofluorescence microscopy showed co-localization of μ-opioid receptors with Cav-3 in both sarcolemmal and intracellular membranes of the adult myocyte [6]. Ventricular myocardium actually contains the highest levels of preproenkephalin (enkephalin precursor) mRNA in the body, surpassing that for the central nervous system [7]. Myocardial expression of opioids and ORs is consistent with opioidergic regulation of cardiovascular function and myocardial stress resistance. This review focuses specifically on the roles of OR in myocardial cytoprotection.
2. Opioid Receptor-Mediated Cardioprotection
Endogenous opioids possess autocrine/paracrine functions within the heart and vessels. For example, OR activity inhibits excitation-contraction coupling, modulates vascular tone, may play a role in cardiogenesis, and exerts potent cytoprotective actions in the heart. Recent work indicates endogenous opioids play a role in protecting cardiac tissues from ischemia-reperfusion (I/R) injury [8], and in mediating ischaemic preconditioning [9-11]. Other studies suggest opioids function as ‘mediators’ rather than ‘triggers’ of acute preconditioning [12].
3. Involvement in Pre- and Post-Conditioning
Two of the most intensely studied protective modalities are the conditioning responses—pre- and postconditioning. Preconditioning was discovered by Murry et al. [13], and refers to induction of both acute and delayed protective states in response to a transient episode of ischemia prior to prolonged insult. The transient ischemia can be replaced by transient agonism of GPCRs implicated in this response [14]. Protection against infarction with postconditioning was established by Vinten-Johannsen and colleagues, who documented protective actions of brief episodic ischemia during the first minutes of reperfusion following sustained insult [15], extending earlier observations of electrophysiological protection with intermittent reperfusion [16]. These responses have garnered considerable interest as potentially clinically relevant protective stimuli [17], underpinning extensive interrogation of underlying mechanisms. Despite some conflicting findings, these studies identify roles for opioids and ORs in induction or mediation of conditioning responses.
Pre-ischemic OR agonism mimics ischemic preconditioning [18], antagonists of ORs counter the protection with preconditioning when applied prior to the ischemic preconditioning stimulus, in an acute setting [19] or during the index ischemia in a delayed preconditioning model [20]. Thus, there is some support not only for a role for ORs in the initial trigger phase of preconditioning, but also in subsequent mediation of protection during subsequent ischemia-reperfusion.
Consistent with mechanistic links between preconditioning and more recently studied postconditioning, evidence also supports an essential role for ORs in postconditioning. Beneficial effects of ischemic post-conditioning are replicated by OR activation, and countered by δ-OR antagonism [21]. Furthermore, Zatta et al. [22] presented evidence implicating both μ- and δ-ORs in cardioprotection afforded by ischemic postconditioning, and showed protection was associated with preservation of myocardial enkephalin levels (particularly the precursor proenkephalin). In contrast, a recent study in a similar model reports that κ- and δ-ORs but not μ-ORs mediated ischemic postconditioning [23]. Reasons underlying these differences are unclear, though may potentially involve dose-dependent selectivity of pharmacological tools employed. Analysis of protection of the brain via remote postcondtioning (triggered in response to ischemia in remote limbs or organs) also supports protection via intrinsic OR activity [24], though this is yet to be established for remote cardiac postconditioning.
As with opioidergic preconditioning, exogenous activation of κ- and δ-ORs at reperfusion affords protective postconditioning [25-28], and underlying mechanisms mirroring those for ischemic conditioning responses. Studies thus support recruitment of the archetypal PI3k and GSK3β signalling axis [26,27,29], phosphorylation of eNOS and NO production [28], regulation of mitochondrial and sarcolemmal KATP channel opening [26,27,29], and inhibition of mPTP function, perhaps through a NO-cGMP-PKG path [21]. However, multiple pathways to cardiac protection have been identified, including the Reperfusion Injury Salvage Kinase (RISK) [30] and Survivor Activating Factor Enhancement (SAFE) [31] paths. In this respect, there is also evidence for JAK-STAT involvement and modulation of BCL-2 expression and apoptosis [32], as in the SAFE signalling model. Whether these different signal paths are distinct or do indeed interact and/or converge on end-effectors is at present unclear.
3.1. Downstream Effectors of Opioid Mediated Cardioprotection
As detailed previously [33,34], conventional models link acute OR activation to protein kinase cascades, reactive oxygen species (ROS) generation, and modulation of mito KATP channel controlling mPTP opening [35-39]. Whether the latter channels are ‘end-effectors’ or proximal to end-effectors is still debated, as is the contribution of sarcolemmal channels [36,40,41].
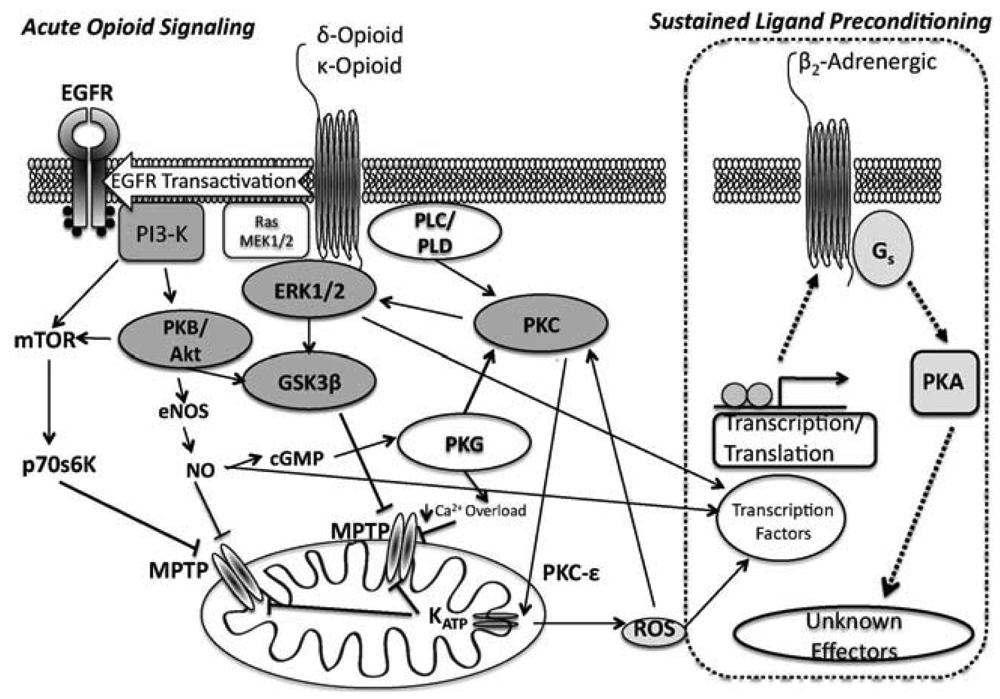
ORs couple to Gi/o proteins to inhibit adenylyl cyclase, with δ- and κ-ORs known to activate PLC [42] and phosphoinositol turnover [43]. Additionally, OR agonism activates tyrosine kinase and PKC, perhaps in parallel [36,44], and leads to opening of both sarcolemmal and mito KATP channels [37,38]. ORs also regulate ion channels via G-protein interactions [45,46]. In terms of cardioprotection, infarct limitation with δ-OR agonism is PKC- and NOS-dependent [44,47], and involves tyrosine kinase (TK) and MAPK signalling [36,44,48]. Acute OR protection during reperfusion is dependent upon PI3-K, target of rapamycin (mTOR), and GSK3β modulation [49]. Collectively, data implicate non-Src-dependent TK, extracellular signal-regulated kinase (ERK1/2) and PI3K/PKC pathways as integral signalling components of acute δ-OR mediated cardioprotection.
Signalling in acute κ-OR dependent cardioprotection is less well defined: κ-OR inhibition of ventricular cardiomyocyte shortening appears to be Gi-dependent [50], and κ-ORs suppress cardiomyocyte cAMP accumulation via a phosphoinositol/Ca2+ path [51]. Downstream effectors affording cardioprotection from κ-OR activation likely mimics that of the δ-OR, (PI3k/Akt, PKC and mito KATP channel dependent, [26,52]) (Figure 1).
There is little information regarding any potential role for the μ-ORs in the setting of cardioprotection. Until recently it was thought that the μ-OR was absent from the mature myocardium [4-6]. Indeed, there is a lack of evidence supporting a role for the μ-OR in preconditioning. However, Zatta et al. [22] recently reported that ischemic postconditioning was mediated through endogenous μ-OR activation. The involvement of μ-OR in conditioning the heart is controversial at best. As such, detail regarding potential mechanisms is absent.
3.2. Caveolae and ORs
Immunohistochemical analysis co-locates μ-ORs and caveolin-3 (Cav-3) in sarcolemmal and intracellular membranes of the adult myocyte, and caveolins appear essential to OR cytoprotection [6]. Caveolae, microenvironments enriched in caveolin, cholesterol and sphingolipids, form characteristic flask-like invaginations of the membrane. Caveolar structural proteins (caveolins-1, -2 and -3) may act as scaffolding molecules to localise and regulate interactions between receptors, signalling components, and effectors, facilitating coordinated regulation of cell function [53]. This not only impact on sarcolemmal targeting, but transduction of protective signals to mitochondria [54]. Importantly, caveolae are critical to I/R tolerance and cardioprotection: protective stimuli increase caveolae formation, cardioprotection is blocked by caveolar disruption, caveolin-3 (cav-3) knockout (KO) eliminates preconditioning [55,56], and overexpression of cav-3 boosts intrinsic I/R tolerance [56].
In adult cardiac myocytes, ORs are localised to cav-3-associated domains [6], and caveolar disruption abolishes the protective effects of δ-OR activation [57]. Interestingly, an in vivo model utilising cav-3 overexpression and knock-out mice confirmed cav-3 dependence of δ-OR protection, and revealed that protection conferred by cav-3 overexpression was negated by naloxone [58]. This supports an integral relationship between caveolae, caveolins and ORs, with δ-ORs specifically implicated in the cytoprotective capacity of caveolae. Caveolar localisation of ORs may also contribute to potential cross-talk between ORs and other protective GPCRs.
4. Opioid Receptor Cross-Talk and Cardioprotection
The OR family engages in cross-talk between its own members and with other receptors. Action of ORs may involve modulation of or dependence upon the function of other receptor types and vice versa. Cross-talk may be indirect through trans-regulation of downstream signalling and the effects of heterologous sensitisation and desensitisation, or more direct, in the context of receptor-receptor interactions and GPCR dimerisation. The ORs are known to form homo- and heterodimers in heterologous cell lines and non-cardiac tissue: δ-ORs can heterodimerise with both κ- and μ-ORs, altering pharmacological properties, and heterodimers may also form between ORs and other GPCRs, including somatostatin, substance P, and α1 and ß2-adrenergic receptors. Specific studies in cardiovascular tissue are lacking, though such complexes could impact on cardiac control and underpin forms of OR cross-talk.
One of the earlier reported forms of myocardial OR cross talk involves δ-OR modulation of ß-adrenergic responsiveness, limiting norepinephrine-induced increases in sarcolemmal L-type Ca2+ current, cytosolic Ca2+ transients, and contraction in isolated ventricular myocytes [59]. These results were confirmed by Pepe et al. [60] in intact hearts, demonstrating cross-talk between δ-OR and ß1-adrenoceptor signaling, with ORs inhibiting adenylyl cyclase via a PTX-sensitive Gi/o protein. The cAMP-independent inotropic effects of ß2-AR agonism appear insensitive to δ-OR activity. In terms of cardioprotection, δ-OR mediated infarct reduction is negated by ß2-adrenoceptor blockade [61]. These investigators also reported an essential requirement for intrinsic cardiac adrenergic cells in δ-OR protection of cardiac myocytes, supporting reliance on endogenous epinephrine/ß2-adrenoceptor signaling. A subsequent study [62] added calcitonin gene-related peptide receptors (CGRP-R) to the interaction between δ-OR and ß-adrenoceptors, reporting synergistic cardioprotection through a δ-OR regulated ß2-adrenoceptorAR/CGRP-R co-signaling in cardiac adrenergic cells. Our own data indicates that mediation of protection with sustained opioid agonism (see below) is ß2-adrenoceptor dependent [63]. ß-adrenoceptor and OR cross-talk appears to involve modulation of G-protein signaling and kinase activity, but could also involve dimerisation: both δ- and κ-ORs physically associate with ß2-adrenoceptors when co-expressed in HEK-293 cells [64]. As a result, δ-OR activation mediates ß2-adrenoceptor internalisation and inhibits ß2-adrenoceptor triggered ERK1/2 activation.
Our prior work highlights cardioprotective cross-talk between OR and adenosine receptors. In an initial study [65], the infarct sparing effects of OR stimulation in rats were abolished by an adenosine A1 receptor (A1AR) antagonist, and the reverse was also found to be true (OR blockade attenuated cardioprotection in response to A1AR activation). In a subsequent study, the protective effects of increased endogenous adenosine (following adenosine kinase inhibition) were found to be dependent upon δ-OR activity [66]. The basis of this cross-talk, which evidences an essential role for both receptors in responses to either, remains to be established. There is evidence of positive cross-talk between AR and ORs in other models, which may reflect modulation of endogenous ligand levels and distal signalling: Kaster et al. [67] report that anti-depressant actions of A1 and A2ARs involve κ- and μ-OR activities; and remote conditioning effects of intrathecal ORs require both central and peripheral AR activities [68]. While these effects may involve modulation of endogenous ligand signaling, AR/OR receptor complexes could contribute. There is evidence that ARs are essential for synergistic signaling between δ-OR, μ-ORs, CB1 and D2 receptors in nervous tissue (with A1 or A2A activity and Gi-βγ required for this synergistic action) [69,70]. The mechanism underlying AR-dependent hypersensitisation of co-expressed OR receptors is not known, but appears to centre on priming of adenylate cyclase and increased signaling activation. However, while studies in heterologous cell lines support receptor-receptor interactions between A1ARs and δ-ORs, their nature is distinct from responses observed in native cardiac tissue: in a CHO cell model, co-expressed A1ARs heterologously desensitize δ-OR mediated kinase signaling, and induce phosphorylation of δ-ORs [71]. Our data, in contrast, supports an essential requirement for both AR and OR activities in cardioprotection. In recent studies in murine HL-1 cells [72], we also find that intracellular kinase signaling (ERK1/2) and AR and OR mRNA expression are similarly co-regulated via A1ARs and δ-ORs, with antagonism of either receptor alone negating responses to both. Providing a link between the two receptors, signal activation by either A1AR or δ-ORs appears commonly dependent upon EGFR function. Indeed, δ-OR post-conditioning of the myocardium has been shown to depend upon transactivation of the EGFR [73]. Thus, ORs and ARs may both engage a common EGFR-dependent pathway to activate downstream protective signals, which may contribute to observed cross-talk.
In addition to these forms of cross-talk, one can consider δ-ORs as signaling intermediates involved in transducing cardioprotection. It was recently demonstrated that epicatechin, an antioxidant flavonoid with no known direct receptor-mediated activity, produced profound δ-OR dependent protection against infarct development [74]. Protective effects of epicatechin were associated with increased phosphorylation of pro-survival kinases (Src, Akt, IκBα) and decreased pro-apoptotic protein expression, both effects countered by δ-OR antagonism. Similarly, infarct sparing actions of the epoxyeicosatrienoic acid 11,12-EET, for which a specific EET membrane receptor is yet to be identified, are reversed by either δ- or κ-OR inhibition [75]. Irrespective of the specific molecular basis of these cross-talk effects, such reports further highlight the importance of intrinsic OR activity in transduction of cardioprotection.
5. Sustained Opioidergic Preconditioning
In 2004, we described a protective phenomenon dubbed chronic morphine preconditioning [76], which we more accurately label as sustained ligand-activated protection (SLP), since we show the response involves selective δ-opioid receptor (δ-OR) activation [77]. SLP can be induced by 3–5 days of δ-OR activation, mediating protection that exceeds that with preconditioning or postconditioning and persists for at 5 days (perhaps ≥7 days) [77]. Signalling is unique vs. that for preconditioning or postconditioning [63].
From a clinical standpoint, generation of sustained protected states (as in SLP) has advantages [78]. Precise timing of treatment relative to I/R becomes less critical, and the need for ongoing therapy is reduced. Prolonged protection might be particularly useful in prophylactic therapy in high-risk patients, and for limiting time-dependent progression of injury during or after surgery (for example). Temporal properties of classic preconditioning are sub-optimal: despite rapid induction, the powerful initial window is brief (1–2 h), while the later sustained window is less efficacious and lasts only 2–3 days. With postconditioning, protection arises rapidly yet does not impact on ischaemic injury, the window of opportunity for protection is likely narrow, and it is unclear if protection is long-lasting (or effective in aged hearts).
Downey's group initially addressed the possibility of sustained cardioprotection (via continuous A1 adenosine receptor agonism) [79], and Dana et al. [80] subsequently showed it was possible to generate persistent protection for 10 days with repetitive A1 receptor agonism. Inagaki et al. [81] described sustained protection following 10-day infusion of a PKC-ε activator. We document protection for at least 5 days following acute irreversible δ-OR agonism [82]. Thus, prolonged protected states can be generated in cardiac tissue, though are yet to be exploited clinically to limit injury.
The SLP response [76,77,83] affords protection exceeding that for conventional preconditioning, and persists for up to 7 days after removal of the initiating stimulus [77]. We showed that SLP depends upon signalling distinct from so-called RISK paths [30,63] that mediate preconditioning and postconditioning, likely explaining retention of SLP efficacy with aging [34] vs. failure of the latter responses [84-88]. Inhibition of downstream kinases (including PI3-K/Akt, PKC) and mito KATP channels all fail to block mediation of SLP during acute I/R [63]. Rather, mediation of SLAP is Gs- vs. Gi-dependent, requires protein kinase A (PKA), and depends upon ß2-adrenoceptor activity [63] (Figure 1). These findings are consistent with emerging roles for PKA in cardioprotection [89-91]. ß2-adrenoceptor involvement is interesting, since ß2-adrenoceptors favour cell survival, limiting I/R injury and apoptosis in a Gi/PI3-K (vs. Gs) dependent manner [92,93]. Inhibitory effects of ß2-adrenoceptor antagonism against SLP protection may reflect shifts in effector coupling or PKA activation in SLP hearts.
Certainly this is documented for the ß2 receptor, though not strictly in accordance with these observations. For example, receptor phosphorylation by PKA alters G-protein selectivity of ß2-adrenergic receptors, favoring coupling to Gi vs. Gs protein, and reversing the effects of the receptor on cAMP generation [94]. Phosphorylation-dependent switching of G-protein coupling allows the receptor to engage alternate signaling (e.g., Gi-dependent MAPK activation). Cardiomyocyte ß2 (but not ß1) receptors favor cell survival via pertussis toxin sensitive Gi signaling, PI3K and Akt.
In contrast to the mediation phase (i.e., the period of ischemia-reperfusion when protection is expressed), the induction of the SLP phenotype is δ-opioid receptor mediated and is induced in a PI3K-dependent/PKA-independent manner [77]. This is interesting, as it supports distinct phases to the response, with PI3K/Akt involvement during induction vs. mediation, and PKA involvement during mediation. It seems that sustained OR agonism induces changes in signaling that may switch OR responses from Gi to Gs dependent mechanisms, and involves PI3K- and PKA signalling (albeit, temporally distinct). These findings are also somewhat consistent with the role for OR signalling in hibernation.
6. Opioids and Hibernation
Opioids, and in particular the δ-receptor sub-type, are strongly implicated in mammalian hibernation. Hibernation can be induced or reversed by δ-opioid agonists and antagonists, respectively, even in species that do not normally hibernate. Hibernation is thought to be triggered by changes in serum levels of a δ-opioid like peptide, termed ‘hibernation induction trigger’ (HIT) [95]. HIT and the δ-opioid peptide DADLE can induce hibernation like states in non-hibernating mammals, and mammalian hibernation is associated with an improvement in tissue resistance to stressors such as hypoxia.
Many parallels exist between SLP and hibernation: hibernation is normally triggered by endogenous δ-opioid agonism, which can also induce hibernation in non-hibernating primates [96,97]; δ-opioid-triggered hibernation increases cellular stress resistance [98,99]; hibernating and anoxia-tolerant species specifically harness PKA-dependent signaling [100,101]; and repression of Akt may suppress energy-costly anabolic/growth processes to maintain cell viability over extended hibernation periods [102,103]. The protected SLP phenotype is induced by prolonged δ-opioid agonism, involves PI3K dependent signals with early and late repression of Akt expression (both total and phosphorylated) [77], leading to sustained PKA-dependent cardioprotection. We also unexpectedly found that prolonged PI3K/Akt inhibition with wortmannin induces some ischemic tolerance. Together, these speculative data hint at cardioprotection in response to sustained PI3K/Akt repression, consistent with the role for Akt in δ-opioid-mediated cytoprotective hibernation.
7. Summary
The opioid system of peptides and receptors have been shown to evoke profound cytoprotective states, from intrinsic/endogenous examples such as hibernation, through to exogenous pharmacological manipulation of receptors such as a post-conditioning mimetic. While the mechanisms may not be fully understood, they appear to mirror those of ischemic preconditioning (involving a signaling axis incorporating PI3k, GSK3β, KATP channels and the mPTP). Acute opioid-mediated protection also appears dependent upon activated adenosine receptors. Moreover, opioids can confer an extended window of cardioprotection. As opioids are currently used both post-operatively and for both acute and chronic pain, a long period of drug development before opioids will be approved for use as cardioprotective agents would not be required.

References
- Pugsley, M.K. The diverse molecular mechanisms responsible for the actions of opioids on the cardiovascular system. Pharmacol. Ther. 2002, 93, 51–75. [Google Scholar]
- Barron, B.A.; Jones, C.E.; Caffrey, J.L. Pericardial repair depresses canine cardiac catecholamines and met-enkephalin. Regul. Peptides 1995, 59, 313–320. [Google Scholar]
- Eliasson, T.; Mannheimer, C.; Waagstein, F.; Andersson, B.; Bergh, C.H.; Augustinsson, L.E.; Hedner, T.; Larson, G. Myocardial turnover of endogenous opioids and calcitonin-gene-related peptide in the human heart and the effects of spinal cord stimulation on pacing-induced angina pectoris. Cardiology 1998, 89, 170–177. [Google Scholar]
- Ventura, C.; Bastagli, L.; Bernardi, P.; Caldera, C.M.; Guarnieri, C. Opioid receptors in rat cardiac sarcolemma: effect of phenylephrine and isoproterenol. Biochim. Biophys. Acta 1989, 987, 69–74. [Google Scholar]
- Wittert, G.; Hope, P.; Pyle, D. Tissue distribution of opioid gene expression in the rat. Biochem. Biophys. Res. Commun. 1996, 218, 877–888. [Google Scholar]
- Head, B.P.; Patel, H.H.; Roth, D.M.; Lai, N.C.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol Chem. 2005, 280, 31036–31044. [Google Scholar]
- Howells, R.D.; Kilpatrick, D.L.; Bailey, L.C.; Noe, M.; Udenfriend, S. Proenkephalin mRNA in rat heart. Proc. Natl. Acad. Sci. USA 1986, 83, 1960–1963. [Google Scholar]
- Romano, M.A.; Seymour, E.M.; Berry, J.A.; McNish, R.A.; Boiling, S.F. Relative contribution of endogenous opioids to myocardial ischemic tolerance. J. Surg. Res. 2004, 118, 32–37. [Google Scholar]
- Schultz, J.J.; Hsu, A.K; Gross, G.J. Ischemic preconditioning and morphine-induced cardioprotection involve the δ-opioid receptor in the intact rat heart. J. Mol. Cell. Cardiol. 1997, 29, 2187–2195. [Google Scholar]
- Schultz, J.E.; Hsu, A.K; Gross, G.J. Ischemic preconditioning in the intact rat heart is mediated by δ1- but not μ- or κ-opioid receptors. Circulation 1998, 97, 1282–1289. [Google Scholar]
- Schulz, R.; Gres, P.; Heusch, G. Role of endogenous opioids in ischemic preconditioning but not in short-term hibernation in pigs. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2175–H2181. [Google Scholar]
- Genade, S.; Moolman, J.A.; Lochner, A. Opioid receptor stimulation acts as mediator of protection in ischemic preconditioning. Cardiovasc JS Afr 2001, 12, 8–16. [Google Scholar]
- Murry, C.E.; Jennings, R.; Reimer, K.A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar]
- Kharbanda, R.K. Cardiac conditioning: a review of evolving strategies to reduce ischaemia-reperfusion injury. Heart 2010, 96, 1179–1186. [Google Scholar]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar]
- Na, H.S; Kim, Y.I.; Yoon, Y.W.; Han, H.C.; Nahm, S.H.; Hong, S.K. Ventricular premature beat-driven intermittent restoration of coronary blood flow reduces the incidence of reperfusion-induced ventricular fibrillation in a cat model of regional ischemia. Am. Heart J. 1996, 132, 78–83. [Google Scholar]
- Yang, X.C.; Liu, Y.; Wang, L.F.; Cui, L.; Wang, T.; Ge, Y.G.; Wang, H.S.; Li, W.M.; Xu, L.; Ni, Z.H.; Liu, S.H.; Zhang, L.; Jia, H.M.; Vinten-Johansen, J.; Zhao, Z.Q. Reduction in myocardial infarct size by postconditioning in patients after percutaneous coronary intervention. J. Invasive Cardiol. 2007, 19, 424–430. [Google Scholar]
- Schultz, J.E.; Hsu, A.K.; Gross, G.J. Morphine mimics the cardioprotective effect of ischemic preconditioning via a glibenclamide-sensitive mechanism in the rat heart. Circ. Res. 1996, 78, 1100–1104. [Google Scholar]
- Schultz, J.E.; Rose, E.; Yao, Z.; Gross, G.J. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am. J. Physiol. 1995, 268, H2157–H2161. [Google Scholar]
- Fryer, R.M.; Hsu, A.K.; Eells, J.T.; Nagase, H.; Gross, G.J. Opioid-induced second window of cardioprotection: potential role of mitochondrial KATP channels. Circ. Res. 1999, 84, 846–851. [Google Scholar]
- Jang, Y.; Xi, J; Wang, H.; Mueller, R.A.; Norfleet, E.A.; Xu, Z. Postconditioning prevents reperfusion injury by activating delta-opioid receptors. Anesthesiology 2008, 108, 243–250. [Google Scholar]
- Zatta, A.J.; Kin, H.; Yoshishige, D.; Jiang, R.; Wang, N.; Reeves, J.G.; Mykytenko, J.; Guyton, R.A.; Zhao, Z.Q.; Caffrey, J.L.; Vinten-Johansen, J. Evidence that cardioprotection by postconditioning involves preservation of myocardial opioid content and selective opioid receptor activation. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1444–H1151. [Google Scholar]
- Wong, G.T.; Li, R.; Jiang, L.L.; Irwin, M.G. Remifentanil post-conditioning attenuates cardiac ischemia-reperfusion injury via kappa or delta opioid receptor activation. Acta Anaesthesiol. Scand. 2010, 54, 510–518. [Google Scholar]
- Zhou, Y.; Fathali, N.; Lekic, T.; Ostrowski, R.P.; Chen, C.; Martin, R.D.; Tang, J.; Zhang, J.H. Remote limb ischemic postconditioning protects against neonatal hypoxic-ischemic brain injury in rat pups by the opioid receptor/akt pathway. Stroke 2011, 42, 439–444. [Google Scholar]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. GSK3beta inhibition and K(ATP) channel opening mediate acute opioid-induced cardioprotection at reperfusion. Basic Res. Cardiol. 2007, 102, 341–349. [Google Scholar]
- Peart, J.N.; Gross, E.R.; Reichelt, M.E.; Hsu, A.; Headrick, J.P.; Gross, G.J. Activation of kappa-opioid receptors at reperfusion affords cardioprotection in both rat and mouse hearts. Basic Res. Cardiol. 2008, 103, 454–463. [Google Scholar]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Acute methadone treatment reduces myocardial infarct size via the delta-opioid receptor in rats during reperfusion. Anesth. Analg. 2009, 109, 1395–1402. [Google Scholar]
- Tong, G.; Sun, Z.; Wei, X.; Gu, C.; Kaye, A.D.; Wang, Y.; Li, J.; Zhang, Q.; Guo, H.; Yu, S.; Yi, D.; Pei, J. U50,488H postconditioning reduces apoptosis after myocardial ischemia and reperfusion. Life Sci. 2011, 88, 31–38. [Google Scholar]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ. Res. 2004, 94, 960–966. [Google Scholar]
- Hausenloy, D.J.; Yellon, D.M. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004, 61, 448–460. [Google Scholar]
- Lecour, S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J. Mol. Cell. Cardiol. 2009, 47, 32–40. [Google Scholar]
- You, L.; Li, L.; Xu, Q.; Ren, J.; Zhang, F. Postconditioning reduces infarct size and cardiac myocyte apoptosis via the opioid receptor and JAK-STAT signaling pathway. Mol. Biol. Rep. 2011, 38, 437–443. [Google Scholar]
- Gross, G.J. Role of opioids in acute and delayed preconditioning. J. Mol. Cell. Cardiol. 2003, 35, 709–718. [Google Scholar]
- Peart, J.N.; Gross, E.R.; Gross, G.J. Opioid-induced preconditioning: Recent advances and future perspectives. Vasc. Pharmacol. 2005, 42, 211–218. [Google Scholar]
- Cohen, M.V.; Baines, C.P.; Downey, J.M. Ischemic preconditioning: From adenosine receptor to K-ATP channel. Ann. Rev. Physiol. 2000, 62, 79–109. [Google Scholar]
- Fryer, R.M.; Wang, Y.; Hsu, A.K.; Gross, G.J. Essential activation of PKC-delta in opioid-initiated cardioprotection. Am. J. Physiol. 2001, 280, H1346–H1353. [Google Scholar]
- Patel, H.H.; Hsu, A.K.; Peart, J.N.; Gross, G.J. Sarcolemmal KATP channel triggers opioid-induced delayed cardioprotection in the rat. Circ. Res. 2002, 91, 186–188. [Google Scholar]
- Peart, J.N.; Gross, G.J. Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H81–H89. [Google Scholar]
- Xi, J; Tian, W; Zhang, L; Jin, Y; Xu, Z. Morphine prevents the mitochondrial permeability transition pore opening through NO/cGMP/PKG/Zn2+/GSK-3beta signal pathway in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H601–H607. [Google Scholar]
- Gross, E.R.; Peart, J.N.; Hsu, A.K.; Grover, G.J.; Gross, G.J. KATP opener-induced delayed cardioprotection: involvement of sarcolemmal and mitochondrial KATP channels, free radicals and MEK1/2. J. Mol. Cell. Cardiol. 2003, 35, 985–992. [Google Scholar]
- Patel, H.H.; Gross, E.R.; Peart, J.N.; Hsu, A.K.; Gross, G.J. Sarcolemmal KATP channel triggers delayed ischemic preconditioning in rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H445–H447. [Google Scholar]
- Lee, J.W.; Joshi, S.; Chan, J.S.C.; Wong, Y.H. Differential coupling of μ-, δ-, and κ-opioid receptors to Gα16-mediated stimulation of phospholipase C. J. Neurochem. 1998, 70, 2203–2211. [Google Scholar]
- Schultz, J.E.; Gross, G.J. Opioids and cardioprotection. Pharmacol. Ther. 2001, 89, 123–137. [Google Scholar]
- Fryer, R.M.; Schultz, J.E.; Gross, G.J. Pretreatment with tyrosine kinase inhibitors partially attenuates ischemic preconditioning in rat hearts. Am. J. Physiol. 1998, 275, H2009–H2015. [Google Scholar]
- Gross, R.A.; Moises, H.C.; Uhler, M.D.; Macdonald, R.L. Dynorphin A and cAMP-dependent protein kinase independently regulate neuronal calcium currents. Proc. Natl. Acad. Sci. USA 1990, 87, 7025–7029. [Google Scholar]
- North, R.A.; Williams, J.T.; Surprenant, A.; Christie, M.J. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc. Natl. Acad. Sci. USA 1987, 84, 5487–5491. [Google Scholar]
- Maslov, L.N.; Lishmanov, Y.B.; Oeltgen, P.R.; Barzakh, E.I.; Krylatov, A.V.; Govindaswami, M.; Brown, S.A. Activation of peripheral delta2 opioid receptors increases cardiac tolerance to ischemia/reperfusion injury Involvement of protein kinase C, NO-synthase, KATP channels and the autonomic nervous system. Life Sci. 2009, 84, 657–663. [Google Scholar]
- Fryer, R.M.; Wang, Y.; Hsu, A.K.; Nagase, H.; Gross, G.J. Dependence of 1-opioid receptor-induced cardioprotection on a tyrosine kinase-dependent but not a Src-dependent pathway. J. Pharmacol. Exp. Ther. 2001, 299, 477–482. [Google Scholar]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ. Res. 2004, 94, 960–966. [Google Scholar]
- Wenzlaff, H.; Stein, B.; Teschemacher, H. Diminution of contractile response by kappa-opioid receptor agonists in isolated rat ventricular cardiomyocytes is mediated via a pertussis toxin-sensitive G protein. Nauyn-Schmiederberg's Arch. Pharmacol. 1998, 358, 360–366. [Google Scholar]
- Zhang, W.M.; Wong, T.M. Suppression of cAMP by phosphoinositol/Ca2+ pathway in the cardiac k-opioid receptor. Am. J. Physiol. 1998, 274, C82–C87. [Google Scholar]
- Wong, T.M.; Wu, S. Roles of kappa opioid receptors in cardioprotection against ischemia: the signaling mechanisms. Sheng Li Xue Bao 2003, 55, 115–120. [Google Scholar]
- Insel, P.A.; Head, B.P.; Ostrom, R.S.; Patel, H.H.; Swaney, J.S.; Tang, C.M.; Roth, D.M. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann. NY Acad. Sci. 2005, 1047, 166–172. [Google Scholar]
- Quinlan, C.L.; Costa, A.D.; Costa, C.L.; Pierre, S.V.; Dos Santos, P.; Garlid, K.D. Conditioning the heart induces formation of signalosomes that interact with mitochondria to open mitoKATP channels. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H953–H961. [Google Scholar]
- Horikawa, Y.T.; Patel, H.H.; Tsutsumi, Y.M.; Jennings, M.M.; Kidd, M.W.; Hagiwara, Y.; Ishikawa, Y.; Insel, P.A.; Roth, D.M. Caveolin-3 expression and caveolae are required for isofluorane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2008, 44, 123–130. [Google Scholar]
- Tsutsumi, Y.M.; Horikawa, Y.T.; Jennings, M.M.; Kidd, M.W.; Niesman, I.R.; Yokoyama, U.; Head, B.P.; Hagiwara, Y.; Ishikawa, Y.; Miyanohara, A.; et al. Cardiac-specific overexpression of caveolin-3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation 2008, 118, 1979–1988. [Google Scholar]
- Patel, H.H.; Head, B.P.; Petersen, H.N.; Niesman, I.R.; Huang, D.; Gross, G.J.; Insel, P.A.; Roth, D.M. Protection of adult rat cardiac myocytes from ischemic cell death: role of caveolar microdomains and delta-opioid receptors. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H344–H350. [Google Scholar]
- Tsutsumi, Y.M.; Kawaraguchi, Y.; Niesman, I.R.; Patel, H.H.; Roth, D.M. Opioid-induced preconditioning is dependent on caveolin-3 expression. Anesth. Analg. 2010, 111, 1117–1121. [Google Scholar]
- Xiao, R.P.; Pepe, S.; Spurgeon, H.A.; Capogrossi, M.C.; Lakatta, E.G. Opioid peptide receptor stimulation reverses beta-adrenergic effects in rat heart cells. Am. J. Physiol. 1997, 272, H797–H805. [Google Scholar]
- Pepe, S.; Xiao, R.P.; Hohl, C.; Altschuld, R.; Lakatta, E.G. ‘Cross talk’ between opioid peptide and adrenergic receptor signaling in isolated rat heart. Circulation 1997, 95, 2122–2129. [Google Scholar]
- Huang, M.H.; Wang, H.Q.; Roeske, W.R.; Birnbaum, Y.; Wu, Y.; Yang, N.P.; Lin, Y.; Ye, Y.; McAdoo, D.J.; Hughes, M.G.; et al. Mediating delta-opioid-initiated heart protection via the beta2-adrenergic receptor: role of the intrinsic cardiac adrenergic cell. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H376–H384. [Google Scholar]
- Huang, M.H.; Nguyen, V.; Wu, Y.; Rastogi, S.; Lui, C.Y.; Birnbaum, Y.; Wang, H.Q.; Ware, D.L.; Chauhan, M.; Garg, N.; et al. Reducing ischaemia/reperfusion injury through delta-opioid-regulated intrinsic cardiac adrenergic cells: adrenopeptidergic co-signalling. Cardiovasc Res. 2009, 84, 452–460. [Google Scholar]
- Peart, J.N.; Gross, G.J. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1746–H1753. [Google Scholar]
- Jordan, B.A.; Trapaidze, N.; Gomes, I.; Nivarthi, R.; Devi, L.A. Oligomerization of opioid receptors with beta 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2001, 98, 343–348. [Google Scholar]
- Peart, J.N.; Gross, G.J. Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H81–H89. [Google Scholar]
- Peart, J.N.; Gross, G.J. Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res. Cardiol. 2005, 100, 328–336. [Google Scholar]
- Kaster, M.P.; Budni, J.; Santos, A.R.; Rodrigues, A.L. Pharmacological evidence for the involvement of the opioid system in the antidepressant-like effect of adenosine in the mouse forced swimming test. Eur. J. Pharmacol. 2007, 576, 91–98. [Google Scholar]
- Yao, L.; Wong, G.T.; Xia, Z.; Irwin, M.G. Interaction between spinal opioid and adenosine receptors in remote cardiac preconditioning: effect of intrathecal morphine. J. Cardiothorac Vasc. Anesth. 2010. in press. [Google Scholar]
- Yao, L.; McFarland, K.; Fan, P.; Jiang, Z.; Ueda, T.; Diamond, I. Adenosine A2a blockade prevents synergy between mu-opiate and cannabinoid CB1 receptors and eliminates heroin-seeking behavior in addicted rats. Proc. Natl. Acad. Sci. USA 2006, 103, 7877–7882. [Google Scholar]
- Yao, L.; Fan, P.; Jiang, Z.; Mailliard, W.S.; Gordon, A.S.; Diamond, I. Addicting drugs utilize a synergistic molecular mechanism in common requiring adenosine and Gi-beta gamma dimers. Proc. Natl. Acad. Sci. USA 2003, 100, 14379–14384. [Google Scholar]
- Cheng, Y.; Tao, Y.M.; Sun, J.F.; Wang, Y.H.; Xu, X.J.; Chen, J.; Chi, Z.Q.; Liu, J.G. Adenosine A(1) receptor agonist N(6)-cyclohexyl-adenosine induced phosphorylation of delta opioid receptor and desensitization of its signaling. Acta Pharmacol. Sin. 2010, 31, 784–790. [Google Scholar]
- Williams-Pritchard, G.A.; Peart, J.N.; Headrick, J.P. Abstract 373: Transcriptional and cell signaling cross-talk between adenosine and diploid receptors in cardiac cells. Circulation 2008, 118, S293. [Google Scholar]
- Förster, K.; Kuno, A.; Solenkova, N.; Felix, S.B.; Krieg, T. The delta-opioid receptor agonist DADLE at reperfusion protects the heart through activation of pro-survival kinases via EGF receptor transactivation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1604–H1608. [Google Scholar]
- Panneerselvam, M.; Tsutsumi, Y.M.; Bonds, J.A.; Horikawa, Y.T.; Saldana, M.; Dalton, N.D.; Head, B.P.; Patel, P.M.; Roth, D.M.; Patel, H.H. Dark chocolate receptors: epicatechin-induced cardiac protection is dependent on delta-opioid receptor stimulation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1604–H1609. [Google Scholar]
- Gross, G.J.; Baker, J.E.; Hsu, A.; Wu, H.E.; Falck, J.R.; Nithipatikom, K. Evidence for a role of opioids in epoxyeicosatrienoic acid-induced cardioprotection in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2201–H2207. [Google Scholar]
- Peart, J.N.; Gross, G.J. Morphine-tolerant mice exhibit a profound and persistent cardioprotective phenotype. Circulation 2004, 109, 1219–1222. [Google Scholar]
- Peart, J.N.; See Hoe, L.E.; Gross, G.J.; Headrick, J.P. Sustained ligand-activated preconditioning via δ-opioid receptors. J. Pharmacol. Exp. Ther. 2011, 336, 274–281. [Google Scholar]
- Peart, J. N.; Headrick, J. P. Sustained cardioprotection: Exploring unconventional modalities. Vasc. Pharmacol. 2008, 49, 63–70. [Google Scholar]
- Tsuchida, A.; Thompson, R.; Olsson, R.A.; Downey, J.M. The anti-infarct effect of an adenosine A1-selective agonist is diminished after prolonged infusion as is the cardioprotective effect of ischaemic preconditioning in rabbit heart. J. Mol. Cell. Cardiol. 1994, 26, 303–311. [Google Scholar]
- Dana, A.; Baxter, G.F.; Walker, J.M.; Yellon, D.M. Prolonging the delayed phase of myocardial protection: repetitive adenosine A1 receptor activation maintains a preconditioned state. J. Am. Coll. Cardiol. 1998, 31, 1142–1149. [Google Scholar]
- Inagaki, K.; Begley, R.; Ikeno, F.; Mochly-Rosen, D. Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation 2005, 111, 44–50. [Google Scholar]
- Gross, E.R.; Peart, J.N.; Hsu, A.K.; Auchampach, J.A.; Gross, G.J. Extending the cardioprotective window using a novel delta-opioid agonist fentanyl isothiocyanate via the PI3-kinase pathway. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2744–H2749. [Google Scholar]
- Peart, J.N.; Gross, G.J. Chronic exposure to morphine produces a marked cardioprotective phenotype in aged mouse hearts. Exp. Gerontol. 2004, 39, 1021–1026. [Google Scholar]
- Bartling, B.; Friedrich, I.; Silber, R.E.; Simm, A. Ischemic preconditioning is not cardioprotective in senescent human myocardium. Ann. Thorac. Surg. 2003, 76, 105–111. [Google Scholar]
- Headrick, J.P. Aging impairs functional, metabolic and ionic recovery from ischemia reperfusion and hypoxia-reoxygenation. J. Mol. Cell. Cardiol. 1998, 30, 1415–1430. [Google Scholar]
- Headrick, J.P.; Willems, L.; Ashton, K.J.; Holmgren, K.; Matherne, G.P. Ischaemic tolerance in aged myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. J. Physiol. 2003, 549, 823–833. [Google Scholar]
- Peart, J.N.; Gross, E.R.; Headrick, J.P.; Gross, G.J. Impaired p38 MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J. Mol. Cell. Cardiol. 2007, 42, 972–980. [Google Scholar]
- Willems, L.; Ashton, K. J.; Headrick, J. P. Adenosine-mediated cardioprotection in the aging myocardium. Cardiovasc. Res. 2005, 66, 245–255. [Google Scholar]
- Nishida, H.; Sato, T.; Miyazaki, M.; Nakaya, H. Infarct size limitation by adrenomedullin: protein kinase A but not PI3-kinase is linked to mitochondrial KCa channels. Cardiovasc. Res. 2008, 77, 398–405. [Google Scholar]
- Redel, A.; Lange, M.; Jazbutyte, V.; Lotz, C.; Smul, T.M.; Roewer, N.; Kehl, F. Activation of mitochondrial large-conductance calcium-activated K+ channels via protein kinase A mediates desflurane-induced preconditioning. Anesth. Analg. 2008, 106, 384–391. [Google Scholar]
- Sanada, S.; Asanuma, H.; Tsukamoto, O.; Minamino, T.; Node, K.; Takashima, S.; Fukushima, T.; Ogai, A.; Shinozaki, Y.; Fujita, M.; et al. Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 2004, 110, 51–57. [Google Scholar]
- Foerster, K.; Groner, F.; Matthes, J.; Koch, W.J.; Birnbaumer, L.; Herzig, S. Cardioprotection specific for the G protein Gi2 in chronic adrenergic signaling through beta 2-adrenoceptors. Proc. Natl. Acad. Sci. USA 2003, 100, 14475–14480. [Google Scholar]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The beta2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar]
- Bruce, D.S.; Cope, G.W.; Elam, T.R.; Ruit, K.A.; Oeltgen, P.R.; Su, T.P. Opioids and hibernation. I. Effects of naloxone on bear HIT'S depression of guinea pig ileum contractility and on induction of summer hibernation in the ground squirrel. Life Sci. 1987, 41, 2107–2113. [Google Scholar]
- Oeltgen, P.R.; Walsh, J.W.; Hamann, S.R.; Randall, D.C.; Spurrier, W.A.; Myers, R.D. Hibernation “trigger”: opioid-like inhibitory action on brain function of the monkey. Pharmacol. Biochem. Behav. 1982, 17, 1271–1274. [Google Scholar]
- Oeltgen, P.R.; Nilekani, S.P.; Nuchols, P.A.; Spurrier, W.A.; Su, T.P. Further studies on opioids and hibernation: delta opioid receptor ligand selectively induced hibernation in summer-active ground squirrels. Life Sci. 1988, 43, 1565–1574. [Google Scholar]
- Bolling, S.F.; Tramontini, N.L.; Kilgore, K.S.; Su, T.P.; Oeltgen, P.R.; Harlow, H.H. Use of “natural” hibernation induction triggers for myocardial protection. Ann. Thorac. Surg. 1997, 64, 623–627. [Google Scholar]
- Hong, J.; Sigg, D.C.; Coles, J.A., Jr.; Oeltgen, P.R.; Harlow, H.J.; Soule, C.L.; Iaizzo, P.A. Hibernation induction trigger reduces hypoxic damage of swine skeletal muscle. Muscle Nerve 2005, 32, 200–207. [Google Scholar]
- Storey, K.B. Metabolic adaptations supporting anoxia tolerance in reptiles: recent advances. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1996, 113, 23–35. [Google Scholar]
- Holden, C.P.; Storey, K.B. Protein kinase A from bat skeletal muscle: a kinetic study of the enzyme from a hibernating mammal. Arch. Biochem. Biophys. 1998, 358, 243–250. [Google Scholar]
- Cai, D.; McCarron, R.M.; Yu, E.Z.; Li, Y.; Hallenbeck, J. Akt phosphorylation and kinase activity are down-regulated during hibernation in the 13-lined ground squirrel. Brain Res. 2004, 1014, 14–21. [Google Scholar]
- Abnous, K.; Dieni, C.A.; Storey, K.B. Regulation of Akt during hibernation in Richardson's ground squirrels. Biochim. Biophys. Acta 2008, 1780, 185–193. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Williams-Pritchard, G.; Headrick, J.P.; Peart, J.N. Myocardial Opioid Receptors in Conditioning and Cytoprotection. Pharmaceuticals 2011, 4, 470-484. https://doi.org/10.3390/ph4030470
Williams-Pritchard G, Headrick JP, Peart JN. Myocardial Opioid Receptors in Conditioning and Cytoprotection. Pharmaceuticals. 2011; 4(3):470-484. https://doi.org/10.3390/ph4030470
Chicago/Turabian StyleWilliams-Pritchard, Grant, John P. Headrick, and Jason N. Peart. 2011. "Myocardial Opioid Receptors in Conditioning and Cytoprotection" Pharmaceuticals 4, no. 3: 470-484. https://doi.org/10.3390/ph4030470
APA StyleWilliams-Pritchard, G., Headrick, J. P., & Peart, J. N. (2011). Myocardial Opioid Receptors in Conditioning and Cytoprotection. Pharmaceuticals, 4(3), 470-484. https://doi.org/10.3390/ph4030470