Beta-Blockers and Oxidative Stress in Patients with Heart Failure

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Oxidative stress has been implicated in the pathogenesis of heart failure. Reactive oxygen species (ROS) are produced in the failing myocardium, and ROS cause hypertrophy, apoptosis/cell death and intracellular Ca2+ overload in cardiac myocytes. ROS also cause damage to lipid cell membranes in the process of lipid peroxidation. In this process, several aldehydes, including 4-hydroxy-2-nonenal (HNE), are generated and the amount of HNE is increased in the human failing myocardium. HNE exacerbates the formation of ROS, especially H2O2 and ·OH, in cardiomyocytes and subsequently ROS cause intracellular Ca2+ overload. Treatment with beta-blockers such as metoprolol, carvedilol and bisoprolol reduces the levels of oxidative stress, together with amelioration of heart failure. This reduction could be caused by several possible mechanisms. First, the beta-blocking effect is important, because catecholamines such as isoproterenol and norepinephrine induce oxidative stress in the myocardium. Second, anti-ischemic effects and negative chronotropic effects are also important. Furthermore, direct antioxidative effects of carvedilol contribute to the reduction of oxidative stress. Carvedilol inhibited HNE-induced intracellular Ca2+ overload. Beta-blocker therapy is a useful antioxidative therapy in patients with heart failure.1. Introduction
Reactive oxygen species (ROS) induce ‘oxidative stress’ unless the prepared antioxidant mechanisms compensate for the ROS load [1]. Oxidative stress has been implicated in the pathogenesis of cardiovascular disease [2,3], including heart failure [4,5]. Increased oxidative stress results from an imbalance between ROS load and antioxidative mechanisms. Catecholamines [6-8], angiotensin II [9-11], tumor necrosis factor-alpha (TNF-alpha) [9], tachycardia and ischemia [12] induce generation of ROS in the myocardium. Activities of some antioxidant enzymes such as paraoxonase-1 (PON-1) in serum and manganese superoxide dismutase (MnSOD) in the myocardium, if not all enzymes, are diminished in patients with heart failure [7,13-15]. Thus, oxidative stress levels are elevated in the failing myocardium [6,16-18], and ROS causes hypertrophy, apoptosis/cell death and intracellular Ca2+ overload in cardiac myocytes [7,9,10,19-21]. In this review, we will focus on the involvement of oxidative stress in patients with heart failure and its reduction by beta-blocker therapy.
2. Involvement of Oxidative Stress in Heart Failure
In 1991, Belch et al. reported that concentrations of a plasma lipid peroxide, malondialdehyde, were significantly higher in the patients with congestive heart failure than in controls [4]. There was a significant negative correlation between malondialdehyde and left ventricular ejection fraction. Mallat et al. reported that pericardial levels of 8-iso-prostaglandin F2a (8-iso-PGF2a), a specific nonenzymatic peroxidation product of arachidonic acid, increase with increase in the functional severity of heart failure and are associated with ventricular dilatation [22].
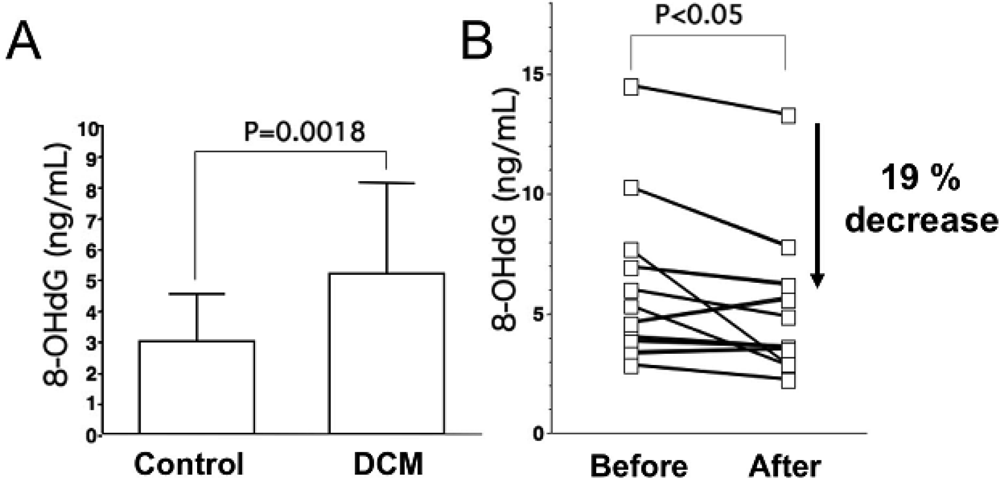
As for oxidative stress markers other than lipid peroxides, levels of 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage, are elevated in serum and urine of patients with heart failure, and urinary 8-OHdG reflects the clinical severity of CHF on the basis of symptomatic status and cardiac dysfunction (Figure 1A) [18,23]. Results of these studies indicate that oxidative stress is involved in severity of heart failure.
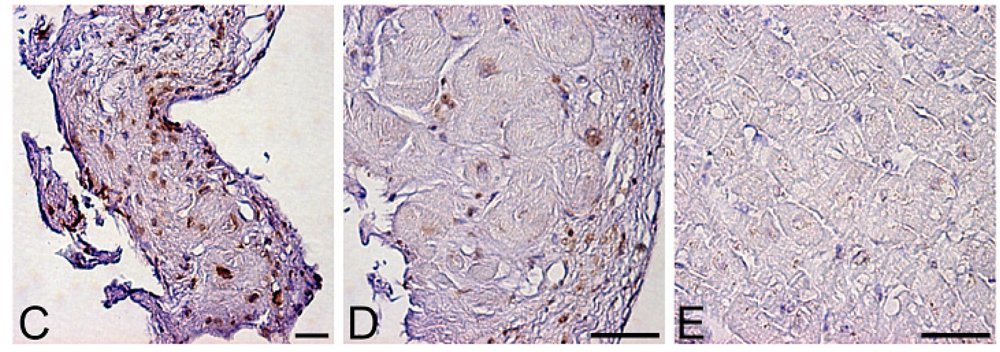
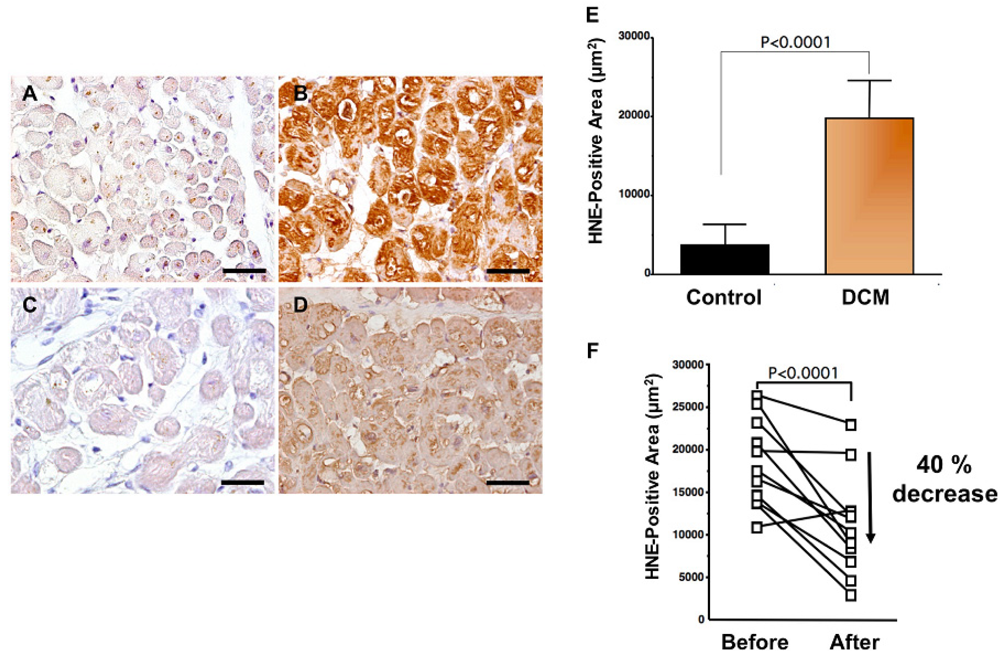
Levels of oxidative stress are also elevated in the myocardium of patients with heart failure. ROS cause damage to lipid cell membranes in the process of lipid peroxidation. In this process, several aldehydes, including 4-hydroxy-2-nonenal (HNE), are generated and the amount of HNE is increased in the human failing myocardium [Figures 2(B,E)] [6,17]. The presence of 8-OHdG has also been detected in nuclei of cardiac myocytes in patients with dilated cardiomyopathy (DCM) [Figures 1(C,D) [18]. Therefore, levels of oxidative stress are elevated in both body fluid including serum, urine and pericardial effusion and myocardium of patients with heart failure.
HNE is recognized not only as a reliable marker of oxidative stress but also as a toxic aldehyde to many types of cells [1,24-26]. HNE exhibits cytopathological effects, such as enzyme inhibition and inhibition of DNA, RNA and protein synthesis. HNE exacerbates heart failure. Administration of HNE was found to cause contractile failure and to elicit proarrhythmic effects in hearts [27,28]. HNE also has pro-oxidant properties.
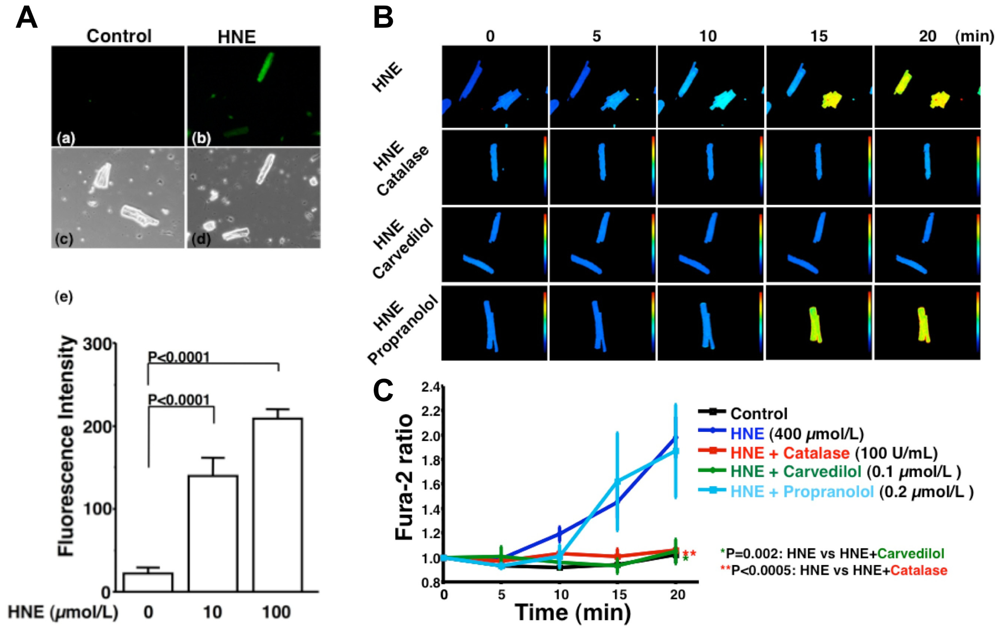
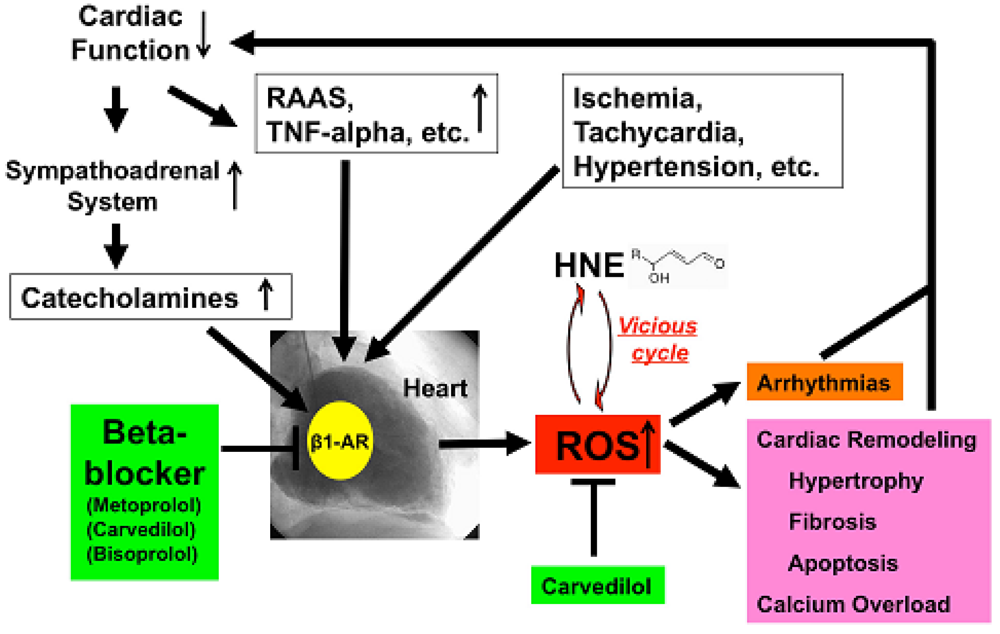
HNE can markedly induce intracellular production of ROS in cultured rat hepatocytes and human neuroblastoma cells [26,29,30]. HNE also exacerbates the formation of ROS, especially H2O2 and ·OH, in cardiomyocytes and subsequently ROS cause intracellular Ca2+ overload (Figure 3) [21]. Therefore, HNE exacerbates heart failure by increasing oxidative stress in the myocardium. Angiotensin II [9-11], catecholamines [6-8], tumor necrosis factor-alpha (TNF-alpha) [9] and ischemia [12] induce generation of ROS in the myocardium (Figure 4). ROS cause HNE formation in the process of lipid peroxidation. HNE inversely induces generation of ROS. In this way, a vicious cycle of oxidative stress is formed in the failing myocardium (Figure 5). Terminating this vicious cycle may be important to reduce oxidative stress in the failing myocardium.
Different oxidative stress levels stimulate cellular proliferation, trigger apoptosis or produce necrosis in various cells [1,31]. Experimental studies have shown that ROS can exert a graded effect on the cardiac myocyte phenotype [7,32]. Lower levels of oxidative stress stimulate hypertrophy and higher levels of oxidative stress induce apoptosis [7,32]. Higher rates of ROS production contribute to the transition from compensatory left-ventricular hypertrophy to heart failure [33,34]. We previously reported that oxidative stress was elevated in myocardia of patients with hypertrophic cardiomyopathy and that the levels were correlated with left ventricular dilatation and systolic dysfunction [17]. Thus, graded effects of ROS on the myocyte phenotype may be present in clinical settings. Further studies are needed to clarify this point.
3. Reduction of Oxidative Stress by Beta-blocker Therapy
3.1. Reduction Effect of a Beta-blocker on Oxidative Stress in Patients with Heart Failure
Results of many clinical trials, including the US Carvedilol Heart Failure Study, CIBIS-II and III, MERIT-HF, CARMEN, MUCHA, COMET, COPERNICS and REVERT have supported the efficacy of three beta-blockers (carvedilol, metoprolol and bisoprolol) in the treatment of heart failure. These studies have revealed that both ß1 adrenoreceptor blockers (metoprolol and bisoprolol) and a nonselective ß1 and ß2 adrenoreceptor blocker (carvedilol) ameliorate cardiac function and mortality in patients with heart failure. One of the mechanisms is thought to be reduction effect of a beta-blocker on oxidative stress. Kukin et al. reported that both metoprolol, a β1-selective blocker, and carvedilol, an α and β blocker with antioxidant activity, reduced plasma lipid peroxidation in patients with heart failure, together with amelioration of heart failure [35]. Chin et al. reported that β-blockers (bisoprolol and carvedilol), reduced the levels of serum lipid hydroperoxides in patients with CHF [36]. The serum levels of 8-OHdG in patients with DCM significantly decreased by 19% (Figure 1B) [18]. Thus, treatment with a beta-blocker can reduce systemic levels of oxidative stress, along with amelioration of heart failure.
A beta-blocker decreases elevated oxidative stress not only in serum or plasma but also in the human failing myocardium [6]. Endomyocardial biopsy samples from 11 patients with DCM were examined before and after treatment (mean, 9 ± 4 months) with carvedilol (5 to 30 mg/day; mean dosage, 22 ± 8 mg/day). After treatment with carvedilol, myocardial HNE-modified protein levels decreased by 40% along with amelioration of cardiac function (Figures 2B,D,F) [6]. Since HNE is a cytotoxic product, the reducing effects of carvedilol may play a critical role in amelioration of heart failure.
3.2. Mechanism of Reduction of Oxidative Stress by Beta-blocker Therapy
This reduction can be caused by several possible mechanisms.
3.2.1. Class Effect of Beta-Blockers
- a.
ß1-blocking effect. Isoproterenol induces lipid peroxidation [8]. Apoptosis stimulated by ß1-adrenergic receptors (ß1-AR) may also be mediated by ROS production [7]. Therefore, ß1-adrenergic receptor blockers are useful for catecholamine-induced oxidative stress.
- b.
Anti-ischemic properties. Anti-ischemic properties may also be important since ischemia has been shown to increase HNE formation in the heart [12].
- c.
Negative chronotropic effect. Tachycardia induces ROS generation in mitochondria of the myocardium [16]. Therefore, a negative chronotropic effect may be important.
- d.
Anti-hypertensive effect. Mechanical strain in cardiac myocytes increases ROS production [37,38]. Lowering blood pressure by a beta-blocker reduces after-load of the heart and finally may reduce ROS production.
3.2.2. Specific Effect of Carvedilol
- e.
Direct antioxidative property of carvedilol. The direct antioxidative property of carvedilol may contribute to the reduction of oxidative stress. Carvedilol inhibited Fe2+-initiated lipid peroxidation in vitro, but propranolol did not [39]. The mechanism of inhibition is via scavenging free radicals [39]. Carvedilol prevented hydroxyl radical-induced cardiac contractile dysfunction in human myocardial tissue, but metoprolol did not [40]. These results suggest the possible importance of the use of carvedilol.
- f.
Inhibitory effects of carvedilol on ROS generation by leukocytes. Carvedilol inhibits ROS generation by leukocytes [41].
- g.
α-blocking effect of carvedilol. Xiao et al. reported that ventricular myocytes express components of an NAD(P)H oxidase that appear to be involved in α1-AR-stimulated hypertrophic signaling via ROS-mediated activation of Ras-MEK1/2-ERK1/2. Therefore, the α-blocking effect of carvedilol may reduce oxidative stress [42].
3.2.3. Future Perspective of Mechanism Analysis
Emerging studies have revealed that β adrenergic receptor polymorphism may have an impact on response of β blocker treatment in patients with heart failure [43,44]. For example, there is a polymorphism at amino acid residue 389 (Arg/Gly) in human adrenergic β1-receptor gene (ADRB2). Mialet Perez et al. reported that homozygosity for Arg389 was associated with improvement in ventricular function during carvedilol treatment in 224 patients with heart failure [43]. A large clinical trial and further experimental examinations are needed to clarify this response. Determination of whether antioxidative effects of β blockers are different between Arg389 and Gly389 may be important.
Endothelial function is impaired in patients with heart failure [45] and is also related to elevated oxidative stress [46]. The third-generation β blocker nebivolol improves left ventricular dysfunction and have an effect on NO-mediated endothelial function in mice with extensive anterior myocardial infarction [47]. Nebivolol inhibits superoxide formation by vascular NADPH oxidase activation in angiotensin II-treated rats [48] and by vascular NOS III uncoupling in Watanabe heritable hyperlipidemic rabbits [49] and prevents endothelial dysfunction. Further studies are needed to clarify the beneficial effects of nebivolol in clinical settings [50,51].
4. Conclusions
Beta-blocker as a Possible Antioxidant in Treatment for Heart Failure
Depressed cardiac function causes activation of the sympathoadrenal system (ANS) and rennin-angiotensin-aldosterone system (RAAS) and elaboration of cytokines such as TNF-alpha [52]. All of these ischemia, tachycardia or hypertension increased ROS generation in the human failing myocardium (Figure 5). Catecholamines generate ROS via β1-AR, and beta-blockers, including metoprolol, carvedilol and bisoprolol, can therefore decrease ROS generation in the heart. Furthermore, carvedilol can scavenge redundant ROS and can terminate the vicious cycle such as oxidative stress caused by the cytotoxic aldehyde, HNE. ROS cause remodeling, hypertrophy, fibrosis, apoptosis and calcium overload and induce arrhythmia [53], which in turn depresses cardiac function. Beta-blockers such as metoprolol, bisoprolol and carvedilol can stop those vicious cycles in patients with heart failure via indirect or direct anti-oxidative properties.
Antioxidants to ameliorate cardiovascular diseases including heart failure have not generally yielded favorable results [54]. Furthermore, small or adequate degrees of stimulation by ROS are physiological and are needed in the bio-defense system or anti-tumorigenesis [54,55]. Thus, total blocking of oxidative stress in patients with heart failure is not needed or it may be harmful. In our study, carvedilol decreased the serum levels of 8-OHdG by 19% (Figure 1B) [18] and myocardial HNE-modified protein levels by 40% along with amelioration of cardiac function in patients with heart failure [Figures 2B,D,F] [6]. Since the therapy is safe and reduces patient mortality, the antioxidative properties of a beta-blocker may be sufficient to treat patients with heart failure. Further investigations are needed to clarify this point. In conclusion, beta-blocker therapy is an antioxidative therapy that is useful and safe in patients with heart failure.


A. Serum levels of 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage, in patients with dilated cardiomyopathy (DCM). DCM patients had significantly elevated serum levels of 8-OHdG compared with those in control subjects. Data are given as mean ± SD. B. Decrease in serum 8-OHdG levels due to treatment with carvedilol. Serum 8-OHdG levels decreased by 19% during treatment with carvedilol. C, D and E, immunohistochemical examination of 8-OHdG in myocardial biopsy samples. C. Representative immunohistochemical staining (brown) of a myocardial biopsy sample from a patient with DCM in a low-power field. Positive staining (brown) for 8-OHdG was distinct in the nuclei of cardiac myocytes. D. Representative staining of a myocardial biopsy sample from a patient with DCM in a high-power field. E. Representative staining in a myocardial biopsy sample from a control subject. Bar = 50 μm.

A to D. Immunohistochemical examination of HNE-modified protein in myocardial biopsy samples. A. Representative immunohistochemical staining in a myocardial biopsy sample from a control subject. There is no positive staining in cardiac myocytes; B. Representative staining (brown) in a myocardial biopsy sample from a dilated cardiomyopathy (DCM) patient before carvedilol treatment. Positive staining (brown) for HNE-modified protein was distinct in the cytosol of cardiac myocytes; C. Negative control sections incubated without a primary antibody; D. Representative staining in a myocardial biopsy sample from the same DCM patient 9 months after the beginning of carvedilol treatment. HNE-positive area is diminished. Bar = 50 μm; E. Expression levels of HNE-modified protein (HNE-positive area) in patients with DCM. DCM patients had significantly elevated myocardium levels of HNE-modified protein compared with those in control subjects. Data are given as mean ± SD; F. Decrease in HNE-positive area attributable to treatment with carvedilol. During treatment with carvedilol, the HNE-positive area decreased by 40%.

A. Generation of ROS induced by HNE exposure. Analysis of dichlorofluorescein (DCF) fluorescence. DCF is a non-specific detector of the production of intracellular ROS such as H2O2, ·OH and hydroperoxides (ROOH). (a–d), These are representative living cardiac myocytes observed by fluorescence microscopy (a and b) and phase contrast microscopy (c and d). Isolated rat cardiac myocytes on glass-base dishes were treated with a diluent (control) (a and c), HNE (100 μmol/L) (b and d). Treatment time was 10 minutes. (e), DCF fluorescence intensity per cell (n = 13 cells). Treatment with HNE (10 to 100 μmol/L) for ten minutes also induced the production of ROS (H2O2, OH or ROOH) in isolated rat ventricular myocytes in a dose-dependent manner, as assessed by DCF fluorescence. Each point is the mean ± SE. B and C. Inhibitory effects of antioxidants on the increase in fura-2 ratio due to HNE exposure; B. These are representative fura-2 ratio images in living cardiac myocytes observed by fluorescence microscopy. Isolated rat cardiac myocytes on glass-base dishes were treated with HNE (400 μmol/L), HNE and catalase (100 U/mL), HNE and carvedilol (0.1 μmol/L), or HNE and propranolol (0.2 μmol/L). Treatment time was 20 minutes; C. Antioxidants, catalase (n = 6) or carvedilol (n = 6), significantly inhibited the increase in fura-2 ratio. Propranolol did not inhibit the increase in fura-2 ratio (n = 7). Each point is the mean ± SE. HNE (400 μmol/L) exposure increased [Ca2+]i of isolated cardiac myocytes as assessed by fura-2 ratio in a time-dependent manner. Catalase (100 U/mL), an antioxidative enzyme, significantly attenuated the increase in [Ca2+]i induced by HNE. Furthermore, carvedilol (0.1 μmol/L), a beta-blocker with potent antioxidant activity, significantly attenuated the increase in [Ca2+]i induced by HNE, but propranolol (0.2 μmol/L) had no effect on [Ca2+]i increase. These results indicate that ROS mediate HNE-induced intracellular Ca2+ overload.

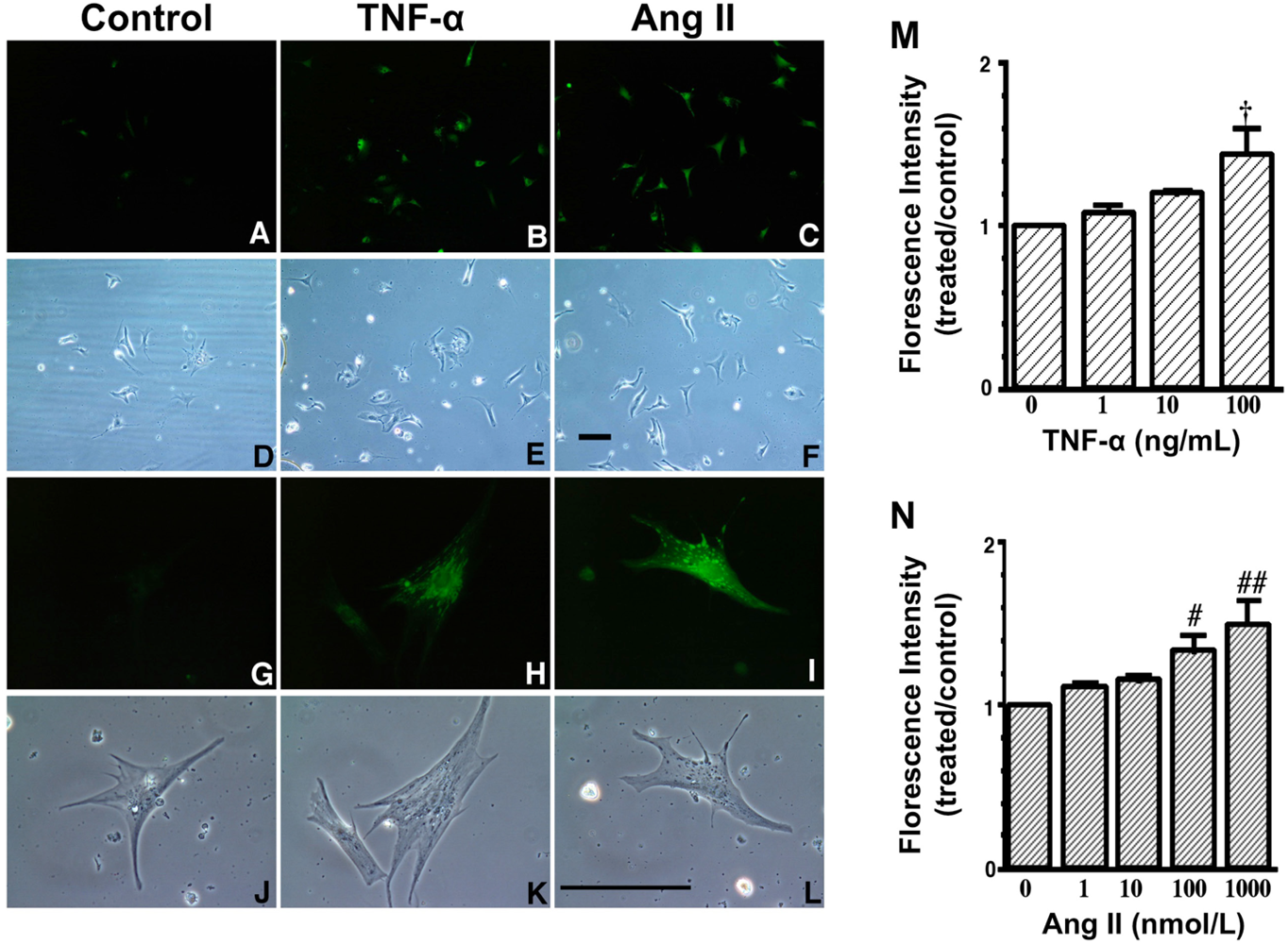
A to L. Increase in fluorescence with 2′,7′- dichlorofluorescin diacetate (DCFH-DA) due to TNF-α and Ang II exposure. These are representative living cardiac myocytes observed by fluorescence microscopy (A–C, G–I) and phase contrast microscopy (D–F, J–L). On culture day 4, cultured cardiac myocytes on glass cover slips were treated with a diluent (control) (left: A, D, G and J), TNF-α (10 ng/mL) (middle: B, E, H and K) or Ang II (100 nmol/L) (right: C, F, I and L) and with DCFH-DA (5 μmol/L). Treatment time was 1 hour. Upper two lanes (A–F) at low magnification and lower two lanes (G–L) at high magnification. Bar = 100 μm. M and N, Dose-dependent increase in fluorescence due to TNF-α and Ang II exposure. On culture day 4, cultured cardiac myocytes were treated with TNF-α (1 to 100 ng/mL), Ang II (1 to 1000 nmol/L) or a diluent without TNF-α and Ang II (control) and simultaneously with 2′,7′- dichlorofluorescin diacetate (DCFH-DA) (1 μmol/L). After 1 hour of incubation, cells were collected and fluorescence intensity per culture well was measured. Each point is the mean ± SE (n = 5 experiments). In each experiment, a treated-to-control ratio was calculated from the fluorescence intensity of five experiments. † P < 0.005, # P < 0.01, ## P < 0.0005 vs control cultures treated with diluent only. These data indicate that both TNF-α and Ang II generate ROS in cardiac myocytes.

References
- Toyokuni, S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol. Int. 1999, 49, 91–102. [Google Scholar]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. Nad(p)h oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar]
- Fukai, T.; Folz, R.J.; Landmesser, U.; Harrison, D.G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc. Res. 2002, 55, 239–249. [Google Scholar]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar]
- McMurray, J.; Chopra, M.; Abdullah, I.; Smith, W.E.; Dargie, H.J. Evidence of oxidative stress in chronic heart failure in humans. Eur. Heart J. 1993, 14, 1493–1498. [Google Scholar]
- Nakamura, K.; Kusano, K.; Nakamura, Y.; Kakishita, M.; Ohta, K.; Nagase, S.; Yamamoto, M.; Miyaji, K.; Saito, H.; Morita, H.; et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002, 105, 2867–2871. [Google Scholar]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell Cardiol. 2002, 34, 379–388. [Google Scholar]
- Singal, P.K.; Beamish, R.E.; Dhalla, N.S. Potential oxidative pathways of catecholamines in the formation of lipid peroxides and genesis of heart disease. Adv. Exp. Med. Biol. 1983, 161, 391–401. [Google Scholar]
- Nakamura, K.; Fushimi, K.; Kouchi, H.; Mihara, K.; Miyazaki, M.; Ohe, T.; Namba, M. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin ii. Circulation 1998, 98, 794–799. [Google Scholar]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci.USA 2006, 103, 7432–7437. [Google Scholar]
- Kakishita, M.; Nakamura, K.; Asanuma, M.; Morita, H.; Saito, H.; Kusano, K.; Nakamura, Y.; Emori, T.; Matsubara, H.; Sugaya, T.; et al. Direct evidence for increased hydroxyl radicals in angiotensin ii-induced cardiac hypertrophy through angiotensin ii type 1a receptor. J. Cardiovasc. Pharmacol. 2003, 42, S67–S70. [Google Scholar]
- Eaton, P.; Li, J.M.; Hearse, D.J.; Shattock, M.J. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am. J. Physiol. 1999, 276, H935–H943. [Google Scholar]
- Tang, W.H.; Wu, Y.; Mann, S.; Pepoy, M.; Shrestha, K.; Borowski, A.G.; Hazen, S.L. Diminished antioxidant activity of high-density lipoprotein-associated proteins in systolic heart failure. Circ. Heart Fail. 2011, 4, 59–64. [Google Scholar]
- Sam, F.; Kerstetter, D.L.; Pimental, D.R.; Mulukutla, S.; Tabaee, A.; Bristow, M.R.; Colucci, W.S.; Sawyer, D.B. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card. Fail. 2005, 11, 473–480. [Google Scholar]
- Dieterich, S.; Bieligk, U.; Beulich, K.; Hasenfuss, G.; Prestle, J. Gene expression of antioxidative enzymes in the human heart: Increased expression of catalase in the end-stage failing heart. Circulation 2000, 101, 33–39. [Google Scholar]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex i is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar]
- Nakamura, K.; Kusano, K.F.; Matsubara, H.; Nakamura, Y.; Miura, A.; Nishii, N.; Banba, K.; Nagase, S.; Miyaji, K.; Morita, H.; et al. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J. Card. Fail. 2005, 11, 117–123. [Google Scholar]
- Kono, Y.; Nakamura, K.; Kimura, H.; Nishii, N.; Watanabe, A.; Banba, K.; Miura, A.; Nagase, S.; Sakuragi, S.; Kusano, K.F. Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circ. J. 2006, 70, 1001–1005. [Google Scholar]
- Josephson, R.A.; Silverman, H.S.; Lakatta, E.G.; Stern, M.D.; Zweier, J.L. Study of the mechanisms of hydrogen peroxide and hydroxyl free radical-induced cellular injury and calcium overload in cardiac myocytes. J. Biol. Chem. 1991, 266, 2354–2361. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin d reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar]
- Nakamura, K.; Miura, D.; Kusano, K.F.; Fujimoto, Y.; Sumita-Yoshikawa, W.; Fuke, S.; Nishii, N.; Nagase, S.; Hata, Y.; Morita, H.; et al. 4-hydroxy-2-nonenal induces calcium overload via the generation of reactive oxygen species in isolated rat cardiac myocytes. J. Card. Fail. 2009, 15, 709–716. [Google Scholar]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin f2alpha in pericardial fluid of patients with heart failure: A potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539. [Google Scholar]
- Kobayashi, S.; Susa, T.; Tanaka, T.; Wada, Y.; Okuda, S.; Doi, M.; Nao, T.; Yoshiga, Y.; Yamada, J.; Okamura, T.; et al. Urinary 8-hydroxy-2′-deoxyguanosine reflects symptomatic status and severity of systolic dysfunction in patients with chronic heart failure. Eur. J. Heart Fail. 2011, 13, 29–36. [Google Scholar]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar]
- Lee, S.H.; Oe, T.; Blair, I.A. Vitamin c-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science 2001, 292, 2083–2086. [Google Scholar]
- Uchida, K. 4-hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar]
- Haenen, G.R.; Plug, H.J.; Vermeulen, N.P.; Timmerman, H.; Bast, A. Contribution of 4-hydroxy-2,3-trans-nonenal to the reduction of beta-adrenoceptor function in the heart by oxidative stress. Life Sci. 1989, 45, 71–76. [Google Scholar]
- Bhatnagar, A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ. Res. 1995, 76, 293–304. [Google Scholar]
- Uchida, K.; Shiraishi, M.; Naito, Y.; Torii, Y.; Nakamura, Y.; Osawa, T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J. Biol. Chem 1999, 274, 2234–2242. [Google Scholar]
- Kondo, M.; Oya-Ito, T.; Kumagai, T.; Osawa, T.; Uchida, K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J. Biol. Chem. 2001, 276, 12076–12083. [Google Scholar]
- Dypbukt, J.M.; Ankarcrona, M.; Burkitt, M.; Sjoholm, A.; Strom, K.; Orrenius, S.; Nicotera, P. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting rinm5f cells. The role of intracellular polyamines. J. Biol. Chem. 1994, 269, 30553–30560. [Google Scholar]
- Siwik, D.A.; Tzortzis, J.D.; Pimental, D.R.; Chang, D.L.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ. Res. 1999, 85, 147–153. [Google Scholar]
- Dhalla, A.K.; Hill, M.F.; Singal, P.K. Role of oxidative stress in transition of hypertrophy to heart failure. J. Am. Coll. Cardiol. 1996, 28, 506–514. [Google Scholar]
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in ventricular remodelling. Lancet 2006, 367, 356–367. [Google Scholar]
- Kukin, M.L.; Kalman, J.; Charney, R.H.; Levy, D.K.; Buchholz-Varley, C.; Ocampo, O.N.; Eng, C. Prospective, randomized comparison of effect of long-term treatment with metoprolol or carvedilol on symptoms, exercise, ejection fraction, and oxidative stress in heart failure. Circulation 1999, 99, 2645–2651. [Google Scholar]
- Chin, B.S.; Langford, N.J.; Nuttall, S.L.; Gibbs, C.R.; Blann, A.D.; Lip, G.Y. Anti-oxidative properties of beta-blockers and angiotensin-converting enzyme inhibitors in congestive heart failure. Eur J. Heart Fail. 2003, 5, 171–174. [Google Scholar]
- Cheng, W.; Li, B.; Kajstura, J.; Li, P.; Wolin, M.S.; Sonnenblick, E.H.; Hintze, T.H.; Olivetti, G.; Anversa, P. Stretch-induced programmed myocyte cell death. J. Clin. Invest. 1995, 96, 2247–2259. [Google Scholar]
- Pimentel, D.R.; Amin, J.K.; Xiao, L.; Miller, T.; Viereck, J.; Oliver-Krasinski, J.; Baliga, R.; Wang, J.; Siwik, D.A.; Singh, K.; et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ. Res. 2001, 89, 453–460. [Google Scholar]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol. Exp. Ther. 1992, 263, 92–98. [Google Scholar]
- Flesch, M.; Maack, C.; Cremers, B.; Baumer, A.T.; Sudkamp, M.; Bohm, M. Effect of beta-blockers on free radical-induced cardiac contractile dysfunction. Circulation 1999, 100, 346–353. [Google Scholar]
- Dandona, P.; Karne, R.; Ghanim, H.; Hamouda, W.; Aljada, A.; Magsino, C.H., Jr. Carvedilol inhibits reactive oxygen species generation by leukocytes and oxidative damage to amino acids. Circulation 2000, 101, 122–124. [Google Scholar]
- Xiao, L.; Pimentel, D.R.; Wang, J.; Singh, K.; Colucci, W.S.; Sawyer, D.B. Role of reactive oxygen species and nad(p)h oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am. J. Physiol. Cell. Physiol. 2002, 282, C926–C934. [Google Scholar]
- Mialet Perez, J.; Rathz, D.A.; Petrashevskaya, N.N.; Hahn, H.S.; Wagoner, L.E.; Schwartz, A.; Dorn, G.W.; Liggett, S.B. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat. Med. 2003, 9, 1300–1305. [Google Scholar]
- Dorn, G.W., II. Adrenergic signaling polymorphisms and their impact on cardiovascular disease. Physiol. Rev. 2010, 90, 1013–1062. [Google Scholar]
- Katz, S.D.; Biasucci, L.; Sabba, C.; Strom, J.A.; Jondeau, G.; Galvao, M.; Solomon, S.; Nikolic, S.D.; Forman, R.; LeJemtel, T.H. Impaired endothelium-mediated vasodilation in the peripheral vasculature of patients with congestive heart failure. J. Am. Coll. Cardiol. 1992, 19, 918–925. [Google Scholar]
- Katz, S.D. Mechanisms and implications of endothelial dysfunction in congestive heart failure. Curr. Opin. Cardiol. 1997, 12, 259–264. [Google Scholar]
- Sorrentino, S.A.; Doerries, C.; Manes, C.; Speer, T.; Dessy, C.; Lobysheva, I.; Mohmand, W.; Akbar, R.; Bahlmann, F.; Besler, C.; et al. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional beta1-blockade. J. Am. Coll. Cardiol. 2011, 57, 601–611. [Google Scholar]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by nadph oxidase and endothelial dysfunction in angiotensin ii-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar]
- Mollnau, H.; Schulz, E.; Daiber, A.; Baldus, S.; Oelze, M.; August, M.; Wendt, M.; Walter, U.; Geiger, C.; Agrawal, R.; et al. Nebivolol prevents vascular nos iii uncoupling in experimental hyperlipidemia and inhibits nadph oxidase activity in inflammatory cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 615–621. [Google Scholar]
- Munzel, T.; Gori, T. Nebivolol: The somewhat-different beta-adrenergic receptor blocker. J. Am. Coll. Cardiol. 2009, 54, 1491–1499. [Google Scholar]
- Pedersen, M.E.; Cockcroft, J.R. The vasodilatory beta-blockers. Curr. Hypertens. Rep. 2007, 9, 269–277. [Google Scholar]
- Braunwald, E.; Bristow, M.R. Congestive heart failure: Fifty years of progress. In Circulation; 2000; Volume 102, pp. IV14–IV23. [Google Scholar]
- Tanaka, M.; Nakamura, K.; Kusano, K.F.; Morita, H.; Ohta-Ogo, K.; Miura, D.; Miura, A.; Nakagawa, K.; Tada, T.; Murakami, M.; et al. Elevated oxidative stress is associated with ventricular fibrillation episodes in patients with brugada-type electrocardiogram without scn5a mutation. Cardiovasc. Pathol. 2011, 20, e37–e42. [Google Scholar]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med 2011. in press. [Google Scholar]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced nrf2 transcription promotes ros detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakamura, K.; Murakami, M.; Miura, D.; Yunoki, K.; Enko, K.; Tanaka, M.; Saito, Y.; Nishii, N.; Miyoshi, T.; Yoshida, M.; et al. Beta-Blockers and Oxidative Stress in Patients with Heart Failure. Pharmaceuticals 2011, 4, 1088-1100. https://doi.org/10.3390/ph4081088
Nakamura K, Murakami M, Miura D, Yunoki K, Enko K, Tanaka M, Saito Y, Nishii N, Miyoshi T, Yoshida M, et al. Beta-Blockers and Oxidative Stress in Patients with Heart Failure. Pharmaceuticals. 2011; 4(8):1088-1100. https://doi.org/10.3390/ph4081088
Chicago/Turabian StyleNakamura, Kazufumi, Masato Murakami, Daiji Miura, Kei Yunoki, Kenki Enko, Masamichi Tanaka, Yukihiro Saito, Nobuhiro Nishii, Toru Miyoshi, Masashi Yoshida, and et al. 2011. "Beta-Blockers and Oxidative Stress in Patients with Heart Failure" Pharmaceuticals 4, no. 8: 1088-1100. https://doi.org/10.3390/ph4081088
APA StyleNakamura, K., Murakami, M., Miura, D., Yunoki, K., Enko, K., Tanaka, M., Saito, Y., Nishii, N., Miyoshi, T., Yoshida, M., Oe, H., Toh, N., Nagase, S., Kohno, K., Morita, H., Matsubara, H., Kusano, K. F., Ohe, T., & Ito, H. (2011). Beta-Blockers and Oxidative Stress in Patients with Heart Failure. Pharmaceuticals, 4(8), 1088-1100. https://doi.org/10.3390/ph4081088