Multiple Facets of cAMP Signalling and Physiological Impact: cAMP Compartmentalization in the Lung


Abstract
:1. Introduction

2. Spatio-Temporal Nature of Compartmentalized cAMP Signalling: Paradigm Shifts

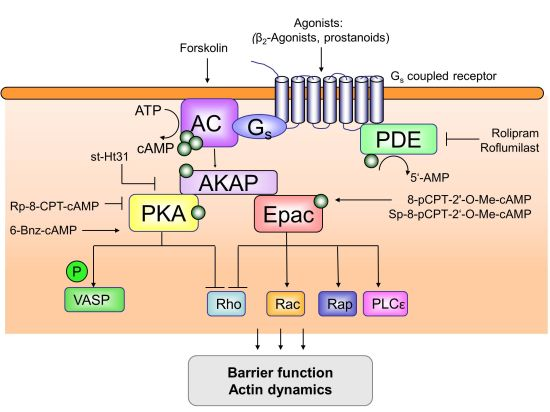{kind=link}
{kind=link}
{kind=link}
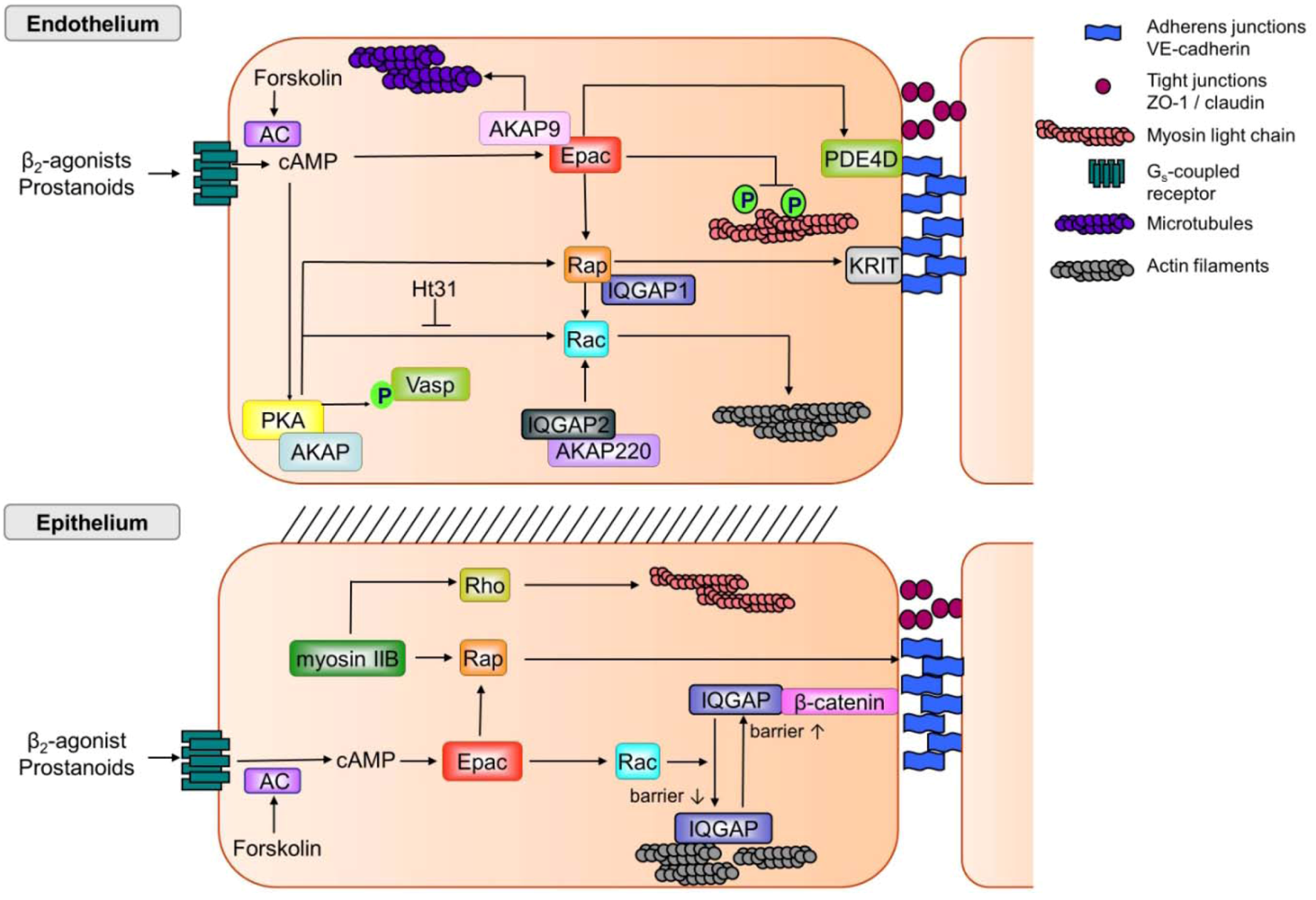{kind=link}
| Epac | PKA | AKAP | PDE | AC | small GTPases | |
|---|---|---|---|---|---|---|
| Bronchial epithelium | Epac1 [128] Epac 1 & 2 [129] | PKA [29] | AKAP9 [130] | ++ PDE4, PDE1 [131,132] +- PDE3, PDE5 [131,132] PDE4D [133] PDE3A [134] PDE7A1&2 [135] | AC9 [136,137] AC1, 4, 7, 8 [138] sAC [139] | Rap [140,141,142] Rac [29,141,142,143,144,145] Rap1 [129,143] Rap2 |
| Vascular endothelium | Epac1 [146,147,148,149] | PKA [147] | AKAP9 [149] Gravin [150] | PDE4D [22,147,151] PDE4 [135] PDE3 [135] | Membrane bound [2] Soluble AC [2] AC2, 3, 5, 6 [138] | Rap [143,146,147,149,152] Rac [146,147,152,153,154,155,156,157] Rac1 [158] RRas [147,159] |
| Airway smooth muscle cells | Epac1 [95,107,129] Epac2 [95,107,129] | PKA [107] | AKAP5, 9, 12 [160] Gravin, ezrin [117,150] | PDE1C, 3, 5A, 7 [135,161] PDE7A1&2 [135] | 7 membrane bound subtypes [118] 1, 3-7, 9 [115] 2, 6, 7, 9 [113] | RhoA [87] Rac1 [87] Rap1 [95] Rap2 [95] |
| Vascular smooth muscle cells | Epac1 [162] Epac2 [162] | PKA [162] | AKAP12 [163] | PDE1(C), 3(A), 5 [135] PDE7A1&2 [135] 1A, 1C, 2A, 3A, 3B, 4A, 4B, 4C, 4D, 5A, 7A, 7B, 8A, 9A, 9B, 10A and 11A [164] | AC1, 2, 3, 4, 6, 7, 9 [165] 2, 3, 5, 6, 7, 8 [138] | Rap1 [166] Rac1 [167] RhoA [168, 169] |
| Pulmonary fibroblasts | Epac [72] Epac1 [170] Epac2* [170] *only mRNA not protein | PKA [122] | AKAP9 [122] | PDE4A, B, D [119] 3A&B, 4A5, 4B2, 4C1, 4D3, 7 [171] | 6 membrane bound subtypes [118] | Rho A [172] Rac1 [172] Rac2 [172] Rap1 [173] |
| Inflammatory cells | Epac1 [109] | PKA [109] | Ezrin [174] AKAP9 [175] | PDE4B2 [176] PDE1B, 3A, 7A1, 2, 3 [135] 7A1&7A2 [177] | AC [178] 1, 2, 6, 9 [179] 4, 5, 6, 7, 9, sAC [180] sAC [181] | Rap [182] Ras [182] Rac1 [183] Rho [183] |
3. Cellular Diversity in cAMP Responses: Compartmentalization?
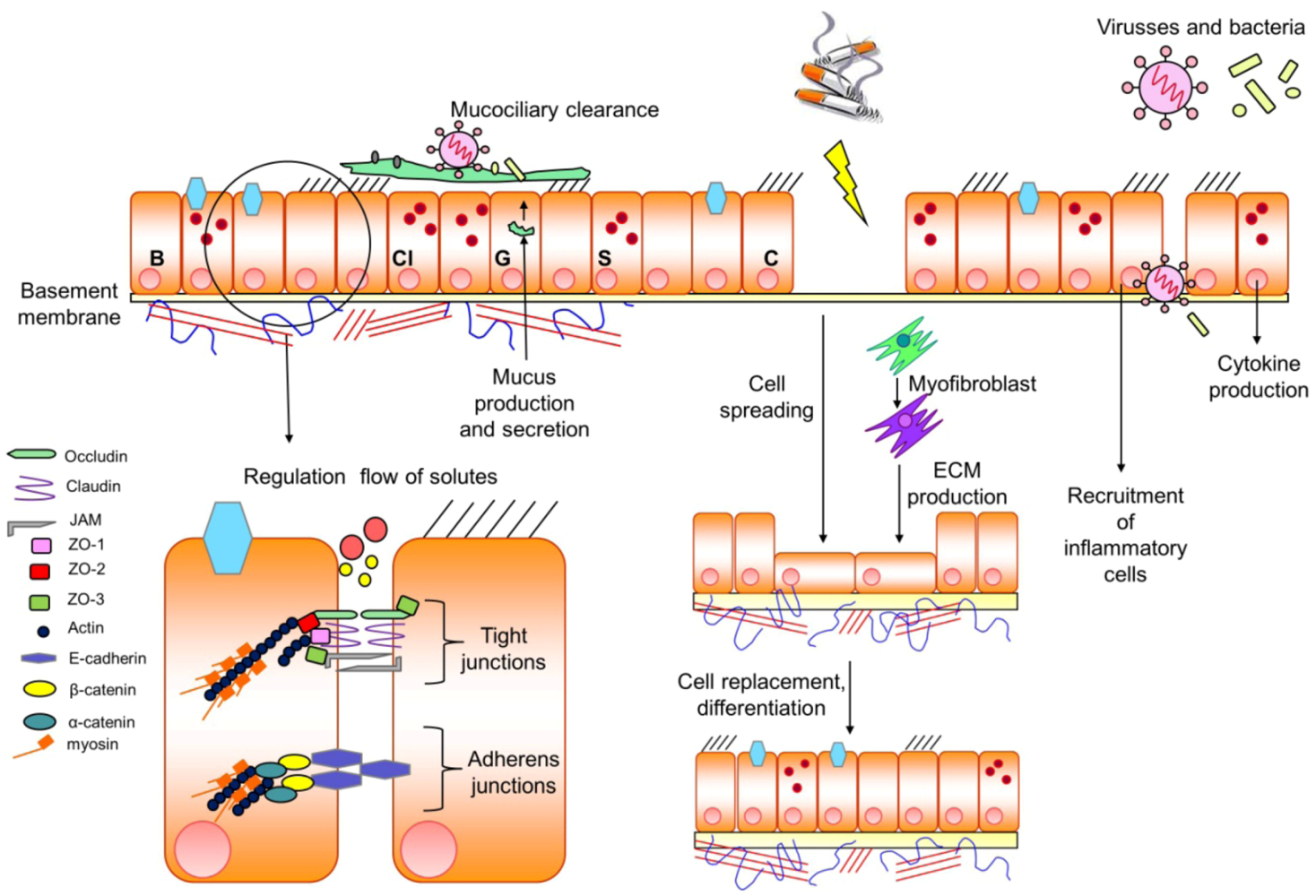
4. Regulation of Epithelial Barrier Function: Tight Junctions versus Adherens Junctions
 such as cigarette smoke
such as cigarette smoke  a (persistent) damage of the epithelial barrier, a process being compensated by cell spreading and production of extracellular matrix (ECM) by myofibroblasts to gain cell replacement and differentiation of distinct cells within the epithelium. In addition, toxic particles such as viruses and/or bacteria within the epithelial barrier will induce the recruitment of inflammatory cells and the production of cytokines to diminish the entrance of the devastating particles. For further details see text.
such as cigarette smoke a (persistent) damage of the epithelial barrier, a process being compensated by cell spreading and production of extracellular matrix (ECM) by myofibroblasts to gain cell replacement and differentiation of distinct cells within the epithelium. In addition, toxic particles such as viruses and/or bacteria within the epithelial barrier will induce the recruitment of inflammatory cells and the production of cytokines to diminish the entrance of the devastating particles. For further details see text.
a (persistent) damage of the epithelial barrier, a process being compensated by cell spreading and production of extracellular matrix (ECM) by myofibroblasts to gain cell replacement and differentiation of distinct cells within the epithelium. In addition, toxic particles such as viruses and/or bacteria within the epithelial barrier will induce the recruitment of inflammatory cells and the production of cytokines to diminish the entrance of the devastating particles. For further details see text.
such as cigarette smoke a (persistent) damage of the epithelial barrier, a process being compensated by cell spreading and production of extracellular matrix (ECM) by myofibroblasts to gain cell replacement and differentiation of distinct cells within the epithelium. In addition, toxic particles such as viruses and/or bacteria within the epithelial barrier will induce the recruitment of inflammatory cells and the production of cytokines to diminish the entrance of the devastating particles. For further details see text.
5. Novel Aspects of Barrier Functioning

6. Conclusions
Acknowledgements
Conflict of interest
References
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research -- still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar]
- Sayner, S.L. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L667–L678. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef]
- McCahill, A.C.; Huston, E.; Li, X.; Houslay, M.D. PDE4 associates with different scaffolding proteins: modulating interactions for certain diseases. Handb. Exp. Pharmacol 2008, 186, 125–166. [Google Scholar] [CrossRef]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef]
- Patel, H.H.; Murray, F.; Insel, P.A. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb. Exp. Pharmacol 2008, 167–184. [Google Scholar]
- Patel, H.H.; Murray, F.; Insel, P.A. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol Toxicol. 2008, 48, 359–391. [Google Scholar] [CrossRef]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar]
- Zaccolo, M. cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef]
- Zaccolo, M. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharmacol. 2011, 11, 649–655. [Google Scholar] [CrossRef]
- Stangherlin, A.; Zaccolo, M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol Heart Circ. Physiol. 2012, 302, H379–H390. [Google Scholar] [CrossRef]
- Beene, D.L.; Scott, J.D. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 2007, 19, 192–198. [Google Scholar] [CrossRef]
- Skroblin, P.; Grossmann, S.; Schafer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of protein kinase A anchoring. Int. Rev. Cell Mol. Biol. 2010, 283, 235–330. [Google Scholar] [CrossRef]
- Wong, W.; Scott, J.D. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Tröger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef]
- Ostroveanu, A.; van der Zee, E.; Eisel, U.L.; Schmidt, M.; Nijholt, I.M. Exchange protein directly activated by cyclic AMP2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus 2010, 20, 1018–1026. [Google Scholar] [CrossRef]
- Aye, T.-T.; Soni, S.; van Veen, T.A.B.; van der Heyden, M.A.G.; Cappadona, S.; Varro, A.; de Weger, R.A.; de Jonge, N.; Vos, M.A.; Heck, A.J.R.; Scholten, A. Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol. 2012, 52, 511–518. [Google Scholar] [CrossRef]
- Kovanich, D.; van der Heyden, M.A.; Aye, T.T.; van Veen, T.A.; Heck, A.J.; Scholten, A. Sphingosine kinase interacting proteins is an A-kinase anchroing protein specific for type I cAMP-dependent protein kinase. Chembiochem. 2010, 3, 963–971. [Google Scholar]
- Noda, K.; Zhang, J.; Fukuhara, S.; Kunimotot, S.; Yoshimura, M.; Mochizuki, N. Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through a- and b-catenins in cyclic AMP-Epac1-Rap1 signal-activated endothelial cells. Mol. Cell. Biol. 2010, 21, 584–596. [Google Scholar]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; Pare, P.D. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459. [Google Scholar] [CrossRef]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef]
- van den Berge, M.; ten Hacken, N.H.; Cohen, J.; Douma, W.R.; Postma, D.S. Small airway disease in asthma and COPD: clinical implications. Chest 2011, 139, 412–423. [Google Scholar]
- Rogers, D.F.; Barnes, P.J. Trends in clinical practice: Treatment of airway mucus hypersecretion. Ann. Med. 2006, 38, 116–125. [Google Scholar] [CrossRef]
- Lai, H.; Rogers, D.F. New pharmacotherapy for airway mucus hypersecretion in asthma and COPD: Targeting intracellular signaling pathways. J. Aerosol. Med. Pulm. Drug Deliv. 2010, 23, 219–231. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Otaki, F.; Bonsu, P.; Dang, D.T.; Teater, M.; Strulovici-Barel, Y.; Salit, J.; Harvey, B.G.; Crystal, R.G. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell. Mol. Life Sci. 2011, 68, 877–892. [Google Scholar] [CrossRef]
- Lai, H.Y.; Rogers, D.F. Mucus hypersecretion in asthma: intracellular signalling pathways as targets for pharmacotherapy. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 67–76. [Google Scholar] [CrossRef]
- Heijink, I.H.; Brandenburg, S.M.; Postma, D.S.; van Oosterhout, A.J.M. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur. Respir. J. 2012, 39, 419–428. [Google Scholar] [CrossRef]
- Lapperre, T.S.; Sont, J.K.; van Schadewijk, A.; Gosman, M.M.E.; Postma, D.S.; Bajema, I.M.; Timens, W.; Maua, T.; Hiemstra, P.S.; the GLUCOLD Study group. Smoking cessation and bronchial epithelial remodelling in COPD: a cross-sectional study. Respir. Res. 2007, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Voynow, J.A.; Rubin, B.K. Mucins, mucus, and sputum. Chest 2009, 135, 505–512. [Google Scholar] [CrossRef]
- Hurst, J.R.; Vestbo, J.; Anzueto, A.; Locantore, N.; Mullerova, H.; Tal-Singer, R.; Miller, B.; Lomas, D.A.; Agusti, A.; Macnee, W.; Calverley, P.; Rennard, S.; Wouters, E.F.; Wedzicha, J.A. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N. Engl. J. Med. 2010, 363, 1128–1138. [Google Scholar]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar]
- Cooper, P.R.; Kurten, R.C.; Zhang, J.; Nicholls, D.J.; Dainty, I.A.; Panettieri, R.A. Formoterol and salmeterol induce a similar degree of beta2-adrenoceptor tolerance in human small airways but via different mechanisms. Br. J. Pharmacol. 2011, 163, 521–532. [Google Scholar]
- Penn, R.B. Embracing emerging paradigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 149–169. [Google Scholar] [CrossRef]
- Bateman, E.D.; Hurd, S.S.; Barnes, P.J.; Bousquet, J.; Drazen, J.M.; FitzGerald, M.; Gibson, P.; Ohta, K.; O'Byrne, P.; Pedersen, S.E.; Pizzichini, E.; Sullivan, S.D.; Wenzel, S.E.; Zar, H.J. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J. 2008, 31, 143–178. [Google Scholar] [CrossRef]
- Penn, R.B. Agonizing over agonism: should asthmatics turn their beta-receptors on or off? Proc. Natl. Acad. Sci. USA 2009, 106, 2095–2096. [Google Scholar] [CrossRef]
- Yan, H.; Deshpande, D.A.; Misior, A.M.; Miles, M.C.; Saxena, H.; Riemer, E.C.; Pascual, R.M.; Panettieri, R.A.; Penn, R.B. Anti-mitogenic effects of {beta}-agonists and PGE2 on airway smooth muscle are PKA dependent. FASEB J. 2011, 25, 389–397. [Google Scholar]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H.; Toews, G.B.; Sisson, T.H.; Moore, B.B.; Peters-Golden, M. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Invest. 2010, 120, 1950–1960. [Google Scholar]
- Huang, S.K.; Fisher, A.S.; Scruggs, A.M.; White, E.S.; Hogaboam, C.M.; Richardson, B.C.; Peters-Golden, M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am. J. Pathol. 2010, 177, 2245–2255. [Google Scholar]
- Stumm, C.L.; Wettlaufer, S.H.; Jancar, S.; Peters-Golden, M. Airway remodeling in murine asthma correlates with a defect in PGE2 synthesis by lung fibroblasts. Am. J. Physiol Lung Cell Mol. Physiol. 2011, 301, L636–L644. [Google Scholar] [CrossRef]
- Sears, M.R. Safe use of long-acting b-agonists: what have we learnt? Expert. Opin. Drug. Saf. 2012, 10, 767–778. [Google Scholar] [CrossRef]
- Bos, J.L. Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell. Biol. 2003, 4, 733–738. [Google Scholar] [CrossRef]
- Gloerich, M.; Bos, J.L. Epac: defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 355–375. [Google Scholar] [CrossRef]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanisms of b-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de, F.T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS. Biol. 2009, 7, 1–8. [Google Scholar]
- Ferrandon, S.; Feinstein, T.N.; Castro, M.; Wang, B.; Bouley, R.; Potts, J.T.; Gardella, T.J.; Vilardaga, J.-P. Sustained cyclic AMP production by parathroid hormone receptor endocytosis. Nat. Chem. Biol. 2009, 5, 734–742. [Google Scholar] [CrossRef]
- Calebiro, D.; Nikolaev, V.O.; Lohse, M.J. Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J. Mol. Endocrinol. 2010, 45, 1–8. [Google Scholar]
- Murphy, J.E.; Padilla, B.E.; Hasdemir, B.; Cottrell, G.S.; Bunnett, N.W. Endosomes: a legitimate platform for the signaling train. Proc. Natl. Acad. Sci. USA 2009, 106, 17615–17622. [Google Scholar]
- Platta, H.W.; Stenmark, H. Endocytosis and signaling. Curr. Opin. Cell Biol. 2011, 23, 393–403. [Google Scholar] [CrossRef]
- Puthenveedu, M.A.; Lauffer, B.; Temkin, P.; Vistein, R.; Carlton, P.; Thorn, K.; Taunton, J.; Weiner, O.D.; Parton, R.G.; von Zastrow, M. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell 2010, 143, 761–773. [Google Scholar] [CrossRef]
- Kendall, R.T.; Strungs, E.G.; Rachidi, S.M.; Lee, M.-H.; El-Shewy, H.M.; Luttrell, D.K.; Janech, M.G.; Luttrell, L.M. The b-arrestin pathway-selective type 1A angiotensin receptor (AT1a) agonist [Sar1, IL4, Ile8] angiotensin II regulates a robust G protein-independent signaling network. J. Biol. Chem. 2011, 286, 19880–19891. [Google Scholar]
- Walters, R.W.; Shukla, A.K.; Kovacs, J.J.; Violin, J.D.; DeWire, S.M.; Lam, C.M.; Chen, J.R.; Muehlbauer, M.J.; Whalen, E.J.; Lefkowitz, R.J. b-arrestin 1 mediates nicotinic acid-induced flushing, but not its antilipolytic effects, in mice. J. Clin. Invest. 2009, 119, 1312–1321. [Google Scholar]
- Rajagopal, S.; Ahn, S.; Gowen-MacDonald, W.; Lam, C.M.; DeWire, S.M.; Violin, J.D.; Lefkowitz, R.J. Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 2011, 80, 367–377. [Google Scholar] [CrossRef]
- Nguyen, L.P.; Lin, R.; Parra, S.; Omoluabi, O.; Hanania, N.A.; Tuvim, M.J.; Knoll, B.J.; Dickey, B.F.; Bond, R.A. Beta2-adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc. Natl. Acad. Sci. USA 2009, 106, 2435–2440. [Google Scholar]
- Billington, C.K.; Hall, I.P. Novel cyclic AMP Signalling Paradigms: Therapeutic Implications for Airway Disease. Br. J. Pharmacol. 2011, 166, 401–410. [Google Scholar] [CrossRef]
- Walker, J.K.; Penn, R.B.; Hanania, N.A.; Dickey, B.F.; Bond, R.A. New perspectives regarding beta(2) -adrenoceptor ligands in the treatment of asthma. Br. J. Pharmacol. 2011, 163, 18–28. [Google Scholar] [CrossRef]
- Lovgren, A.K.; Kovacs, J.J.; Xie, T.; Potts, E.N.; Li, Y.; Foster, W.M.; Liang, J.; Meltzer, E.B.; Jiang, D.; Lefkowitz, R.J.; Noble, P.W. beta-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci. Transl. Med. 2011, 3, 74–ra23. [Google Scholar] [CrossRef]
- Manson, M.E.; Corey, D.A.; White, N.M.; Kelley, T.J. cAMP-mediated regulation of cholesterol accumulation in cystic fibrosis and Niemann-Pick type C cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L809–L819. [Google Scholar] [CrossRef]
- Li, J.; Ghio, A.J.; Cho, S.H.; Brinckerhoff, C.E.; Simon, S.A.; Liedtke, W. Diesel exhaust particles activate the matrix-metalloproteinase-1 gene in human bronchial epithelia in a beta-arrestin-dependent manner via activation of RAS. Environ. Health Perspect. 2009, 117, 400–409. [Google Scholar]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; Shenoy, S.K.; Gygi, S.P.; Lefkowitz, R.J. Distinct phosphorylation sites on the b(2)-adrenergic receptor establish a barcode that encodes differential functions of b-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef]
- Khasai, A.W.; Xiao, K.; Rajagopal, S.; Ahn, S.; Shukla, A.K.; Sun, J.; Oas, T.G.; Lefkowitz, R.J. Multiple ligand-specific conformations of the b2-adrenergic receptor. Nat. Chem. Biol. 2011, 7, 692–700. [Google Scholar] [CrossRef]
- Xiao, K.; Sun, J.; Kim, J.; Rajagopal, S.; Zhai, B.; Villen, J.; Haas, W.; Kovacs, J.J.; Shukla, A.K.; Hara, M.R.; Hernandez, M.; Lachmann, A.; Zhao, S.; Lin, Y.; Cheng, Y.; Mizuno, K.; Maáyan, A.; Gygi, S.P.; Lefkowitz, R.J. Global phosphorylation analysis of b-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc. Natl. Acad. Sci. US A 2010, 107, 15299–15304. [Google Scholar]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate b2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Modi, A.S.; Shukla, A.K.; Xiao, K.; Berthouze, M.; Ahn, S.; Wilkinson, K.D.; Miller, W.E.; Lefkowitz, R.J. b-Arrestin-dependent signaling and trafficking of 7-transmembrane receptor is reciprocally regulated by the deubititinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. USA 2009, 106, 6650–6655. [Google Scholar]
- Billington, C.K.; Hall, I.P. Real time analysis of beta(2)-adrenoceptor-mediated signaling kinetics in human primary airway smooth muscle cells reveals both ligand and dose dependent differences. Respir. Res. 2011, 12, 89. [Google Scholar] [CrossRef]
- Lamyel, F.; Warnken-Uhlich, M.; Seemann, W.K.; Mohr, K.; Kostenis, E.; Ahmedat, A.S.; Smit, M.; Gosens, R.; Meurs, H.; Miller-Larsson, A.; Racke, K. The beta2-subtype of adrenoceptors mediates inhibition of pro-fibrotic events in human lung fibroblasts. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 384, 133–145. [Google Scholar] [CrossRef]
- Giembycz, M.A.; Newton, R. Beyond the dogma: novel beta2-adrenoceptor signalling in the airways. Eur. Respir. J. 2006, 27, 1286–1306. [Google Scholar] [CrossRef]
- Lynch, M.J.; Baillie, G.S.; Mohamed, A.; Li, X.; Maisonneuve, C.; Klussmann, E.; Van, H.G.; Houslay, M.D. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J. Biol. Chem. 2005, 280, 33178–33189. [Google Scholar]
- Lin, F.; Wang, H.; Malbon, C.C. Gravin-mediated formation of signaling complexes in beta2-adrenergic receptor desensitization and resensitization. J. Biol. Chem. 2000, 275, 19025–19034. [Google Scholar]
- Anderson, G.P. Current issues with beta2-adrenoceptor agonists: pharmacology and molecular and cellular mechanisms. Clin. Rev. Allergy Immunol. 2006, 31, 119–130. [Google Scholar] [CrossRef]
- Cooper, D.M. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem. Soc Trans. 2005, 33, 1319–1322. [Google Scholar] [CrossRef]
- Delint-Ramirez, I.; Willoughby, D.; Hammond, G.V.R.; Ayling, L.J.; Cooper, D.M.F. Palmitylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J. Biol. Chem. 2011, 286, 32962–32975. [Google Scholar]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef]
- Nijholt, I.M.; Dolga, A.M.; Ostroveanu, A.; Luiten, P.G.; Schmidt, M.; Eisel, U.L. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell Signal. 2008, 20, 1715–1724. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases--the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Tasken, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef]
- Zambon, A.C.; Zhang, L.; Minovitsky, S.; Kanter, J.R.; Prabhakar, S.; Salomonis, N.; Vranizan, K.; Dubchak, I.; Conklin, B.R.; Insel, P.A. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc. Natl. Acad. Sci. USA 2005, 102, 8561–8566. [Google Scholar]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef]
- Krugmann, S.; Williams, R.; Stephens, L.; Hawkins, P.T. ARAP3 is a PI3K- and RAP-regulated GAP for RhoA. Curr. Biol. 2004, 14, 1380–1384. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Maarsingh, H.; Elzinga, C.R.; Schuur, J.; Menzen, M.; Halayko, A.J.; Meurs, H.; Schmidt, M. Epac as a novel effector of airway smooth muscle relaxation. J. Cell Mol. Med. 2011, 15, 1551–1562. [Google Scholar] [CrossRef]
- Zieba, B.J.; Artamonov, M.V.; Jin, L.; Momotani, K.; Ho, R.; Franke, A.S.; Neppl, R.L.; Stevenson, A.S.; Khromov, A.S.; Chrzanowska-Wodnicka, M.; Somlyo, A.V. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J. Biol. Chem. 2011, 286, 16681–16692. [Google Scholar]
- Li, Y.; Asuri, S.; Rebhun, J.F.; Castro, A.F.; Paranavitana, N.C.; Quilliam, L.A. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J. Biol. Chem. 2006, 281, 2506–2514. [Google Scholar]
- Lopez De Jesus, M.; Stope, M.B.; Oude Weernink, P.A.; Mahlke, Y.; Borgermann, C.; Ananaba, V.N.; Rimmbach, C.; Rosskopf, D.; Michel, M.C.; Jakobs, K.H.; Schmidt, M. Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J. Biol. Chem. 2006, 281, 21837–21847. [Google Scholar]
- Oestreich, E.A.; Wang, H.; Malik, S.; Kaproth-Joslin, K.A.; Blaxall, B.C.; Kelley, G.G.; Dirksen, R.T.; Smrcka, A.V. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 2007, 282, 5488–5495. [Google Scholar]
- Schmidt, M.; Evellin, S.; Weernink, P.A.; von, D.F.; Rehmann, H.; Lomasney, J.W.; Jakobs, K.H. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat. Cell Biol. 2001, 3, 1020–1024. [Google Scholar] [CrossRef]
- Han, L.; Stope, M.B.; de Jesus, M.L.; Oude Weernink, P.A.; Urban, M.; Wieland, T.; Rosskopf, D.; Mizuno, K.; Jakobs, K.H.; Schmidt, M. Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J. 2007, 26, 4189–4202. [Google Scholar] [CrossRef]
- Wang, C.; Gu, Y.; Li, G.W.; Huang, L.Y. A critical role of the cAMP sensor Epac in switching protein kinase signalling in prostaglandin E2-induced potentiation of P2X3 receptor currents in inflamed rats. J. Physiol. 2007, 584, 191–203. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Kistemaker, L.E.; Menzen, M.H.; Elzinga, C.R.; Gosens, R.; Halayko, A.J.; Meurs, H.; Schmidt, M. PKA and Epac cooperate to augment bradykinin-induced interleukin-8 release from human airway smooth muscle cells. Respir. Res. 2009, 10, 88. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Dekkers, B.G.; Prins, A.G.; Menzen, M.H.; Meurs, H.; Schmidt, M.; Maarsingh, H. cAMP inhibits modulation of airway smooth muscle phenotype via the exchange protein activated by cAMP (Epac) and protein kinase A. Br. J. Pharmacol. 2011, 162, 193–209. [Google Scholar] [CrossRef]
- Keiper, M.; Stope, M.B.; Szatkowski, D.; Bohm, A.; Tysack, K.; Vom, D.F.; Saur, O.; Oude Weernink, P.A.; Evellin, S.; Jakobs, K.H.; Schmidt, M. Epac- and Ca2+ -controlled activation of Ras and extracellular signal-regulated kinases by Gs-coupled receptors. J. Biol. Chem. 2004, 279, 46497–46508. [Google Scholar]
- Kiermayer, S.; Biondi, R.M.; Imig, J.; Plotz, G.; Haupenthal, J.; Zeuzem, S.; Piiper, A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol. Biol. Cell 2005, 16, 5639–5648. [Google Scholar] [CrossRef]
- Grandoch, M.; López de Jesús, M.; Oude Weernink, P.A.; Weber, A.-A.; Jakobs, K.H.; Schmidt, M. B cell receptor-induced growth arrest and apoptosis in WEHI-231 immature B lymphoma cells involve cyclic AMP and Epac proteins. Cell Signal. 2009, 4, 609–621. [Google Scholar]
- Kwak, H.J.; Park, K.M.; Choi, H.E.; Chung, K.S.; Lim, H.J.; Park, H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal. 2008, 20, 803–814. [Google Scholar] [CrossRef]
- Mei, F.C.; Qiao, J.; Tsygankova, O.M.; Meinkoth, J.L.; Quilliam, L.A.; Cheng, X. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J. Biol. Chem. 2002, 277, 11497–11504. [Google Scholar]
- Misra, U.K.; Pizzo, S.V. Coordinate regulation of forskolin-induced cellular proliferation in macrophages by protein kinase A/cAMP-response element-binding protein (CREB) and Epac1-Rap1 signaling: effects of silencing CREB gene expression on Akt activation. J. Biol. Chem. 2005, 280, 38276–38289. [Google Scholar] [CrossRef]
- Misra, U.K.; Kaczowka, S.; Pizzo, S.V. The cAMP-activated GTP exchange factor, Epac1 upregulates plasma membrane and nuclear Akt kinase activities in 8-CPT-2-O-Me-cAMP-stimulated macrophages: Gene silencing of the cAMP-activated GTP exchange Epac1 prevents 8-CPT-2-O-Me-cAMP activation of Akt activity in macrophages. Cell Signal. 2008, 20, 1459–1470. [Google Scholar] [CrossRef]
- Hewer, R.C.; Sala-Newby, G.B.; Wu, Y.-J.; Newby, A.C.; Bond, M. PKA and Epac synergistically inhibit smooth muscle cell proliferation. J. Mol. Cell. Cardiol. 2011, 50, 87–98. [Google Scholar] [CrossRef]
- Fuld, S.; Borland, G.; Yarwood, S.J. Elevation of cyclic AMP in Jurkat T-cells provokes distinct transcriptional responses through the protein kinase A (PKA) and exchange protein activated by cyclic AMP (EPAC) pathways. Exp. Cell Res. 2005, 309, 161–173. [Google Scholar] [CrossRef]
- Scheibner, K.A.; Boodoo, S.; Collins, S.; Black, K.E.; Chan-Li, Y.; Zarek, P.; Powell, J.D.; Horton, M.R. The adenosine a2a receptor inhibits matrix-induced inflammation in a novel fashion. Am. J. Respir. Cell Mol. Biol. 2009, 40, 251–259. [Google Scholar]
- Oldenburger, A.; Roscioni, S.S.; Jansen, E.; Menzen, M.H.; Halayko, A.J.; Timens, W.; Meurs, H.; Maarsingh, H.; Schmidt, M. Anti-inflammatory role of the cAMP effectors Epac and PKA: implications in chronic obstructive pulmonary disease. PLoS One 2012, e31574. [Google Scholar]
- Borland, G.; Smith, B.O.; Yarwood, S.J. EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharmacol. 2009, 158, 70–86. [Google Scholar] [CrossRef]
- Grandoch, M.; Roscioni, S.S.; Schmidt, M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal functions. Br. J. Pharmacol. 2010, 159, 265–284. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Elzinga, C.R.; Schmidt, M. Epac: effectors and biological functions. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 377, 345–357. [Google Scholar] [CrossRef]
- Schmidt, M.; Sand, C.; Jakobs, K.H.; Michel, M.C.; Oude Weernink, P.A. Epac and the cardiovascular system. Curr. Opin. Pharmacol. 2007, 7, 193–200. [Google Scholar] [CrossRef]
- Lorenowicz, M.J.; Fernandez-Borja, M.; Hordijk, P.L. cAMP signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1014–1022. [Google Scholar] [CrossRef]
- Billington, C.K.; Hall, I.P.; Mundell, S.J.; Parent, J.L.; Panettieri, R.A., Jr.; Benovic, J.L.; Penn, R.B. Inflammatory and contractile agents sensitize specific adenylyl cyclase isoforms in human airway smooth muscle. Am. J. Respir. Cell Mol. Biol. 1999, 21, 597–606. [Google Scholar]
- Bogard, A.S.; Xu, C.; Ostrom, R.S. Human bronchial smooth muscle cells express adenylyl cyclase isoforms 2, 4, and 6 in distinct membrane microdomains. J. Pharmacol Exp. Ther. 2011, 337, 209–217. [Google Scholar] [CrossRef]
- Xu, D.; Isaacs, C.; Hall, I.P.; Emala, C.W. Human airway smooth muscle expresses 7 isoforms of adenylyl cyclase: a dominant role for isoform V. Am. J. Physiol Lung Cell Mol. Physiol 2001, 281, L832–L843. [Google Scholar]
- Le Jeune, I.; Shepherd, M.; Van, H.G.; Houslay, M.D.; Hall, I.P. Cyclic AMP-dependent transcriptional up-regulation of phosphodiesterase 4D5 in human airway smooth muscle cells. Identification and characterization of a novel PDE4D5 promoter. J. Biol. Chem. 2002, 277, 35980–35989. [Google Scholar]
- Horvat, S.J.; Deshpande, D.A.; Yan, H.; Panettieri, R.A.; Codina, J.; DuBose, T.D.; Xin, W.; Rich, T.C.; Penn, R.B. A-kinase anchoring proteins regulate compartmenalized cAMP signaling in airway smooth muscle. FASEB J. 2012, 9, 3670–3679. [Google Scholar]
- Liu, X.; Ostrom, R.S.; Insel, P.A. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am. J. Physiol Cell Physiol. 2004, 286, C1089–C1099. [Google Scholar]
- Selige, J.; Hatzelmann, A.; Dunkern, T. The differential impact of PDE4 subtypes in human lung fibroblasts on cytokine-induced proliferation and myofibroblast conversion. J. Cell Physiol. 2011, 226, 1970–1980. [Google Scholar] [CrossRef]
- Selige, J.; Tenor, H.; Hatzelmann, A.; Dunkern, T. Cytokine-dependent balance of mitogenic effects in primary human lung fibroblasts related to cyclic AMP signaling and phosphodiesterase 4 inhibition. J. Cell Physiol. 2010, 223, 317–326. [Google Scholar]
- Togo, S.; Liu, X.; Wang, X.; Sugiura, H.; Kamio, K.; Kawasaki, S.; Kobayashi, T.; Ertl, R.F.; Ahn, Y.; Holz, O.; Magnussen, H.; Fredriksson, K.; Skold, C.M.; Rennard, S.I. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L959–L969. [Google Scholar] [CrossRef]
- Okunishi, K.; Sisson, T.H.; Huang, S.K.; Hogaboam, C.M.; Simon, R.H.; Peters-Golden, M. Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase A signaling. J. Biol. Chem. 2011, 286, 32231–32243. [Google Scholar]
- Tasken, K.A.; Collas, P.; Kemmner, W.A.; Witczak, O.; Conti, M.; Tasken, K. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J. Biol. Chem. 2001, 276, 21999–22002. [Google Scholar]
- Calverley, P.M.; Rabe, K.F.; Goehring, U.M.; Kristiansen, S.; Fabbri, L.M.; Martinez, F.J. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009, 374, 685–694. [Google Scholar] [CrossRef]
- Spina, D. PDE4 inhibitors: current status. Br. J. Pharmacol. 2008, 155, 308–315. [Google Scholar] [CrossRef]
- Michalski, J.M.; Golden, G.; Ikari, J.; Rennard, S.I. PDE4: A novel target in the treatment of chronic obstructive pulmonary disease. Clin. Pharmacol. Ther. 2011. [Google Scholar] [CrossRef]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef]
- Qin, Y.; Stokman, G.; Yan, K.; Ramaiahgari, S.; Verbeek, F.; de Graauw, M.; van de Water, B.; Price, L.S. Cyclic AMP signalling protects proximal tubular epithelial cells from cisplatin-induced apoptosis via activation of Epac. Br. J. Pharmacol. 2011. [Google Scholar] [CrossRef]
- Roscioni, S.S. Epac as a novel regulator of airway smooth muscle phenotype and function. Potential implications in asthma and COPD. Ph.D. Degree, University of Groningen, The Netherland, 2010. [Google Scholar]
- Oldenburger, A.; Rijks, W.; Poppinga, W.; Roscioni, S.S.; Heijink, I.H.; Maarsingh, H.; Schmidt, M. Interaction between cigarette smoke and cyclic AMP signaling in human bronchial epithelial function. FASEB J. 2011, 25, 659.13. [Google Scholar]
- Dent, G.; White, S.R.; Tenor, H.; Bodtke, K.; Schudt, C.; Leff, A.R.; Magnussen, H.; Rabe, K.F. Cyclic nucleotide phosphodiesterase in human bronchial epithelial cells: characterization of isoenyzmes and functional effects of PDE inhibitors. Pulm. Pharmacol. Ther. 1998, 11, 47–56. [Google Scholar] [CrossRef]
- Mata, M.; Sarria, B.; Buenestado, A.; Cortijo, J.; Cerda, M.; Morcillo, E.J. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax 2005, 60, 144–152. [Google Scholar] [CrossRef]
- Barnes, A.P.; Livera, G.; Huang, P.; Sun, C.; O'Neal, W.K.; Conti, M.; Stutts, M.J.; Milgram, S.L. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J. Biol. Chem. 2005, 280, 7997–8003. [Google Scholar]
- Penmatsa, H.; Zhang, W.; Yarlagadda, S.; Li, C.; Conoley, V.G.; Yue, J.; Bahouth, S.W.; Buddington, R.K.; Zhang, G.; Nelson, D.J.; Sonecha, M.D.; Manganiello, V.; Wine, J.J.; Naren, A.P. Compartmentalized cyclic adenosine 3', 5'-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol. Biol Cell 2010, 21, 1097–1110. [Google Scholar]
- Halpin, D.M. ABCD of the phosphodiesterase family: interaction and differential activity in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2008, 3, 543–561. [Google Scholar]
- Small, K.M.; Brown, K.M.; Theiss, C.T.; Seman, C.A.; Weiss, S.T.; Liggett, S.B. An Ile to Met polmorphism in the catalytic domain of adenylyl cycalse type 9 confers reduced beta2-adrenergic receptor stimulation. Pharmacogenetics 2003, 13, 535–541. [Google Scholar] [CrossRef]
- Tantisira, K.G.; Small, K.M.; Litonjua, A.A.; Weiss, S.T.; Liggett, S.B. Molecular properties and pharmacogenetics of a polymorphism of adenylyl cyclase type 9 in asthma: interaction between b-agonist and corticosteroid pathways. Hum. Mol. Genet. 2005, 14, 1671–1677. [Google Scholar] [CrossRef]
- Jourdan, K.B.; Mason, N.A.; Long, L.; Philips, P.G.; Wilkins, M.R.; Morrell, N.W. Characterization of adenylyl cyclase isoforms in rat peripheral pulmonary arteries. Am. J. Physiol Lung Cell Mol. Physiol. 2001, 280, L1359–L1369. [Google Scholar]
- Wang, Y.; Lam, C.S.; Wu, F.; Wang, W.; Duan, Y.; Huang, P. Regulation of CFTR channels by HCO(3)--sensitive soluble adenylyl cyclase in human airway epithelial cells. Am. J. Physiol. Cell. Physiol. 2005, 289, C1145–C1151. [Google Scholar] [CrossRef]
- Blazac, F.; Avolio, M.; Degani, S.; Kaverina, I.; Torti, M.; Silengo, L.; Small, J.V.; Retta, S.F. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the croosraods between cadherin and integrin function. J. Cell Sci. 2005, 118, 4765–4783. [Google Scholar] [CrossRef]
- Hage, B.; Meinel, K.; Baum, I.; Giehl, K.; Menke, A. Rac1 activation inhibits E-cadherin-mediated adherens junctions via binding to IQGAP1 in pancreatic carcinoma cells. Cell Commun. Signal. 2009, 7, 23. [Google Scholar] [CrossRef]
- Smutny, M.; Cox, L.H.; Leerberg, J.M.; Kovacs, E.M.; Conti, M.A.; Ferguson, C.; Hamilton, N.A.; Parton, R.G.; Adelstein, R.S.; Yap, A.S. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat. Cell Biol. 2010, 12, 696–702. [Google Scholar] [CrossRef]
- Jeong, H.-W.; Li, Z.; Brown, M.D.; Sacks, D.B. IQGAP1 binds Rap1 and modulates its activity. J. Biol. Chem. 2007, 282, 20752–20762. [Google Scholar] [CrossRef]
- Kuroda, S.; Fukata, M.; Nakagawa, M.; Fuji, K.; Nakmura, T.; Ookubo, T.; Izawa, I.; Nagase, T.; Nomura, N.; Tani, H.; Shoji, I.; Matsuura, Y.; Ynoehara, S.; Kaibuchi, K. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin mediated cell-cell adhesion. Science 1998, 281, 832–835. [Google Scholar] [CrossRef]
- Noritake, J.; Watanbe, T.; Sato, K.; Wang, S.; Kaibuchi, K. IQGAP1: a key regulator of adhesion and migration. J. Cell Sci. 2005, 118, 2085–2092. [Google Scholar] [CrossRef]
- Birukova, A.A.; Burdette, D.; Moldobaeva, N.; Xing, J.; Fu, P.; Birukov, K.G. Rac GTPase is a hub for protein kinase A and epac signaling in endothelial barrier protection by cAMP. Microvascul. Res. 2010, 79, 128–138. [Google Scholar] [CrossRef]
- Rampersad, S.N.; Ovens, J.D.; Huston, E.; Umana, M.B.; Wilson, L.S.; Netherton, S.J.; Lynch, M.J.; Baillie, G.S.; Houslay, M.D.; Maurice, D.H. Cyclic AMP phosphodieseterase 4D (PDE4D) tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeabilbity. J. Biol. Chem. 2010, 285, 33614–33622. [Google Scholar]
- Sehrawat, S.; Cullere, X.; Patel, S.; Italiano, J.; Mayadas, T.N. Role of Epac1, an exchange factor for rap GTPases, in endothelial microtubule dynamics and barrier function. Mol. Biol. Cell 2008, 19, 1261–1270. [Google Scholar]
- Sehrawat, S.; Ernandez, T.; Cullere, X.; Takahashi, M.; Ono, Y.; Komarova, Y.; Mayadas, T.N. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties. Blood 2011, 117, 708–718. [Google Scholar] [CrossRef]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef]
- Suh, H.N.; Han, H.J. Laminin regulates mouse embryonic stem cell migration: involvement of Epac1/Rap1 and Ra1/cdc42. Am. J. Physiol. Cell Physiol. 2010, 298, C1159–C1169. [Google Scholar] [CrossRef]
- Netherton, S.J.; Sutton, J.A.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Both protein kinase A and exchange protein directly activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ. Res. 2007, 101, 768–776. [Google Scholar] [CrossRef]
- Baumer, Y.; Spindler, V.; Werthmann, R.C.; Bünnemann, M.; Waschke, J. Role of Rac1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J. Cell. Physiol. 2008, 220, 716–726. [Google Scholar]
- Pullar, C.E.; Grahn, J.C.; Liu, W.; Isseroff, R.R. b2-Adrenergic receptor activation delays wound healing. FASEB J. 2004, 20, 76–86. [Google Scholar]
- Raymond, D.R.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Numerous distinct PKA-, or Epac-based, signalling complexes allow selective phosphodiesterase 3 and phosphodiesterase 4 coordination of cell adhesion. Cell Signal. 2007, 19, 2507–2512. [Google Scholar] [CrossRef]
- Spindler, V.; Waschke, J. Beta-adrenergic stimulation contributes to maintenance of endothelial barrier functions under baseline conditions. Microcirculation 2011, 18, 118–127. [Google Scholar] [CrossRef]
- Spindler, V.; Peter, D.; Harms, G.S.; Asan, E.; Waschke, J. Ultrastructural analysis reveals cAMP-dependent enhancement of microvascular endothelial barrier functions via Rac1-mediated reorganization of intercellular junctions. Am. J. Pathol. 2011, 178, 2424–2436. [Google Scholar] [CrossRef]
- Schlegel, N.; Waschke, J. VASP is involved in cAMP-mediated rac 1 activation in microvascular endothelial cells. Am. J. Cell Physiol. 2009, 296, C453–C462. [Google Scholar] [CrossRef]
- Cole, A.L.; Subbanagounder, G.; Mukhopadhyay, S.; Berliner, J.A.; Vora, D.K. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1384–1390. [Google Scholar] [CrossRef]
- Poppinga, W.J.; Holtzer, L.J.; Skroblin, P.; Klussmann, E.; Maarsingh, H.; Schmidt, M. A-kinase anchoring proteins (AKAPs) regulate airway smooth muscle sceretory and proliferative functions. Available online: http://www.pA2online.org/abstracts/Vol10Issue1abst004P.pdf accessed on 28 November 2012.
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Purves, G.I.; Kamishima, T.; Davies, L.M.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) mediates cAMP-dependent but protein kinase A-insensitive modulation of vascular ATP-sensitive potassium channels. J. Physiol. 2009, 587, 3639–3650. [Google Scholar] [CrossRef]
- Streb, J.W.; Long, X.; Lee, T.H.; Sun, Q.; Kitchen, C.M.; Georger, M.A.; Slivano, O.J.; Blaner, W.S.; Carr, D.W.; Gelman, I.H.; Miano, J.M. Retinoid-induced expression and activity of an immediate early tumor suppressor gene in vascular smooth muscle cells. PLos One 2011, 6, e18538. [Google Scholar]
- Murray, F.; Patel, H.H.; Suda, R.Y.; Zhang, S.; Thistlethwaite, P.A.; Yuan, J.X.; Insel, P.A. Expression and activity of cAMP phosphodiesterase isoforms in pulmonary artery smooth muscle cells from patients with pulmonary hypertension: role for PDE1. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L294–L303. [Google Scholar]
- El-Haroun, H.; Bradbury, D.; Clayton, A.; Knox, A.J. Interleukin-1beta, transforming growth factor-beta1, and bradykinin attenuate cyclic AMP production by human pulmonary artery smooth muscle cells in response to prostacyclin analogues and prostaglandin E2 by cyclooxygenase-2 induction and downregulation of adenylyl cyclase isoforms 1, 2, and 4. Circ. Res. 2004, 94, 353–361. [Google Scholar] [CrossRef]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Akaike, T.; Jin, M.; Otsu, K.; Ulucan, C.; Wang, X.; Baljinnyam, E.; Takaoka, M.; Sata, M.; Ishikawa, Y. Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration. Am. J. Physiol. Heart. Circ. Physiol. 2008, 295, H1547–H1555. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Djordjevic, T.; Belaiba, R.S.; Fineman, J.; Black, S.; Schreiber, C.; Fratz, S.; Hess, J.; Kietzmann, T.; Gorlach, A. Reciprocal regulation of Rac1 and PAK-1 by HIF-1alpha: a positive-feedback loop promoting pulmonary vascular remodeling. Antioxid. Redox. Signal. 2010, 13, 399–412. [Google Scholar] [CrossRef]
- Bailly, K.; Ridley, A.J.; Hall, S.M.; Haworth, S.G. RhoA activation by hypoxia in pulmonary arterial smooth muscle cells is age and site specific. Circ. Res. 2004, 94, 1383–1391. [Google Scholar] [CrossRef]
- Guilluy, C.; Eddahibi, S.; Agard, C.; Guignabert, C.; Izikki, M.; Tu, L.; Savale, L.; Humbert, M.; Fadel, E.; Adnot, S.; Loirand, G.; Pacaud, P. RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am. J. Respir. Crit. Care. Med. 2009, 179, 1151–1158. [Google Scholar] [CrossRef]
- Haag, S.; Warnken, M.; Juergens, U.R.; Racke, K. Role of Epac1 in mediating anti-proliferative effects of prostanoid EP(2) receptors and cAMP in human lung fibroblasts. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 617–630. [Google Scholar] [CrossRef]
- Kohyama, T.; Liu, X.; Wen, F.Q.; Zhu, Y.K.; Wang, H.; Kim, H.J.; Takizawa, H.; Cieslinski, L.B.; Barnette, M.S.; Rennard, S.I. PDE4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am. J. Respir. Cell Mol. Biol. 2002, 26, 694–701. [Google Scholar]
- Tufvesson, E.; Westergren-Thorsson, G. Biglycan and decorin induce morphological and cytoskeletal changes involving signalling by the small GTPases RhoA and Rac1 resulting in lung fibroblast migration. J. Cell Sci. 2003, 116, 4857–4864. [Google Scholar] [CrossRef]
- Huang, S.K.; Wettlaufer, S.H.; Chung, J.; Peters-Golden, M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am. J. Respir. Cell Mol. Biol. 2008, 39, 482–489. [Google Scholar] [CrossRef]
- Brudvik, K.W.; Tasken, K. Modulation of T cell immune functions by the prostaglandin E(2) - cAMP pathway in chronic inflammatory states. Br. J. Pharmacol. 2012, 166, 411–419. [Google Scholar] [CrossRef]
- El Din El Homasany BS; Volkov, Y.; Takahashi, M.; Ono, Y.; Keryer, G.; Delouvee, A.; Looby, E.; Long, A.; Kelleher, D. The scaffolding protein CG-NAP/AKAP450 is a critical integrating component of the LFA-1-induced signaling complex in migratory T cells. J. Immunol. 2005, 175, 7811–7818. [Google Scholar]
- Wang, P.; Wu, P.; Ohleth, K.M.; Egan, R.W.; Billah, M.M. Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophils. Mol. Pharmacol. 1999, 56, 170–174. [Google Scholar]
- Smith, S.J.; Brookes-Fazakerley, S.; Donnelly, L.E.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am J Physiol Lung Cell Mol. Physiol. 2003, 284, L279–L289. [Google Scholar]
- Mosenden, R.; Tasken, K. Cyclic AMP-mediated immune regulation--overview of mechanisms of action in T cells. Cell Signal. 2011, 23, 1009–1016. [Google Scholar] [CrossRef]
- Chang, L.C.; Wang, C.J.; Lin, Y.L.; Wang, J.P. Expression of adenylyl cyclase isoforms in neutrophils. Biochim. Biophys. Acta 2003, 1640, 53–60. [Google Scholar] [CrossRef]
- Risoe, P.K.; Wang, Y.; Stuestol, J.F.; Aasen, A.O.; Wang, J.E.; Dahle, M.K. Lipopolysaccharide attenuates mRNA levels of several adenylyl cyclase isoforms in vivo. Biochim. Biophys. Acta 2007, 1772, 32–39. [Google Scholar]
- Han, H.; Stessin, A.; Roberts, J.; Hess, K.; Gautam, N.; Kamenetsky, M.; Lou, O.; Hyde, E.; Nathan, N.; Muller, W.A.; Buck, J.; Levin, L.R.; Nathan, C. Calcium-sensing soluble adenylyl cyclase mediates TNF signal transduction in human neutrophils. J. Exp. Med. 2005, 202, 353–361. [Google Scholar]
- Reedquist, K.A.; Tak, P.P. Signal transduction pathways in chronic inflammatory autoimmune disease: small GTPases. Open. Rheumatol. J. 2012, 6, 259–272. [Google Scholar]
- Zhang, J.; Zhu, J.; Bu, X.; Cushion, M.; Kinane, T.B.; Avraham, H.; Koziel, H. Cdc42 and RhoB activation are required for mannose receptor-mediated phagocytosis by human alveolar macrophages. Mol. Biol. Cell 2005, 16, 824–834. [Google Scholar]
- Pierre, S.; Eschenhagen, T.; Geisslinger, G.; Scholich, K. Capturing adenylyl cyclases as potential drug targets. Nat Rev. Drug Discov. 2009, 8, 321–335. [Google Scholar]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Renal Physiol. 2000, 279, F400–F416. [Google Scholar]
- Schmid, A.; Sutto, Z.; Nlend, M.-C.; Horvath, G.; Schmid, N.; Buck, J.; Levin, L.R.; Conner, G.E.; Fregien, N.; Salathe, M. Soluble adenylyl cyclase is localized to cilia and contributes to ciliary beat frequency regulation via production of cAMP. J. Gen. Physiol. 2007, 130, 99–109. [Google Scholar]
- Lopez, E.; Jarreau, P.-H.; Zana, E.; Franco-Montoya, M.-L.; Schmitz, T.; Evain-Brion, D.; Bourbon, J.; Delacourt, C.; Mehats, C. Differential expression of cyclic nucleotide phosphodiesterases 4 in developing rat lung. Develop. Dynam. 2010, 239, 2470–2478. [Google Scholar] [Green Version]
- Hoque, K.M.; Woodward, O.M.; van Rossum, D.B.; Zachos, N.C.; Chen, L.; Leung, G.P.; Guggino, W.B.; Guggino, S.E.; Tse, C.M. Epac1 mediates protein kinase A-independent mechanism of forskolin-activated intestinal chloride secretion. J. Gen. Physiol. 2010, 135, 43–58. [Google Scholar]
- Monterisi, S.; Favia, M.G.L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012, 125, 1106–1117. [Google Scholar]
- Li, C.; Krishnamurthy, P.C.; Penmatsa, H.; Marrs, K.L.; Wang, X.Q.; Zaccolo, M.; Jalink, K.; Li, M.; Nelson, D.J.; Schuetz, J.D.; Naren, A.P. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 2007, 131, 940–951. [Google Scholar]
- Schmidt, M.; Oldenburger, A.; Poppinga, W.; Roscioni, S.S.; Heijink, I.H.; Timens, W.; Skroblin, P.; Klussmann, E.; Maarsingh, H. Cigarette smoke and A-kinase anchoring proteins (AKAPs) in human airway smooth muscle function. FASEB J. 2011, 25, 864.6. [Google Scholar]
- Patel, H.H.; Hamuro, L.L.; Chun, B.J.; Kawaraguchi, Y.; Quick, A.; Olson, G.; Insel, P.A.; Giles, W.R.; Taylor, S.S.; Roth, D.M. Disruption of protein kinase A localization using a trans-activator of transcription (TAT)-conjugated A-kinase-anchoring peptide reduces cardiac function. J. Biol. Chem. 2010, 285, 27632–27640. [Google Scholar]
- Christian, F.; Sazaszák, M.; Friedl, S.; Drewianka, S.; Lorenz, D.; Goncalves, A.; Furkert, J.; Vargas, C.; Schmieder, P.; Götz, F.; Zühlke, K.; Moutty, M.; Göttert, H.; Gáspàr, R.; Noack, C.; Bergmann, M.; Kass, R.; Hampel, K.; Kashin, D.; Genieser, H.-G.; Herberg, F.W.; Willoughby, D.; Cooper, D.M.F.; Baillie, G.S.; Houslay, M.D.; von Kries, J.P.; Zimmermann, B.; Rosenthal, W.; Klussmann, E. Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J. Biol. Chem. 2011, 286, 9079–9096. [Google Scholar]
- Christensen, A.E.; Selheim, F.; de, R.J.; Dremier, S.; Schwede, F.; Dao, K.K.; Martinez, A.; Maenhaut, C.; Bos, J.L.; Genieser, H.G.; Doskeland, S.O. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 2003, 278, 35394–35402. [Google Scholar]
- Poppe, H.; Rybalkin, S.D.; Rehmann, H.; Hinds, T.R.; Tang, X.B.; Christensen, A.E.; Schwede, F.; Genieser, H.G.; Bos, J.L.; Doskeland, S.O.; Beavo, J.A.; Butt, E. Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 2008, 5, 277–278. [Google Scholar]
- Botelho, L.H.; Rothermel, J.D.; Coombs, R.V.; Jastorff, B. cAMP analog antagonists of cAMP action. Methods Enzymol. 1988, 159, 159–172. [Google Scholar]
- Gjertsen, B.T.; Mellgren, G.; Otten, A.; Maronde, E.; Genieser, H.G.; Jastorff, B.; Vintermyr, O.K.; McKnight, G.S.; Doskeland, S.O. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J. Biol. Chem. 1995, 270, 20599–20607. [Google Scholar]
- Holz, G.G.; Chepurny, O.G.; Schwede, F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008, 20, 10–20. [Google Scholar]
- Rehmann, H.; Schwede, F.; Doskeland, S.O.; Wittinghofer, A.; Bos, J.L. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J. Biol. Chem. 2003, 278, 38548–38556. [Google Scholar]
- Laxman, S.; Riechers, A.; Sadilek, M.; Schwede, F.; Beavo, J.A. Hydrolysis products of cAMP analogs cause transformation of Trypanosoma brucei from slender to stumpy-like forms. Proc. Natl. Acad. Sci. USA 2006, 103, 19194–19199. [Google Scholar]
- Shibasaki, T.; Takahashi, H.; Miki, T.; Sunaga, Y.; Matsumura, K.; Yamanaka, M.; Zhang, C.; Tamamoto, A.; Satoh, T.; Miyazaki, J.; Seino, S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Natl. Acad. Sci. USA 2007, 104, 19333–19338. [Google Scholar]
- Suzuki, S.; Yokoyama, U.; Abe, T.; Kiyonari, H.; Yamashita, N.; Kato, Y.; Kurotani, R.; Sato, M.; Okumura, S.; Ishikawa, Y. Differential roles of Epac in regulating cell death in neuronal and myocardial cells. J. Biol. Chem. 2010, 285, 24248–24259. [Google Scholar]
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Diagnosis, Management, and Prevention of COPD. 2010. Available online: http://www.goldcopd.org/ accessed on 28 November 2013.
- Postma, D.S.; Timens, W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 434–439. [Google Scholar] [CrossRef]
- Briggs, M.W.; Sacks, D.B. IQGAPs proteins are integral components of cytoskeletal regulation. EMBO Reports 2003, 4, 571–574. [Google Scholar] [CrossRef]
- Doherty, D.E. The pathophysiology of airway dysfunction. Am. J. Med. 2004, 117 Suppl 12A, 11S–23S. [Google Scholar]
- Rabe, K.F.; Hurd, S.S.; Anzueto, A.; Barnes, P.J.; Buist, S.A.; Calverley, P.; Fukuchi, Y.; Jenkins, C.; Rodriguez-Roisin, R.; van Weel, C.; Zielinski, J. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit Care Med. 2007, 176, 532–555. [Google Scholar] [CrossRef]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur. Respir J. 2003, 22, 672–688. [Google Scholar] [CrossRef]
- Mannino, D.M. COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneity. Chest 2002, 121, 121S–126S. [Google Scholar] [CrossRef]
- Shin, H.J.; Sohn, H.O.; Han, J.H.; Park, C.H.; Lee, H.S.; Hwang, K.J.; Hyun, H.C. Effect of cigarette filters on the chemical composition and in vitro biological activity of cigarette mainstream smoke. Food Chem. Toxicol. 2009, 47, 192–197. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J. Effects of cigarette smoke on the lung and systemic immunity. J. Physiol. Pharmacol. 2008, 59 Suppl 6, 19–34. [Google Scholar]
- Thorley, A.J.; Tetley, T.D. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2007, 2, 409–428. [Google Scholar]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol Lung Cell Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef]
- Kode, A.; Yang, S.R.; Rahman, I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir. Res. 2006, 7, 132. [Google Scholar] [CrossRef]
- Oenema, T.A.; Kolahian, S.; Nanninga, J.E.; Rieks, D.; Hiemstra, P.S.; Zuyderduyn, S.; Halayko, A.J.; Meurs, H.; Gosens, R. Pro-inflammatory mechanisms of muscarinic receptor stimulation in airway smooth muscle. Respir. Res. 2010, 11, 130. [Google Scholar] [CrossRef]
- Pease, J.E.; Sabroe, I. The role of interleukin-8 and its receptors in inflammatory lung disease: Implications for therapy. Am. J. Respir. Med. 2002, 1, 19–25. [Google Scholar] [CrossRef]
- Taylor, J.D. COPD and the response of the lung to tobacco smoke exposure. Pulm. Pharmacol. Ther. 2010, 23, 376–383. [Google Scholar] [CrossRef]
- Cornwell, W.D.; Kim, V.; Song, C.; Rogers, T.J. Pathogenesis of inflammation and repair in advanced COPD. Semin. Respir. Crit. Care Med 2010, 31, 257–266. [Google Scholar] [CrossRef]
- Postma, D.S.; Kerstjens, H.A. Characteristics of airway hyperresponsiveness in asthma and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 158, S187–S192. [Google Scholar]
- Tashkin, D.P.; Altose, M.D.; Connett, J.E.; Kanner, R.E.; Lee, W.W.; Wise, R.A. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group. Am. J. Respir. Crit. Care Med. 1996, 153, 1802–1811. [Google Scholar]
- Tashkin, D.P.; Altose, M.D.; Bleecker, E.R.; Connett, J.E.; Kanner, R.E.; Lee, W.W.; Wise, R. The lung health study: airway responsiveness to inhaled methacholine in smokers with mild to moderate airflow limitation. The Lung Health Study Research Group. Am. Rev. Respir. Dis. 1992, 145, 301–310. [Google Scholar]
- Saetta, M.; Di, S.A.; Turato, G.; Facchini, F.M.; Corbino, L.; Mapp, C.E.; Maestrelli, P.; Ciaccia, A.; Fabbri, L.M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit Care Med. 1998, 157, 822–826. [Google Scholar]
- Bosken, C.H.; Wiggs, B.R.; Pare, P.D.; Hogg, J.C. Small airway dimensions in smokers with obstruction to airflow. Am. Rev. Respir. Dis. 1990, 142, 563–570. [Google Scholar] [CrossRef]
- Cosio, M.G.; Hale, K.A.; Niewoehner, D.E. Morphologic and morphometric effects of prolonged cigarette smoking on the small airways. Am. Rev. Respir. Dis. 1980, 122, 266–271. [Google Scholar]
- Saetta, M.; Turato, G.; Baraldo, S.; Zanin, A.; Braccioni, F.; Mapp, C.E.; Maestrelli, P.; Cavallesco, G.; Papi, A.; Fabbri, L.M. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and chronic airflow limitation. Am. J. Respir. Crit. Care Med. 2000, 161, 1016–1021. [Google Scholar]
- Lambert, R.K.; Wiggs, B.R.; Kuwano, K.; Hogg, J.C.; Pare, P.D. Functional significance of increased airway smooth muscle in asthma and COPD. J. Appl. Physiol. 1993, 74, 2771–2781. [Google Scholar] [CrossRef]
- Jeffery, P.K. Remodeling in asthma and chronic obstructive lung disease. Am. J. Respir. Crit Care Med. 2001, 164, S28–S38. [Google Scholar]
- Jeffery, P.K. Comparison of the structural and inflammatory features of COPD and asthma. Giles F. Filley Lecture. Chest 2000, 117, 251S–260S. [Google Scholar] [CrossRef]
- Barbera, J.A.; Blanco, I. Pulmonary hypertension in patients with chronic obstructive pulmonary disease: advances in pathophysiology and management. Drugs 2009, 69, 1153–1171. [Google Scholar] [CrossRef]
- Barnes, P.J. Mechanisms in COPD: differences from asthma. Chest 2000, 117, 10S–14S. [Google Scholar] [CrossRef]
- Finlay, G.A.; Russell, K.J.; McMahon, K.J.; D'arcy, E.M.; Masterson, J.B.; FitzGerald, M.X.; O'Connor, C.M. Elevated levels of matrix metalloproteinases in bronchoalveolar lavage fluid of emphysematous patients. Thorax 1997, 52, 502–506. [Google Scholar] [CrossRef]
- Ohnishi, K.; Takagi, M.; Kurokawa, Y.; Satomi, S.; Konttinen, Y.T. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab. Invest. 1998, 78, 1077–1087. [Google Scholar]
- Matsuba, K.; Thurlbeck, W.M. The number and dimensions of small airways in emphysematous lungs. Am. J. Pathol. 1972, 67, 265–275. [Google Scholar]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Salazar, L.M.; Herrera, A.M. Fibrotic response of tissue remodeling in COPD. Lung 2011, 189, 101–109. [Google Scholar] [CrossRef]
- Coraux, C.; Roux, J.; Jolly, T.; Birembaut, P. Epithelial cell-extracellular matrix interactions and stem cells in airway epithelial regeneration. Proc. Am. Thorac. Soc. 2008, 5, 689–694. [Google Scholar] [CrossRef]
- Fernandes, D.J.; Bonacci, J.V.; Stewart, A.G. Extracellular matrix, integrins, and mesenchymal cell function in the airways. Curr. Drug Targets 2006, 7, 567–577. [Google Scholar] [CrossRef]
- Jeffery, P.K. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2004, 1, 176–183. [Google Scholar] [CrossRef]
- Mauad, T.; Dolhnikoff, M. Pathologic similarities and differences between asthma and chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 2008, 14, 31–38. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Petecchia, L.; Sabatini, F.; Varesio, L.; Camoirano, A.; Usai, C.; Pezzolo, A.; Rossi, G.A. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest 2009, 135, 1502–1512. [Google Scholar] [CrossRef]
- Giembycz, M.A.; Kaur, M.; Leigh, R.; Newton, R. A Holy Grail of asthma management: toward understanding how long-acting beta(2)-adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids. Br. J. Pharmacol. 2008, 153, 1090–1104. [Google Scholar]
- Barnes, P.J. New therapies for chronic obstructive pulmonary disease. Med. Princ. Pract. 2010, 19, 330–338. [Google Scholar] [CrossRef]
- Pauwels, R.A.; Buist, A.S.; Calverley, P.M.; Jenkins, C.R.; Hurd, S.S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir. Crit. Care Med. 2001, 163, 1256–1276. [Google Scholar]
- Keatings, V.M.; Jatakanon, A.; Worsdell, Y.M.; Barnes, P.J. Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am. J. Respir. Crit Care Med. 1997, 155, 542–548. [Google Scholar]
- Barnes, N.C.; Qiu, Y.S.; Pavord, I.D.; Parker, D.; Davis, P.A.; Zhu, J.; Johnson, M.; Thomson, N.C.; Jeffery, P.K. Antiinflammatory effects of salmeterol/fluticasone propionate in chronic obstructive lung disease. Am. J. Respir. Crit Care Med. 2006, 173, 736–743. [Google Scholar] [CrossRef]
- Kaur, M.; Holden, N.S.; Wilson, S.M.; Sukkar, M.B.; Chung, K.F.; Barnes, P.J.; Newton, R.; Giembycz, M.A. Effect of beta2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. Am. J. Physiol Lung Cell Mol. Physiol. 2008, 295, L505–L514. [Google Scholar] [CrossRef]
- Hallsworth, M.P.; Twort, C.H.; Lee, T.H.; Hirst, S.J. beta(2)-adrenoceptor agonists inhibit release of eosinophil-activating cytokines from human airway smooth muscle cells. Br. J. Pharmacol. 2001, 132, 729–741. [Google Scholar] [CrossRef]
- Shore, S.A.; Moore, P.E. Regulation of beta-adrenergic responses in airway smooth muscle. Respir. Physiol. Neurobiol. 2003, 137, 179–195. [Google Scholar] [CrossRef]
- Banner, K.H.; Press, N.J. PDE3/4 inhibitors as therapeutic agents for chronic obstructive pulmonary disease. Br. J. Pharmacol. 2009, 157, 892–906. [Google Scholar] [CrossRef]
- Gross, N.J.; Giembycz, M.A.; Rennard, S.I. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. J. ChronicObstruct. Pulmon. Dis. 2010, 7, 141–153. [Google Scholar] [CrossRef]
- Giembycz, M.A.; Field, S.K. Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Desig. Develop. Therap. 2010, 4, 147–158. [Google Scholar]
- Press, N.J.; Banner, K.H. PDE4 inhibitors - a review of the current field. Prog. Med. Chem. 2009, 47, 37–74. [Google Scholar] [CrossRef]
- Sowa, G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol. 2012, 2, 120. [Google Scholar]
- Fish, J.E. A primer on the role of microRNAs in endothelial biology and vascular disease. Semin. Nephrol. 2012, 32, 167–175. [Google Scholar] [CrossRef]
- Hirase, T.; Noda, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H499–H505. [Google Scholar] [CrossRef]
- Vestweber, D. Novel insights into leukocyte extravasation. Curr. Opin. Hematol. 2012, 19, 212–217. [Google Scholar] [CrossRef]
- Vestweber, D.; Broermann, A.; Schulte, D. Control of endothelial barrier function by regulating vascular endothelial-cadherin. Curr. Opin. Hematol. 2010, 17, 230–236. [Google Scholar] [CrossRef]
- Vestweber, D.; Winderlich, M.; Cagna, G.; Nottebaum, A.F. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009, 19, 8–15. [Google Scholar] [CrossRef]
- Vestweber, D. Adhesion and signaling molecules that control endothelial cell contacts. Immunol. Rev. 2007, 218, 178–196. [Google Scholar] [CrossRef]
- Davies, D.E. The role of the epithelium in airway remodeling in asthma. Proc. Am. Thorac. Soc. 2009, 6, 678–682. [Google Scholar] [CrossRef]
- Niessen, C.M. Tight junctions/adherens junctions: Basic structure and function. J. Investig Dermatol. 2007, 127, 2525–2532. [Google Scholar] [CrossRef]
- Evans, M.J.; Fanucchi, M.V.; Plopper, C.G.; Hyde, D.M. Postnatal development of the lamina reticularis in primate airways. Anatom. Rec. 2010, 293, 947–954. [Google Scholar] [CrossRef]
- Evans, M.J.; van Winkle, L.S.; Fanucchi, M.V.; Plopper, C.G. Cellular and molecular characteristics of basal cells in airway epithelium. Experiment. Lung Res. 2001, 27, 401–415. [Google Scholar]
- Evans, M.J.; van Winkle, L.S.; Fanucchi, M.V.; Toskala, E.; Luck, E.C.; Sannes, P.L.; Plopper, C.G. Three-dimensional organization of the lamina reticularis in the rat tracheal basement membrane. Am. J. Respir. Cell Mol. Biol. 2000, 22, 393–397. [Google Scholar]
- Anderson, J.M.; van Italie, C.M. Physiology and function of the tight junction. ColdSpringHarb. Perspect. Biol. 2009, 1, a002584. [Google Scholar]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 2008, 1778, 588–600. [Google Scholar] [CrossRef]
- Terry, S.; Nie, M.; Matter, K.; Balda, M.S. Rho signaling and tight junction functions. Physiology 2010, 25, 16–26. [Google Scholar] [CrossRef]
- Corada, M.; Chimeti, S.; Cera, M.R.; Vinci, M.; Salio, M.; Fiordaliso, F.; De Angelis, N.; Villa, A.; Bossi, M.; Staszewsky, L.I.; Vecchi, A.; Parazzoli, D.; Motoike, T.; Latini, R.; Dejana, E. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2005, 102, 10634–10639. [Google Scholar]
- Khandoga, A.; Kessler, J.S.; Meissner, H.; Hanschen, M.; Corada, M.; Motoike, T.; Enders, G.; Dejana, E.; Krombach, F. Junctional adhesion moleculae A deficiency increases hepatic ischmia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood 2005, 106, 725–733. [Google Scholar] [CrossRef]
- Quint, J.K.; Wedzicha, J.A. The neutrophil in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2007, 119, 1065–1071. [Google Scholar] [CrossRef]
- Walters, R.W.; Freimuth, P.; Moninger, T.O.; Ganske, I.; Zabner, J.; Welsh, M.J. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 2002, 110, 789–799. [Google Scholar] [CrossRef]
- Cohen, C.J.; Shieh, J.T.C.; Pickles, R.J.; Okegawa, T.; Hsieh, J.-T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar]
- Zen, K.; Liu, Y.; McCall, I.C.; Wu, T.; Lee, W.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol. Biol. Cell 2005, 16, 2694–2703. [Google Scholar] [CrossRef]
- Giepsmans, B.N.; Ijzendoorn, S.C. Epithelial cell-cell junctions and plasma membrane domains. Biochim. Biophys. Acta 2009, 1788, 820–831. [Google Scholar] [CrossRef]
- Hernandez, S.; Chavez Munguia, B.; Gonzalez-Mariscal, L. ZO-2 silencing in epithelial cells perturbs the gate and fence function of tight junctions and leads to an atypical monolayer architecture. Exp. Cell Res. 2007, 313, 1533–1547. [Google Scholar] [CrossRef]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar]
- González-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junctions components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef]
- Guillemot, L.; Paschoud, S.; Pulimeno, P.; Foglia, A.; Citi, S. The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochim. Biophys. Acta 2008, 1778, 601–613. [Google Scholar] [CrossRef]
- Rodgers, L.S.; Fanning, A.S. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton 2011, 68, 653–660. [Google Scholar] [CrossRef]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef]
- van Itallie, C.M.; Fanning, A.S.; Bridhges, A.; Anderson, J.M. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 2009, 20, 3930–3940. [Google Scholar] [CrossRef]
- Nawijn, M.C.; Hackett, T.L.; Postma, D.S.; van Oosterhout, A.J.M.; Heijink, I.H. E-cadherin: gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 2011, 32, 248–255. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L. Molecular Cell Biology, 4th ed; W.H. Freeman and Company: New York, NY, USA, 2000. [Google Scholar]
- Goeckeler, Z.M.; Wysolmerski, R.B. Myosin phosphatase and cofilin mediate cAMP/cAMP-dependent protein kinase-induced decline in endothelial isometric tension and myosin II regulatory light chain phosphorylation. J. Biol. Chem. 2005, 280, 33083–33095. [Google Scholar] [CrossRef]
- Pritchard, C.A.; Hayes, L.; Wojnowski, L.; Zimmer, A.; Marais, R.M.; Norman, J.C. B-Raf acts via ROCKII/LIMK/Cofilin pathway to maintain actin stress fibres in fibroblasts. Mol. Cell. Biol. 2004, 24, 5937–5952. [Google Scholar] [CrossRef]
- De La Cruz, E.M.; Ostap, E.M. Relating biochemistry and function in the myosin superfamily. Curr. Opin. Cell Biol. 2004, 16, 61–67. [Google Scholar] [CrossRef]
- Pannekoek, W.J.; Kooistra, M.R.; Zwartkruis, F.J.; Bos, J.L. Cell-cell junction formation: The role of Rap1 and Rap1 guanine nucleotide exchange factors. Biochim. Biophys. Acta 2009, 1788, 790–796. [Google Scholar] [CrossRef]
- Retta, S.F.; Balzac, F.; Avolio, M. Rap1: A turnabout for the crosstalk between cadherins and integrins. Eur. J. Cell Biol. 2006, 85, 283–293. [Google Scholar] [CrossRef]
- van Straaten, J.F.; Coers, W.; Noordhoek, J.A.; Huitema, S.; Flipsen, J.T.; Kauffman, H.F.; Timens, W.; Postma, D.S. Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod. Pathol. 1999, 12, 697–705. [Google Scholar]
- Greenlee, K.J.; Werb, Z.; Kheradmand, F. Matrix metalloproteinase in lung: Multiple, multifarious, and multifaceted. Physiol. Rev. 2007, 87, 69–98. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammtion and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef]
- Gueders, M.M.; Foidart, J.M.; Noel, A.; Cataldo, D.D. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: Potential implications in asthma and other lung diseases. Eur. J. Pharmacol. 2006, 533, 133–144. [Google Scholar] [CrossRef]
- Vadenbroucke, R.E.; Dejonckheere, E.; Libert, C. A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur. Respir. J. 2011, 38, 1200–1214. [Google Scholar] [CrossRef]
- Demedts, I.K.; Brusselle, G.G.; Bracke, K.R.; Vemaelen, K.Y.; Pauwels, R.A. Matrix metalloproteinases in asthma and COPD. Curr. Opin. Pharmacol. 2005, 5, 257–263. [Google Scholar] [CrossRef]
- Oikonomid, S.; Kostikas, K.; Tsilioni, I.; Tanou, K.; Gourgoulianis, K.I.; Kiropoulos, T.S. Matrix metalloproteinases in respiratory diseases: from pathogenesis to potential clinical implications. Curr. Med. Chem. 2009, 16, 1214–1228. [Google Scholar] [CrossRef]
- Vial, W.C. Cigarette smoking and lung disease. Am. J. Med. Sci. 1986, 291, 130–142. [Google Scholar] [CrossRef]
- Lawrence, D.W.; Comerford, K.M.; Colgan, S.P. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am. J. Physi. Am. J. Physiol. Cell Physiol. 2002, 282, C1235–C1245. [Google Scholar]
- Lorenowicz, M.J.; Fernandez-Borja, M.; van Stalborch, A.M.; van Sterkenburg, M.A.; Hiemstra, P.S.; Hordijk, P.L. Microtubule dynamics and rac-1 signaling independently regulate barrier function in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1321–L1331. [Google Scholar] [CrossRef]
- Baumer, Y.; Drenckhahn, D.; Waschke, J. cAMP induced Rac1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem. Cell Biol. 2008, 129, 765–778. [Google Scholar]
- Logue, J.S.; Whiting, J.L.; Tunquist, B.; Langeberg, L.K.; Scott, J.D. Anchored protein kinase A recruitment of active Rac GTPase. J. Biol. Chem. 2011, 286, 22113–22121. [Google Scholar]
- Logue, J.S.; Whiting, J.L.; Scott, J.D. Sequestering Rac with PKA confers cAMP control of cytoskeletal remodeling. SmallGTPases 2011, 3, 173–176. [Google Scholar]
- Michel, C.C.; Curry, F.E. Microvascular permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar]
- Metha, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Zemskova, M.A.; Kovalenkov, Y.; Poirier, C.; Verin, A.D. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial (HLMVEC) hyperpermeability. J. Cell. Physiol. 2009, 221, 750–759. [Google Scholar] [CrossRef]
- van Wetering, S.; van Buul, J.D.; Quik, S.; Mul, F.P.J.; Anthony, E.C.; ten Klosster, J.-P.; Collard, J.G.; Hordijk, P.L. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J. CellSci. 2002, 115, 1837–1846. [Google Scholar]
- Birukova, A.A.; Zagranichnaya, T.; Alekseeva, E.; Bokoch, G.M.; Birukov, K.G. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J. Cell Physiol. 2008, 215, 715–724. [Google Scholar] [CrossRef]
- Birukova, A.A.; Fu, P.; Xing, J.; Birukov, K.G. Rap1 mediates protective effects of iloprost against ventilator-induced lung injury. J. Appl. Physiol. 2009, 107, 1900–1910. [Google Scholar] [CrossRef]
- Birukova, A.A.; Zagranichnaya, T.; Fu, P.; Alekseeva, E.; Cheng, X.; Jacobson, J.R.; Birukov, K.G. Prostaglandines PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp. Cell Res. 2007, 313, 2504–2520. [Google Scholar] [CrossRef]
- Xing, J.; Birukova, A.A. ANP attenuates inflammatory signaling and Rho pathway of lung endothelial permeability induced by LPS and TNFa. Microvascul. Res. 2010, 79, 56–62. [Google Scholar] [CrossRef]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef]
- Glading, A.; Han, J.; Stockton, R.A.; Ginsberg, M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007, 179, 247–254. [Google Scholar] [CrossRef]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 4, 881–896. [Google Scholar]
- Gao, S.; Li, C.; Shimokawa, T.; Terashita, T.; Matsuda, S.; Yaoita, E.; Kobayashi, N. Rho-family small GTPases are involved in forskolin-induced cell-cell contact formation of renal glomerular podocytes in vitro. Cell Tissue Res. 2007, 328, 391–400. [Google Scholar] [CrossRef]
- Costantini, T.W.; Deree, J.; Loomis, W.; Putman, J.G.; Choi, S.; Baird, A.; Eliceiri, B.P.; Bansal, V.; Coimbra, R. Phosphodiesterase inhibition attenuates alterations to the tight junction proteins occludin and ZO-1 in immunostimulated caco-2 intestinal monolayers. Life Sci. 2009, 84, 18–22. [Google Scholar] [CrossRef]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef]
- Oldenburger, A.; Rijks, W.F.; Sewbalaksing, V.D.; Poppinga, W.J.; Heijink, I.H.; Maarsingh, H.; Schmidt, M. A-kinase anchoring proteins (AKAPs) as potential novel therapeutic targets to improve cigarette smoke-induced loss of epithelial barrier function. Available online: http://www.pA2online.org/abstracts/Vol10Issue3abst108P.pdf accessed on 28 November 2012.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oldenburger, A.; Maarsingh, H.; Schmidt, M. Multiple Facets of cAMP Signalling and Physiological Impact: cAMP Compartmentalization in the Lung. Pharmaceuticals 2012, 5, 1291-1331. https://doi.org/10.3390/ph5121291
Oldenburger A, Maarsingh H, Schmidt M. Multiple Facets of cAMP Signalling and Physiological Impact: cAMP Compartmentalization in the Lung. Pharmaceuticals. 2012; 5(12):1291-1331. https://doi.org/10.3390/ph5121291
Chicago/Turabian StyleOldenburger, Anouk, Harm Maarsingh, and Martina Schmidt. 2012. "Multiple Facets of cAMP Signalling and Physiological Impact: cAMP Compartmentalization in the Lung" Pharmaceuticals 5, no. 12: 1291-1331. https://doi.org/10.3390/ph5121291
APA StyleOldenburger, A., Maarsingh, H., & Schmidt, M. (2012). Multiple Facets of cAMP Signalling and Physiological Impact: cAMP Compartmentalization in the Lung. Pharmaceuticals, 5(12), 1291-1331. https://doi.org/10.3390/ph5121291