Abstract
Tuberculosis is the second leading cause of infectious deaths globally. Many effective conventional antimycobacterial drugs have been available, however, emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) has overshadowed the effectiveness of the current first and second line drugs. Further, currently available agents are complicated by serious side effects, drug interactions and long-term administration. This has prompted urgent research efforts in the discovery and development of new anti-tuberculosis agent(s). Several families of compounds are currently being explored for the treatment of tuberculosis. This review article presents an account of the existing chemotherapeutics and highlights the therapeutic potential of emerging molecules that are at different stages of development for the management of tuberculosis disease.
1. Introduction
In 1905, Robert Koch, a German physician, was awarded the Nobel Prize for his milestone discovery of Mycobacterium tuberculosis (Mtb), the bacillus of tuberculosis (TB). Despite his groundbreaking discovery, it took more than half a century to find a cure against the bacilli to save millions of human lives. The death of Koch in 1910 prevented him from witnessing the life-saving consequences of his pioneering research. Among these was the first antibiotic for tuberculosis patients, streptomycin, a natural compound. However, the requirement of intravenous administration of streptomycin and development of resistance to it soon necessitated the need of next generation of antibiotics against Mtb. Many antimycobacterial drugs have been discovered since then, classified as first-line and second line drugs. Recent emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) [1], have seriously compromised the usefulness of these current first and second line drugs, once again creating an urgent need for newer, safer and more effective anti-tuberulosis treatments. Adding to this crisis is the limited use of second line drugs for MDR-TB and XDR-TB due to their toxicity and serious side effects. Moreover, recently, totally drug-resistant tuberculosis (TDR-TB) has emerged which is resistant to a wider range of drugs than XDR-TB. Cases of TDR-TB have been reported in several countries including Italy, Iran and India [2].
Bacillus Calmette Guerin (BCG) vaccine, an attenuated strain of M. bovis reliably protects only newborns against Mtb but is ineffective in adult pulmonary TB. The vaccination may also lead to TB-like infection in immunocompromised people [3].
The increasing incidence of MDR-TB, XDR-TB, and TB-HIV coinfection have raised the alarm for the discovery and development of novel anti-tuberculosis agent(s) that do not possess cross-resistance with current antimycobacterial drugs and have minimal toxicity. This article summarizes the features of current anti-tuberculosis drugs and the pharmacological properties of novel compounds that are in the process of development for antimycobacterial therapy.
2. Current Anti-Tuberculosis Drugs
2.1. First Line Drugs
First line anti-tuberculosis drugs include rifampicin, isoniazid, pyrazinamide and ethambutol (Figure 1).
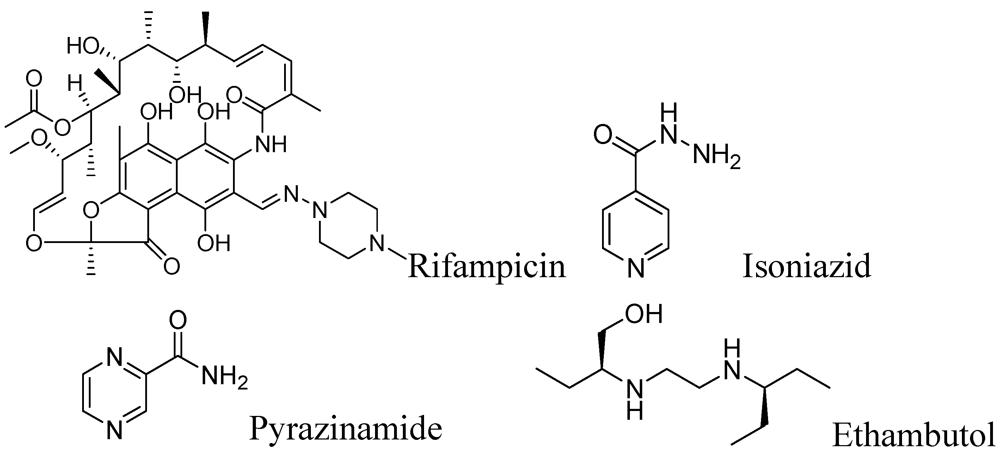
Figure 1.
First line anti-tuberculosis drugs.
Figure 1.
First line anti-tuberculosis drugs.
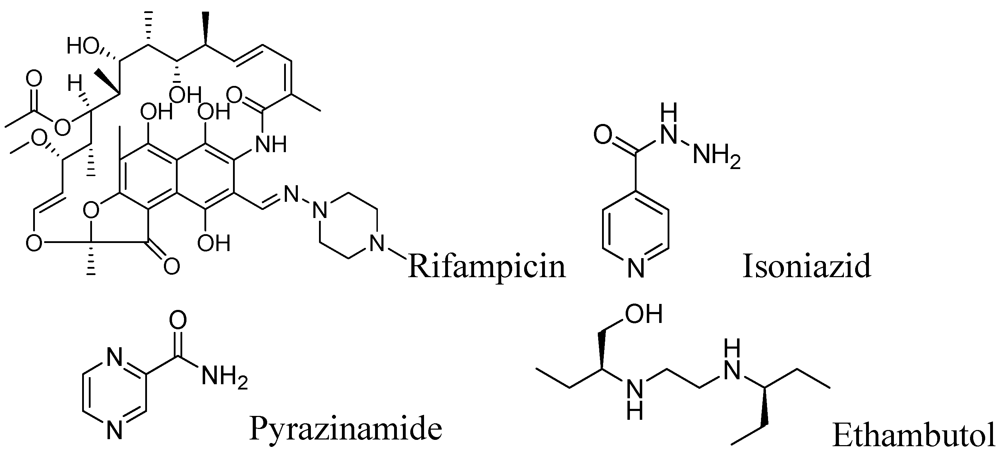
2.1.1. Rifampicin
This drug was discovered in 1966. It possesses very potent in vitro activity against Mtb with an MIC of 0.05–0.5 μg/mL. Rifampicin is highly active against Gram-positive bacteria including Mtb. Unlike many other antibiotics, it is lipid soluble, penetrates cell membranes and kills intracellular bacteria [4]. It acts by the inhibition of DNA-dependent RNA polymerase in bacterial cells by binding to its β-subunit, thus preventing transcription of RNA and subsequent translation of proteins [5,6]. A daily regimen of 10 mg/kg (up to 600 mg/day) orally or an intermittent regimen of 10 mg/kg (up to 600 mg/day) orally, are effective [7]. However, Mtb quickly develops resistance to rifampicin hence the drug is recommended to be used in combination with other antibiotics. Most of the Mtb clinical isolates resistant to rifampicin show mutations in the rpoB gene that encodes the β-subunit of RNA polymerase. These mutations cause conformational changes in the polymerase that result in a low affinity for the drug rendering it ineffective [8]. The side effects include hepatitis with elevation of bile and bilirubin, anaemia, leucopenia, thrombocytopenia, bleeding, fever, eosinophilia, leucopenia, thrombocytopenia, purpura, haemolysis and nephrotoxicity [9]. Interestingly, no serious side effects have been observed in breastfed infants during rifampicin therapy [10,11]. The drugs for possible interactions with rifampicin include 4-aminosalicylic acid (PAS), HIV protease inhibitors, warfarin, oral contraceptives, cyclosporine, itraconazole, digoxin, verapamil, nifedipine, simvastatin, midazolam, clarithromycin, lorazepam atorvastatin, antiretroviral agents, rosiglitazone/pioglitazone, celecoxib, caspofungin [12].
2.1.2. Isoniazid
Isoniazid (INH) was discovered in 1952. It acts as a bactericidal agent with an MIC of 0.01–0.2 μg/mL for fast replicating mycobacteria [13]. It is bacteriostatic to slow-growing or non-dividing mycobacteria like Mtb and therefore, is used to treat latent tuberculosis. Isoniazid is a actually a prodrug and is activated by the mycobacterial enzyme catalase-peroxidase (KatG), which catalyzes the formation of the isonicotinic acyl-NADH complex. Subsequently, this complex binds to the enoyl-acyl carrier protein reductase InhA, and then blocks the natural substrate enoyl-AcpM and fatty acid synthase. This results in inhibition of mycolic acid synthesis which is an essential component in the formation of the mycobacterial cell wall [14,15]. Resistance to isoniazid occurs due to mutations in several genes, including katG, ahpC, inhA, kasA and ndh. In adults, the recommended daily dose of INH is 5 mg/kg/day (max 300 mg daily). For intermittent dosing (twice or three times/week), 19–15 mg/kg/day (max 900 mg/day) is used. The recommended dose for children is 8 to 12 mg/kg/day [7,16]. INH is metabolized in the liver and its metabolites are excreted in the urine [17]. INH chronic toxicity affects the liver, haematologic- and peripheral nervous systems resulting in acute hepatitis, peripheral neuropathy and haemolytic anaemia [18].
2.1.3. Pyrazinamide
Pyrazinamide (PZA) was discovered in 1952. It is mainly bacteriostatic but can be bactericidal for replicating Mtb. It possesses an MIC of 20–100 μg/mL. When used as part of combination therapy, PZA accelerates the sterilizing effect of INH and rifampin [19]. This has enabled reductions in the duration of treatment for susceptible M. tuberculosis isolates from nine to six months and for this reason is used in the first two months of treatment [20]. PZA is also effective for the treatment of tuberculous meningitis [21]. Like isoniazid, PZA is a prodrug. In acidic conditions, the enzyme pyrazinamidase (present in Mtb), converts it to the active form, pyrazinoic acid which subsequently inhibits the enzyme fatty acid synthase (FAS) I, required by the bacterium to synthesize fatty acids [22,23,24]. Mutations of the pyrazinamidase gene (pncA) are responsible for PZA resistance in Mtb [25]. Most alterations occur in a 561 bp region of the open reading frame or in an 82 bp region of its putative promoter [26,27].
The recommended dose of PZA is 20–25 mg/kg daily or 30–40 mg/kg three times a week [7]. Pyrazinamide is metabolized by the liver and the metabolic products are excreted by the kidneys [28]. Some common side effects of PZA include skin rash, nausea, vomiting, hepatotoxicity, anorexia, hyperuricemia, sideroblastic anemia, dysuria, joint pains (arthralgia), urticaria, pruritus, malaise, interstitial nephritis, porphyria and fever [29].
2.1.4. Ethambutol
This drug was discovered in 1961. Ethambutol (EMB) is a bacteriostatic drug against actively growing mycobacteria. It blocks formation of the cell wall of Mtb by inhibiting the enzyme arabinosyl transferase involved in the synthesis of arabinogalactan. Arabinogalactan is an essential component in the formation of the mycolyl-arabinogalactan-peptidoglycan complex of the Mtb cell wall [30,31]. Mutation in gene embB is responsible for resistance to ethambutol [30].
Ethambutol is well absorbed in the gastrointestinal tract, and is efficiently distributed in body tissues and fluids. Fifty percent of the given dose is excreted unchanged in urine [32]. Ethambutol is used at 15–25 mg/Kg once daily dose for 6–8 weeks concurrent with isoniazid therapy [33]. Adverse effects of EMB include peripheral neuropathy, red-green color blindness, arthralgia, hyperuricaemia and optic neuritis [34].
2.2. Second Line Drugs
The available second-line TB drugs can be classified as: (1) polypeptides (e.g., capreomycin); (2) aminoglycosides: (e.g., amikacin); (3) oxazolidinone (e.g., cycloserine); (4) thioamides (e.g., ethionamide); (5) fluoroquinolones (e.g., ciprofloxacin); (6) p-aminosalicylic acid (PAS or P). Some of the second line drugs are summarized in Table 1.

Table 1.
Common second line drugs [35].
| Drug (Discovery) MIC values * | Structure | Daily dose (Max. dose) Route | Adverse effects | Mode of action |
|---|---|---|---|---|
| Capreomycin (1963) MIC 1.25–2.5 μg/mL [ 36,37] |  | 15–30 mg/kg (1 g) IM or IV | Auditory, vestibular, and renal toxicity | Inhibits protein synthesis (binds to ribosomal subunit 16S and 23S rRNA) [38] |
| Amikacin (1972) MIC 4–8 μg/mL [39] |  | 15–30 mg/kg (1 g) IM or IV | Same as capreomycin | Inhibits protein synthesis (binds to the bacterial 30S ribosome) |
| Kanamycin (1957) MIC 1–8 μg/mL | 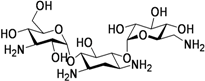 | 15–30 mg/kg (1 g) IM or IV | Same as Capreomycin | Inhibits protein synthesis via S12 ribosomal protein & 16 S RNA. |
| Streptomycin (1944) MIC 2–8 μg/mL | 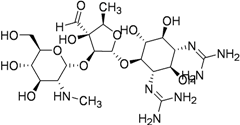 | 15–40 mg/kg (1 g) IM | Renal, ophthalmic and respiratory toxicity | Same as kanamycin |
| Cycloserine (1952) MIC 5–20 μg/mL |  | 15–20 mg/kg (1 g) Oral | Psychosis, Rashes, Convulsions Depression | Inhibition of peptidoglycan synthesis (D-alanine racemase) |
| Ethionamide (1956) MIC 0.6–2.5 μg/mL |  | 15–20 mg/kg (1 g) Oral | GI upset Hepatotoxicity Hypersensitivity | Inhibition of mycolic acid synthesis |
| Clofazimine (1954) MIC 0.12–0.24 μg/mL [40] |  | 100–300 mg/day Oral | Eosinophilic enteritis, GI irritation, discoloration of the skin (upon sun exposure) | Inhibits bacterial proliferation by binding to the guanine bases of bacterial DNA |
| Levofloxacin (1992) MIC 0.50 to 0.75 μg/mL [41] | 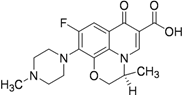 | 500 mg/day Oral | GI upset Dizziness Headache Hypersensitivity Restlessness | Inhibition of DNA replication and transcription by inhibiting DNA gyrase |
| Ofloxacin (1980) Oral, MIC 0.12–2 μg/mL [42] |  | 600–800 mg/day | Same as for levofloxacin | Same as for levofloxacin |
| Ciprofloxacin (1960s) MIC 0.4 to 6.2 μg/mL [43] | 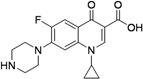 | 750–1,500 mg/day Oral | Same as for levofloxacin | Same as for levofloxacin |
| PAS (1946) MIC 1–8 μg/mL |  | 150 mg/kg (16 g) Oral | Same as for ethionamide, Sodium load | Inhibition of folic acid and iron metabolism (unknown target) |
* MICs (wherever not referenced) are based on Inderlied and Salfinger [44]. IM, intramuscular; IV, intravenous.
3. Drug Discovery Program
This section includes early stage drug discovery, molecules in development, molecules at the pre-clinical stage, molecules in phase I trials, molecules in phase II trials and molecules in phase III trials.
3.1. Early Stage Drug Discovery
After a long pause, the last decade proved to be a golden era in the hunt for new tuberculosis drug(s). Tremendous efforts and high priority research are underway for finding better drugs to combat wild-type and drug-resistant Mtb. Since the last decade, the private sector and government agencies have participated in the fight against this devastating disease. Apart from major financial contributions by corporations, basic and semi-applied researchers are also continuing to make significant progress, despite facing financial constraints. The following section describes some representative examples of different classes of molecules from early stage screening studies carried out in the last decade by these groups.
3.1.1. Nucleosides
The nucleoside class of compounds are well known for their antiviral and anticancer properties. They can be classified as pyrimidine or purine nucleosides.
3.1.1.1. Pyrimidine Nucleosides
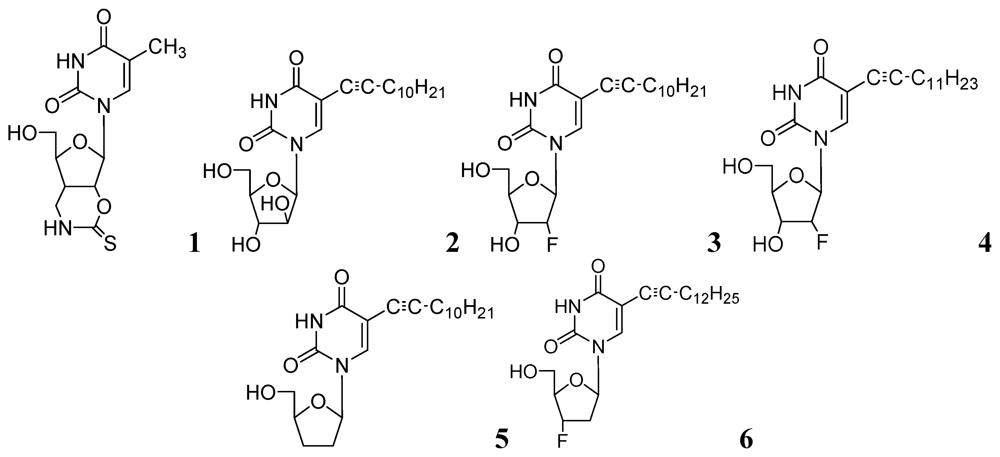
In early 2000s, Vanheusden et al. reported modified nucleoside and nucleotide derivatives as inhibitors of a mycobacterial enzyme thymidine monophosphate kinase (TMPKmt). In 2004, they reported a series of bicyclic analogues of thymidine [45] where compound 1 (Figure 2) demonstrated a Ki of 3.5 μM for TMPKmt with a good selectivity index (SI 200) over its human counterpart TMPKh. In these studies, however, only enzyme inhibition was reported and inhibition of mycobacterial replication was not demonstrated.
The complete genome sequence of Mycobacterium tuberculosis has been determined [46], which identified many of the genes required for encoding enzymes involved in nucleic acid synthesis, and pyrimidine and purine biosynthesis. We hypothesized that modified nucleosides could target several enzymes involved in RNA and DNA metabolism and were the first to investigate and demonstrate potent antimycobacterial activity of 5-substituted pyrimidine nucleoside analogs [47]. The microplate alamar blue assay (MABA) [48] was used to evaluate the antimycobacterial activity of test nucleosides. We observed that the most potent TMPKmt inhibitors reported earlier [49,50,51] did not show antituberculosis activity against mycobacterial replication as determined by MABA assay [47].
Since our initial report in 2005, we (Kumar and colleagues) have made significant contributions in the investigation of pyrimidine nucleosides as new classes of anti-tuberculosis agents. We designed, synthesized and examined a variety of known and unknown pyrimidine nucleosides substituted/unsubstituted at 2-, 4-, 5- and/or 6- positions of the base, and containing various deoxyribose, ribose, arabinose, dideoxyribose and acyclic sugar moieties. We found that 5-alkynyl substituted pyrimidine nucleosides demonstrated the most potent activity against mycobacteria [52,53]. The MIC90 exhibited by compounds of this series (2, 3 and 4, Figure 2) was in the range of 1–5 μg/mL) against Mtb H37Ra. These compounds were also found to retain sensitivity against an RMP-resistant strain of Mtb H37Rv (American Type Culture Collection [ATCC] 35838, resistant to RMP at 2 μg/mL) at similar concentrations. Subsequently, we reported a series of 5-acetylenic derivatives with 2',3-dideoxyuridine, and 3'-fluoro-2',3'-dideoxyuridine. Compound 5 (among 2',3'-dideoxyuridine series) and compound 6 (among 3'-fluoro-2',3'-dideoxyuridine series) exhibited excellent activity against wild-type Mtb H37Ra (MIC 1–2 μg/mL) and a rifampicin-resistant H37Rv strain (ATCC 35838, resistant to RMP at 2 μg/mL) of Mtb [54] (Figure 2).
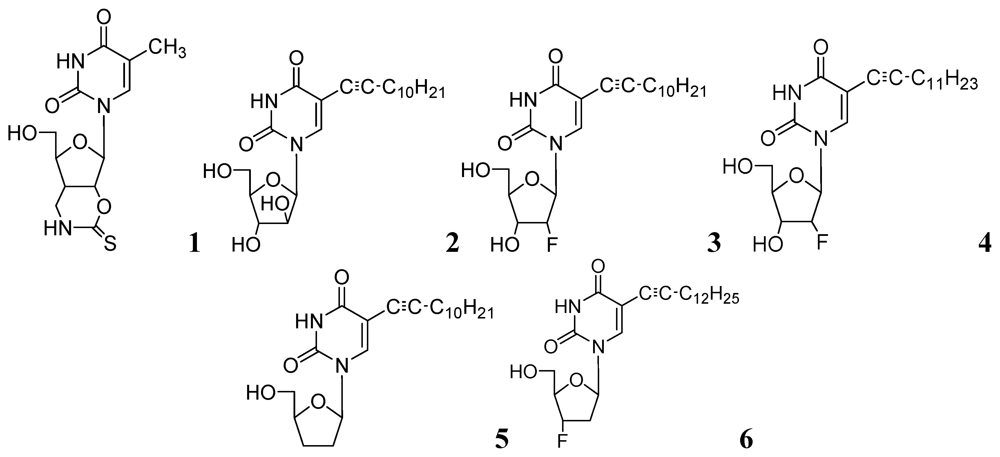
Figure 2.
Pyrimidine nucleosides as anti-tuberculosis agents.
Figure 2.
Pyrimidine nucleosides as anti-tuberculosis agents.
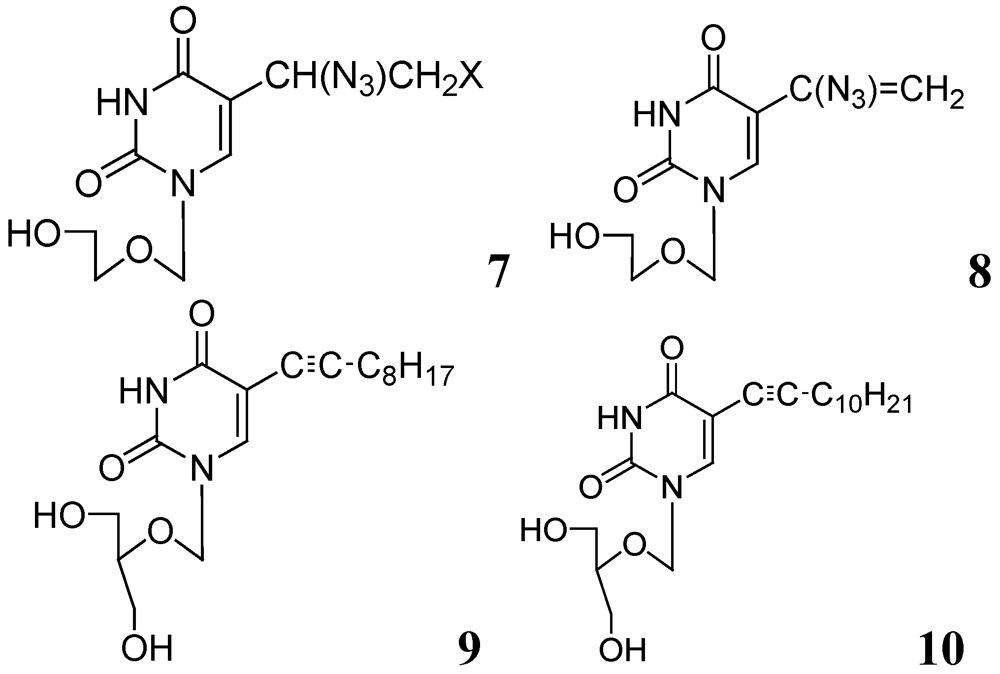
In further studies, we also investigated anti-tuberculosis activities of several 5-substituted acyclic pyrimidine nucleosides against Mtb H37Ra, M. bovis, and M. avium. In this study, 7–10 were moderately active against these mycobacteria [55] (Figure 3).
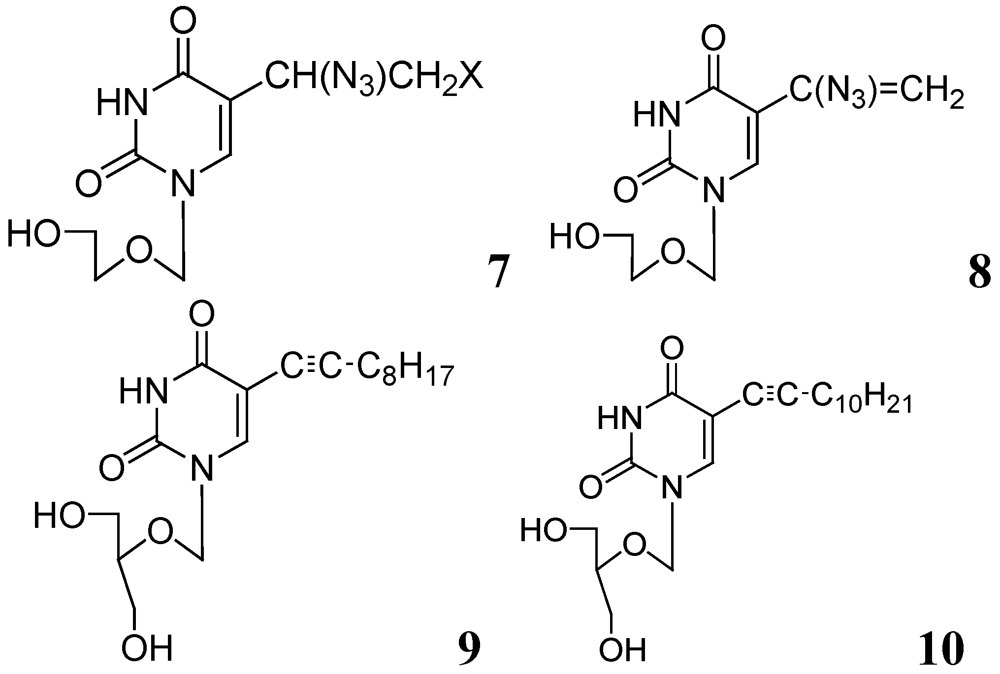
Figure 3.
Acyclic pyrimidine nucleosides as anti-tuberculosis agents.
Figure 3.
Acyclic pyrimidine nucleosides as anti-tuberculosis agents.
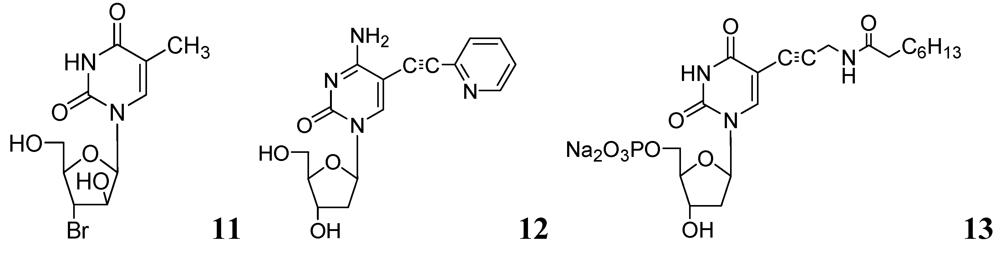
In recent studies, we synthesized and investigated various 2'- or 3'-halogeno derivatives of pyrimidine nucleosides containing uracil, 5-fluorouracil, and a thymine base [56]. In this class, compound 11 was found to be the most effective antituberculosis agent in the in vitro assays against wild-type Mtb strain (H37Ra, MIC50 = 1 μg/mL), and drug-resistant (H37Rv) strains of Mtb (RMP-resistant and INH-resistant, MIC50 = 1–2 μg/mL). The antimycobacterial effect of the most potent compounds was also determined against intracellular mycobacteria in a human monocytic cell-line (THP-1) infected with the Mtb H37Ra strain [57]. Interestingly, compound 11 demonstrated slightly better activity against intramacrophagic mycobacteria than extracellular mycobacteria. In contrast, pyrimidine nucleosides possessing a 5-fluorouracil base were weak inhibitors of Mtb H37Ra.
In the same year our group reported antimycobacterial effects of several 5-alkyl- and 5-alkynyl-furanopyrimidines and related 2'-deoxynucleosides. Compounds with 5-arylalkynyl substituents displayed potent in vitro antitubercular activity against M. bovis and Mtb (MIC 0.5–5 μg/mL). We selected compound 12 to test its potency in a mouse model (BALB/c) of Mtb (H37Ra) infection. At a dose of 50 mg/kg for 5 weeks, statistically significant reduction in mycobacterial load was observed in lungs, livers and spleens of the treated mice. This is the first evidence of antimycobacterial potential of 5-substituted pyrimidine nucleosides in an animal model as a potential new class of antituberculosis agents [58].
Kogler et al. reported a series of 5-substituted -2'-deoxyuridine monophosphate analogs as potential inhibitors of mycobacterial flavin-dependent thymidylate synthase (ThyX). Compound 13 displayed selective inhibition of ThyX (IC50 0.91 μM) but not against the classical mycobacterial thymidylate synthase (ThyA, IC50 > 50 μM) [59] (Figure 4).
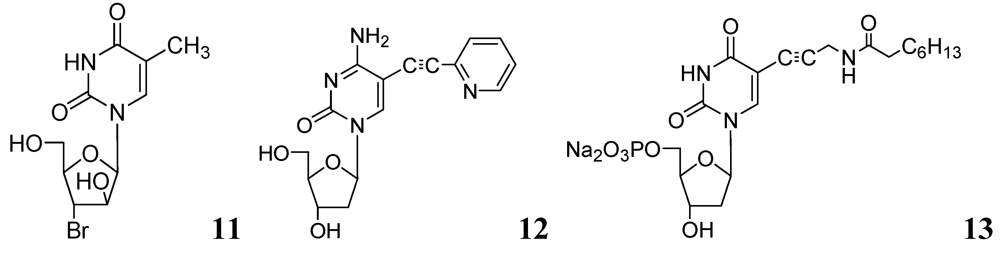
Figure 4.
Some recent pyrimidine nucleosides as anti-tuberculosis agents.
Figure 4.
Some recent pyrimidine nucleosides as anti-tuberculosis agents.
3.1.1.2. Purine Nucleosides
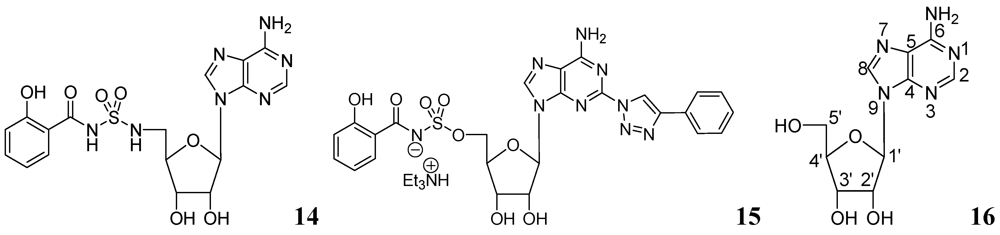
Somu et al. reported a purine nucleoside analog 14 (MIC99 = 0.19 μM) as an inhibitor of siderophore biosynthesis in Mtb under iron-limiting conditions. The authors mentioned that the activity of 14 was due to inhibition of the adenylate-forming enzyme MbtA, which is involved in biosynthesis of the mycobactins [60].
Triazole derivatives of 5'-O-[N-(salicyl)sulfamoyl]adenosine have been investigated as inhibitors of aryl acid adenylating enzymes (AAAE) involved in siderophore biosynthesis by Mtb H37Rv. Enzyme assays were performed at 37 °C with recombinant MbtA expressed in E. coli. Compound 15 (MIC 3.13 μM) was reported as the best candidate [61].
Adenosine (Ado) kinase is a purine salvage enzyme that phosphorylates adenosine to adenosine-monophosphate. A number of adenine nucleosides 16 have been evaluated as substrates and inhibitors of adenosine (Ado) kinase from Mtb. The best substrates were found to be 2-aza-adenosine, 8-aza-9-deazaadenosine and 2-fluoroadenosine, while the most potent compounds were N-1-benzyladenosine (Ki = 0.19 μM), 2-fluoroadenosine (Ki = 0.5 μM), 6-cyclopentyloxy purine riboside (Ki = 0.15 μM) and 7-iodo-7-deazaadenosine (Ki = 0.21 μM). Several of these adenosine analogs exhibited promising MICs [62] (Figure 5).
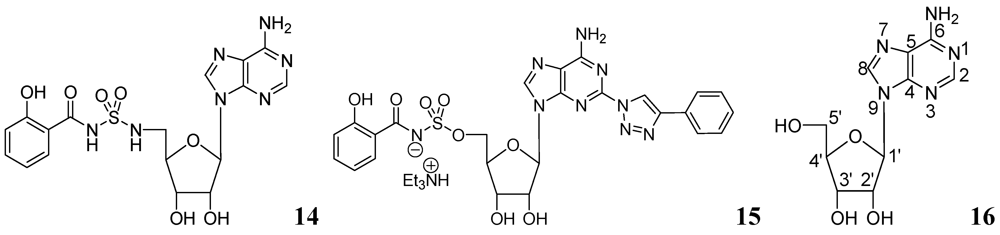
Figure 5.
Purine nucleosides as anti-tuberculosis agents.
Figure 5.
Purine nucleosides as anti-tuberculosis agents.
3.1.2. Carbohydrates
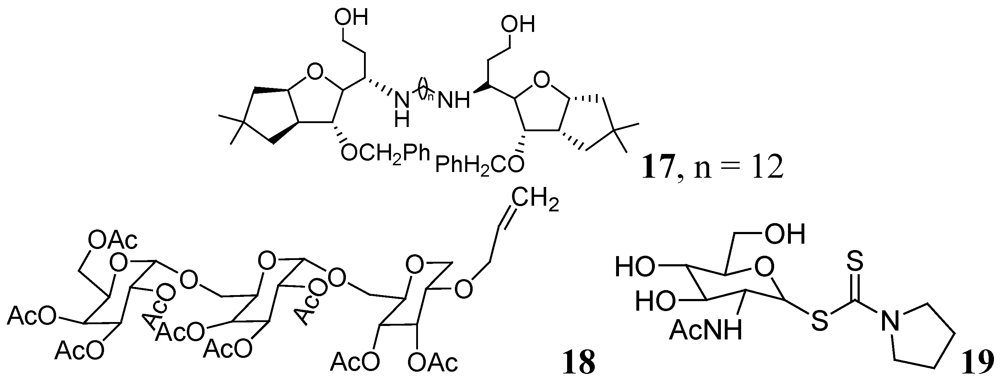
Carbohydrates have been evaluated as antituberculosis agents for a long time. Some selected reports are summarized here. In 2005 bis-glycosylated diamino alcohols were reported by Tripathi et al., where their compound, 17, showed moderate activity against Mtb H37Ra and against Mtb H37Rv. This compound was also active against an MDR strain and showed mild protection in mice at 25 mg/Kg dose [63].
Derivatives of stachyose were reported by Chiba et al. The most active compound in the series against Mtb H37Rv was 18 (OCT359, MIC 3.13 μg/mL) which was also evaluated against various drug-sensitive and -resistant clinical isolates of Mtb. Interestingly, 25 clinical isolates of drug-resistant Mtb and 19 drug-sensitive Mtb were sensitive to OCT359 (MICs ranging from 3.13 to 25 μg/mL) [64].
Recently in this class, compound 19 (OCT313HK, Glc-NAc-PDTC) showed potent anti-tuberculosis activity against wild-type, and clinical isolates of Mtb, including MDR and XDR strains at similar concentrations (MIC 6.25–12.5 μg/mL) [65] (Figure 6).
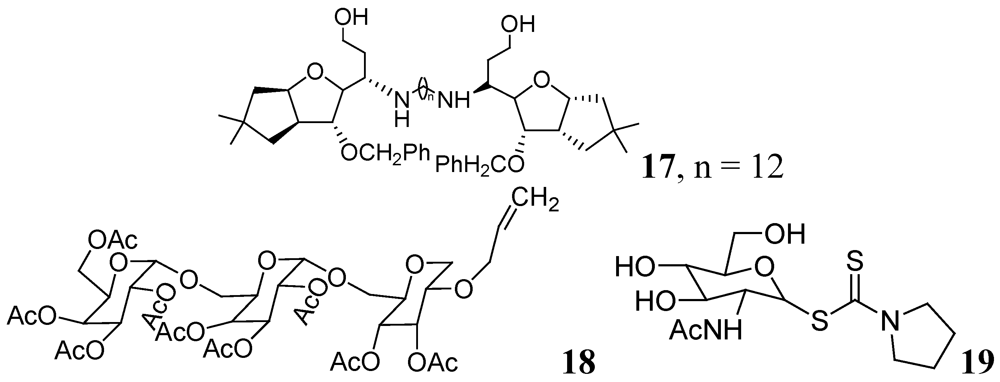
Figure 6.
Carbohydrate derivatives as anti-tuberculosis agents.
Figure 6.
Carbohydrate derivatives as anti-tuberculosis agents.
3.1.3. Heterocyclic Compounds
3.1.3.1. Quinolines and Quinoxalines
Fluoroquinolones have been used as antibiotics (e.g., ciprofloxacin, levofloxacin, ofloxacin). Moxifloxacin and gatifloxacin from this class are in Phase III clinical trials for tuberculosis treatment.
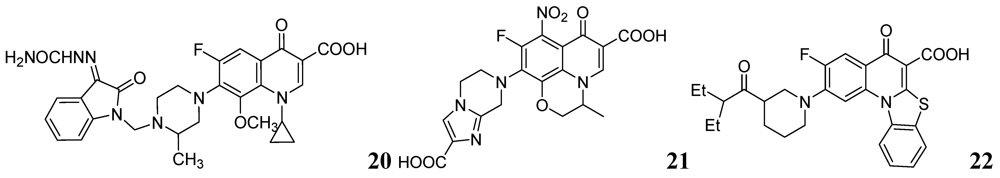
Sriram et al. investigated a series of 7-substituted gatifloxacin derivatives. Their compound, 20, was found to be the most active in vitro studies (MIC value 0.0125 μg/mL) against Mtb and MDR-TB. In an animal model 20 decreased the bacterial loads in the lung and spleen by 3.62- and 3.76-log10, respectively [66].
The same group investigated derivatives of ofloxacin (OFX). Compound 21 exhibited the most potent activity (MIC99 of 0.19 μM and 0.09 μM against Mtb and MDR-TB, respectively) and decreased bacterial loads (strain ATCC 35801) in lung and spleen by 1.91 and 2.91-log10, respectively, at 50 mg/kg dose in a mouse model [67].
Dinakaran et al. further reported derivatives of 2-(sub)-3-fluoro/nitro-5,12-dihydro-5-oxobenzothiazolo[3,2-a]quinoline-6-carboxylic acid. Among them, compound 22 displayed the most potent activity with MICs of 0.18 and 0.08 μM against Mtb and MDR-TB, respectively. In a mouse model of Mtb infection, 22 was effective at 50 mg/kg dose and reduced bacterial loads in lung and spleen tissues by 2.78 and 3.12-log10, respectively [68] (Figure 7).
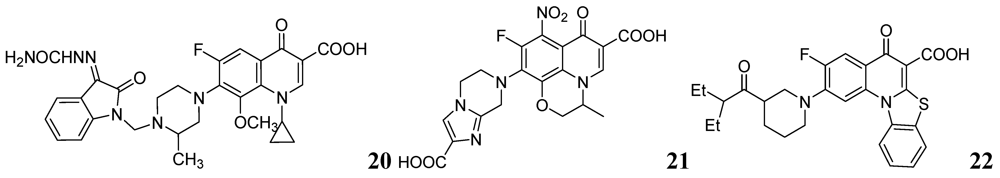
Figure 7.
Fluoroquinolones as anti-tuberculosis agents.
Figure 7.
Fluoroquinolones as anti-tuberculosis agents.
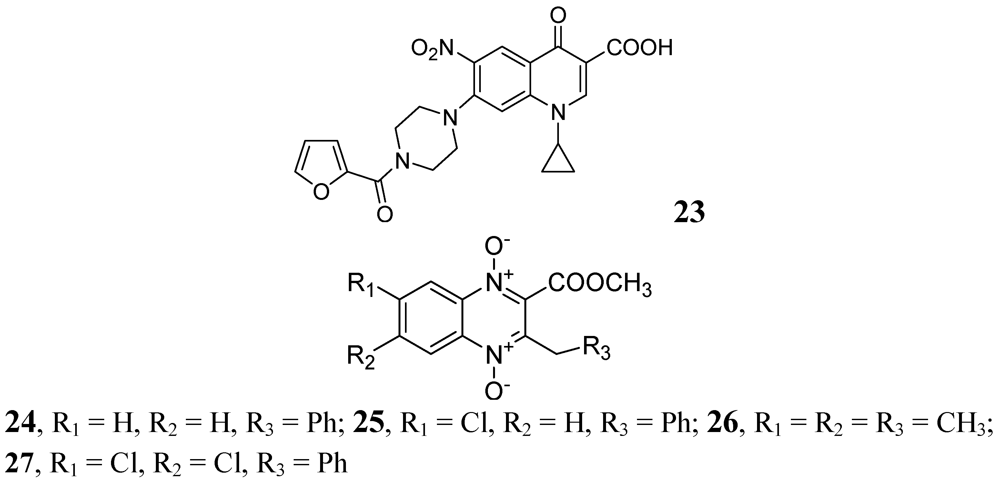
In 2009, Senthilkumar et al. investigated 1-(substituted)-1,4-dihydro-6-nitro-4-oxo-7-(sub-secondary amino)-quinoline-3-carboxylic acids. In vitro, their compound, 23, exhibited MICs of 0.08 and 0.16 μM against Mtb and MDR-TB, respectively. In vivo studies revealed that 23 led to a significant reduction in bacterial load in lung and spleen at 50 mg/kg dose [69].
In 2005, Jaso et al. evaluated a series of 6(7)-substituted quinoxaline-2-carboxylate 1,4-dioxide derivatives against MtbH37Rv. Their compounds 24, (MIC 0.1 μg/mL) and 25, (MIC 0.1 μg/mL) had good antituberculosis activity including intracellular bacteria (EC90 0.15 μg/mL and 0.0005 μg/mL, respectively). Compounds 26 and 27 of the series were also active against drug-resistant strains of Mtb H37Rv with MICs of 0.39–1.56 and 3.13–12.5 μg/mL, respectively [70] (Figure 8).
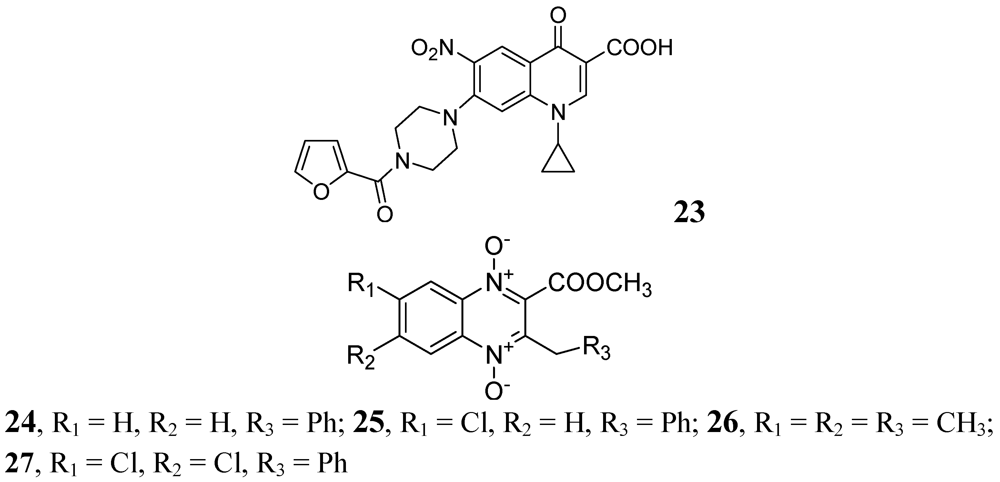
Figure 8.
Quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-tuberculosis agents.
Figure 8.
Quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-tuberculosis agents.
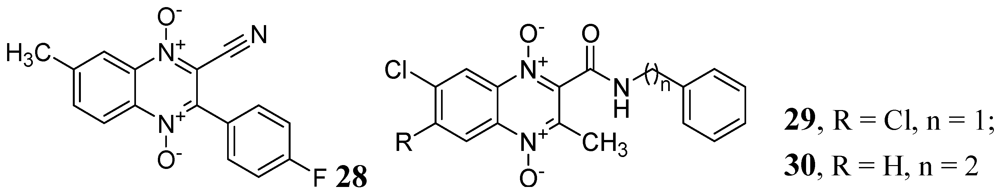
A series of derivatives of 1,4-di-N-oxide-3-phenylquinoxaline was published by Vicente et al. against Mtb H37Rv. Their compound 28 was the most active in the series (MIC < 0.2 μg/mL) [71]. Ancizu et al. also described a series of 1,4-di-N-oxide derivatives of quinoline. Compounds 29 and 30 displayed the most significant inhibition of Mtb H37Rv with MICs < 0.2 μg/mL [72] (Figure 9).
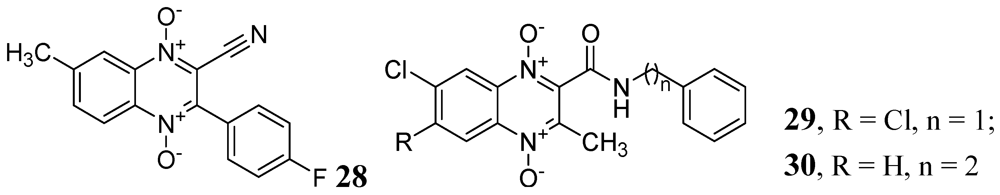
Figure 9.
1,4-di-N-oxide-3-phenylquinoxalines as anti-tuberculosis agents.
Figure 9.
1,4-di-N-oxide-3-phenylquinoxalines as anti-tuberculosis agents.
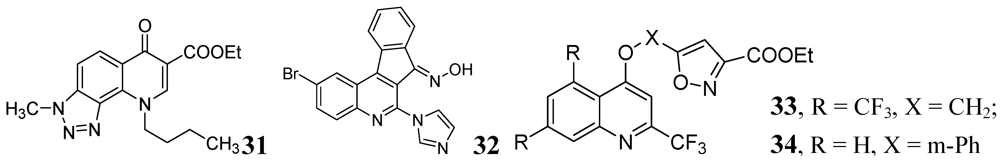
Carta et al. published antituberculosis activity of 3-methyl-9-substituted-6-oxo-6,9-dihydro-3H-[1,2,3]-triazolo[4,5-h]quinolone-carboxylic acids and their esters against wild-type H37Rv and 11 clinically isolated strains of Mtb. The most potent compound in the series was 31 with MIC90 = 0.5 μg/mL [73] (Figure 10).
The indeno[2,1-c]quinoline derivatives described by Upadhayaya et al. were shown to be active with MICs in the range of 0.39–0.78 μg/mL. Ester derivatives in compound 32 retained the activity (MIC of <0.39 μg/mL) [74].
Lilienkampf et al. described quinoline compounds 33 and 34 with MICs of 0.77 μM and 0.95 μM, respectively, against the replicating Mtb. These two compounds also had activity against non-replicating persistent bacteria as well as RMP-, INH-, and streptomycin- resistant Mtb strains [75].
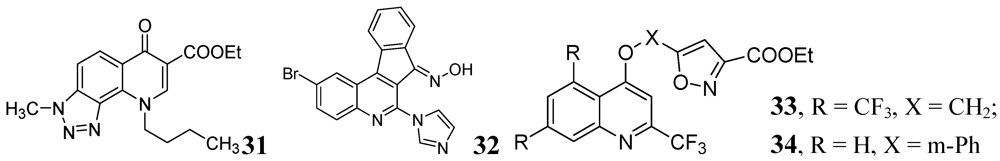
Figure 10.
Some other quinoline derivatives as anti-tuberculosis agents.
Figure 10.
Some other quinoline derivatives as anti-tuberculosis agents.
3.1.3.2. Pyrimidine and Purines
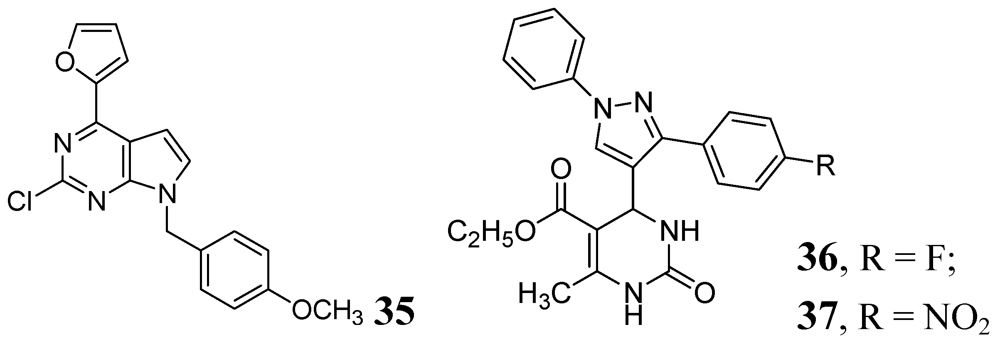
Various purine analogs were synthesized by Khoje et al. Compound 35 emerged as the most potent in the series (MIC 0.11 μM). The five most active compounds of the series were also evaluated against a panel of drug-resistant Mtb strains, where all of them retained activity. However, these compounds did not show good activity against non-replicating Mtb [76]. A series of dihydropyrimidines was examined by Trivedi et al. in 2010. Compounds 36 and 37 were the most potent in the series (MIC of 0.02 μg/mL) [77] (Figure 11).
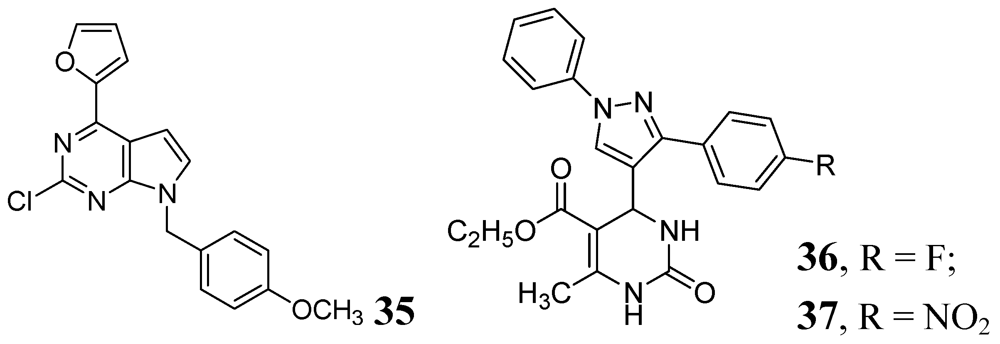
Figure 11.
Pyrimidine and Purine analogs as anti-tuberculosis agents.
Figure 11.
Pyrimidine and Purine analogs as anti-tuberculosis agents.
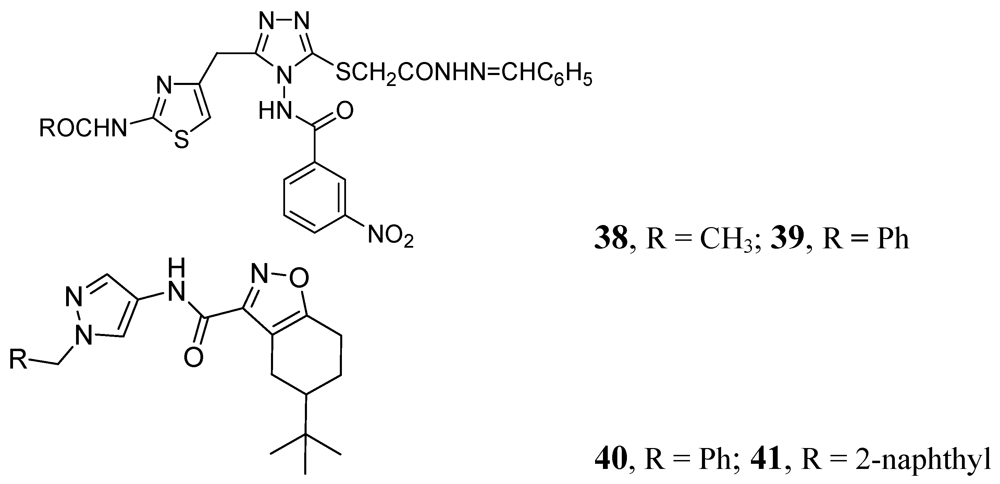
3.1.3.3. Azoles
Many publications have emerged on azoles for anti-TB activity. Some good representatives of this class are described below. In this class, compounds 38 and 39 demonstrated MICs of 0.78 and 0.39 μM, against Mtb H37Rv respectively [78]. Pantothenate is a key precursor of coenzyme A and acyl carrier protein, essential for many intracellular processes. The PS pathway is not present in humans. Velaparthi et al. reported in 2008 compounds 40 and 41 (Figure 12) as the best inhibitors (IC50 of < 100 nM) [79] (Figure 12).
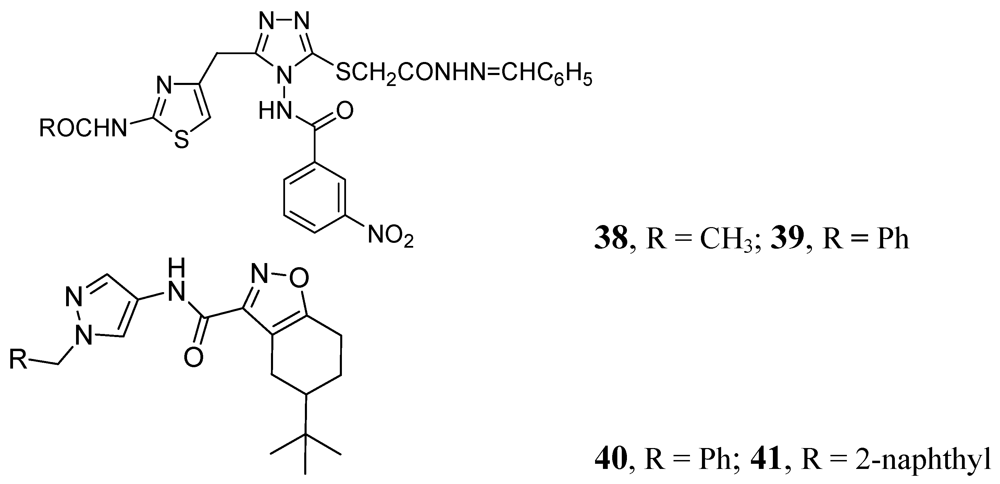
Figure 12.
Azole analogs as anti-tuberculosis agents.
Figure 12.
Azole analogs as anti-tuberculosis agents.
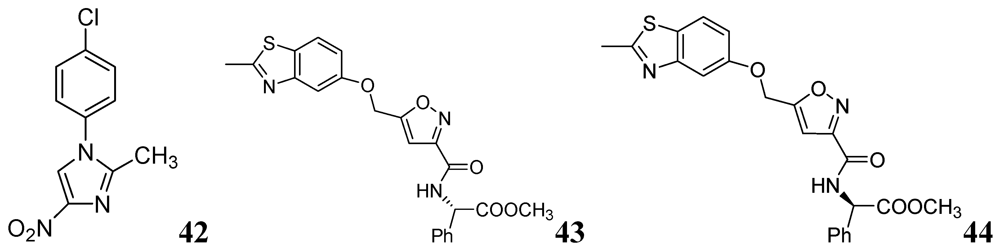
N-Aryl-C-nitroazoles were investigated by Walczak et al. against H37Rv (ATCC 27294) using the MABA assay. Compound 42 exhibited an MIC of 0.39 μg/mL [80]. A series of 2-methylbenzothiazole derivatives was described by Huang et al. Compounds 43 and 44 were found to be potent inhibitors of replicating Mtb H37Rv (MIC 1.4 and 1.9 μM, respectively) [81] (Figure 13).
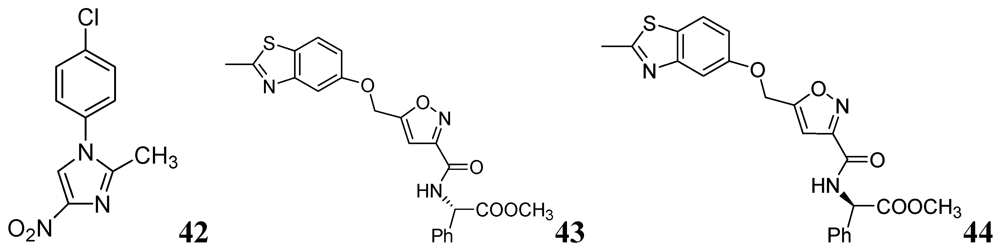
Figure 13.
N-Aryl-C-nitroazoles as anti-tuberculosis agents.
Figure 13.
N-Aryl-C-nitroazoles as anti-tuberculosis agents.
3.1.3.4. Azines
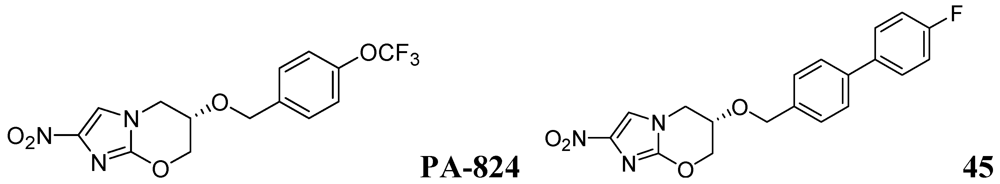
Palmer et al. investigated antitubercular activity of biphenyl analogs of PA-824, which is currently in phase II clinical trial. The most active compound, 45, had MICs of 0.015 and 1.4 μM in MABA and LORA assays, respectively. In a mouse model of acute Mtb infection, seven of the compounds showed substantially (>10-fold) improved efficacies and three of them were >200-fold more effective than PA-824 [82] (Figure 14).
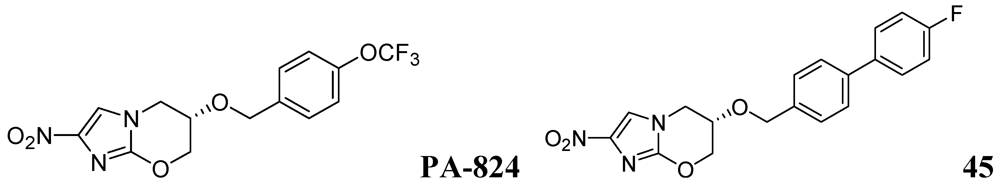
Figure 14.
Azines as anti-tuberculosis agents.
Figure 14.
Azines as anti-tuberculosis agents.
3.1.3.5. Pyridine hydrazides (INH analogs)
Several Schiff bases of INH were synthesized by Hearn et al. that showed good in vitro activity and protected tuberculosis infection in mice. A representative cyclohexanone derivative, 46, displayed an MIC of 0.03 μg/mL against Mtb H37Rv strain Erdman and exhibited reduction in mouse lung of 4.65 log CFU [83]. Lourenco et al. also reported a series of INH derivatives. Compound 47 exhibited significant in vitro activity (MIC 0.31 μg/mL) [84] (Figure 15).
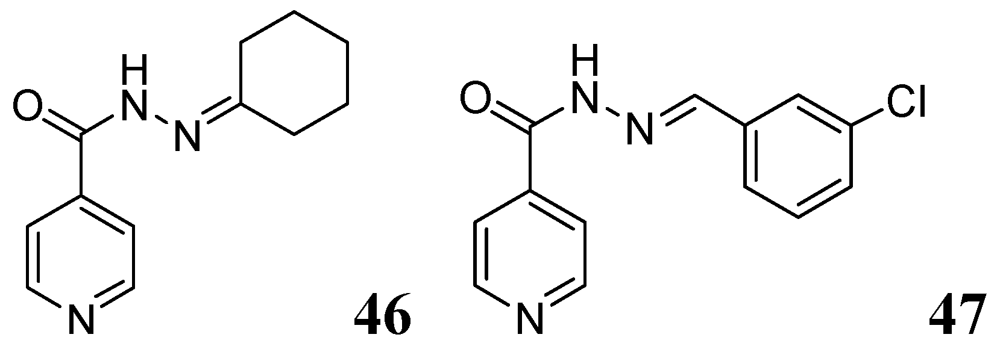
Figure 15.
INH analogs as anti-tuberculosis agents.
Figure 15.
INH analogs as anti-tuberculosis agents.
3.1.3.6. Miscellaneous
3.1.3.6.1. Artemisinin Analog
Artemisinin, commonly known as qinghaosu, is a natural sesquiterpene peroxide with a 1,2,4-trioxane nucleus, and is a highly active antimalarial agent. Miller et al. reported a mycobactin-artemisinin conjugate, 48, with submicromolar activity against different clinical strains of tuberculosis [85] (Figure 16).
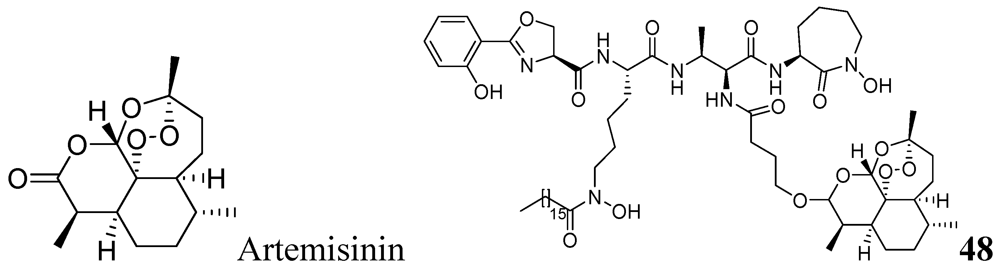
Figure 16.
Artemisinin analog as anti-tuberculosis agents.
Figure 16.
Artemisinin analog as anti-tuberculosis agents.
3.1.3.6.2. Macrolides
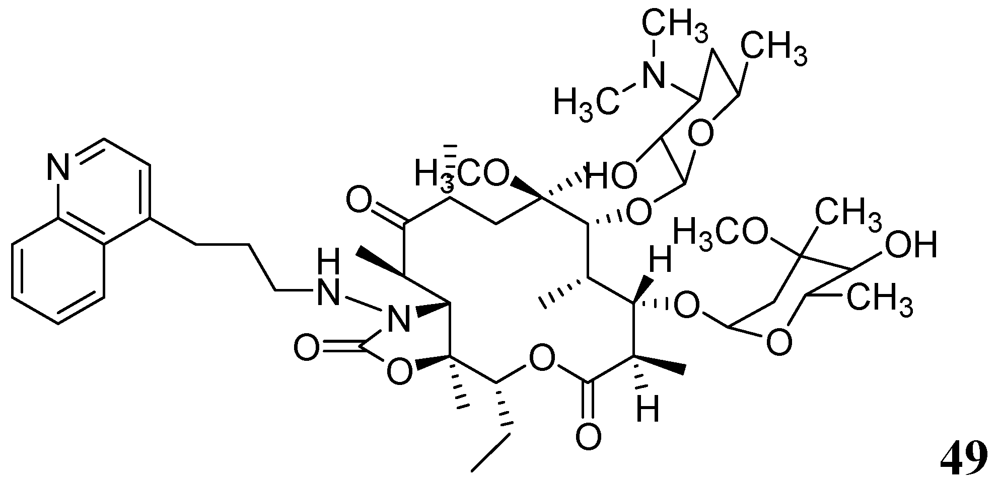
Falzari et al. reported descladinose derivatives of macrolides and ketolides. Many compounds demonstrated submicromolar MICs against Mtb. Compound 49 (RU66252) emerged as a promising inhibitor with an MIC of 0.25 μM [86] (Figure 17).
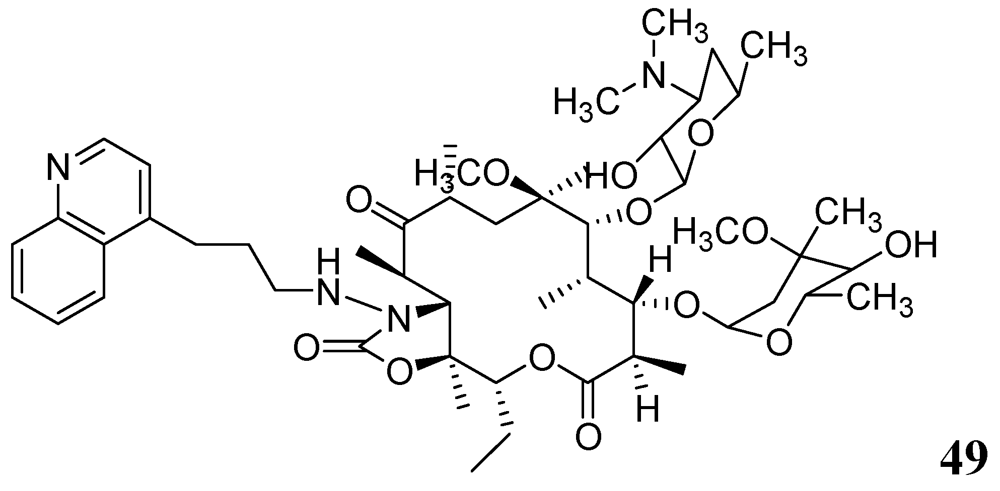
Figure 17.
Macrolide as anti-tuberculosis agents.
Figure 17.
Macrolide as anti-tuberculosis agents.
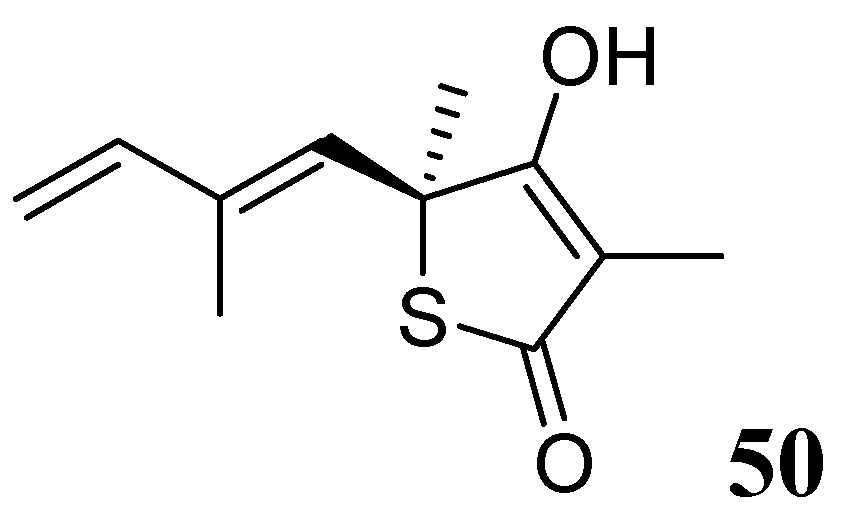
3.1.3.6.3. Thiolactomycin
Thiolactomycin (TLM), 50, is a natural product isolated from Nocardia and Streptomyces species. TLM is an inhibitor of the β-ketoacyl-acyl carrier protein synthase (KAS) enzymes, which are part of the bacterial fatty acid synthase pathway. TLM has MIC of 62.5 μM against Mtb [87,88]. TLM also inhibits human FAS-I enzyme [89], however, its lower affinity (IC50 100 μM) for this enzyme can make it worthy as a selective anti-tuberculosis agent [90] (Figure 18).
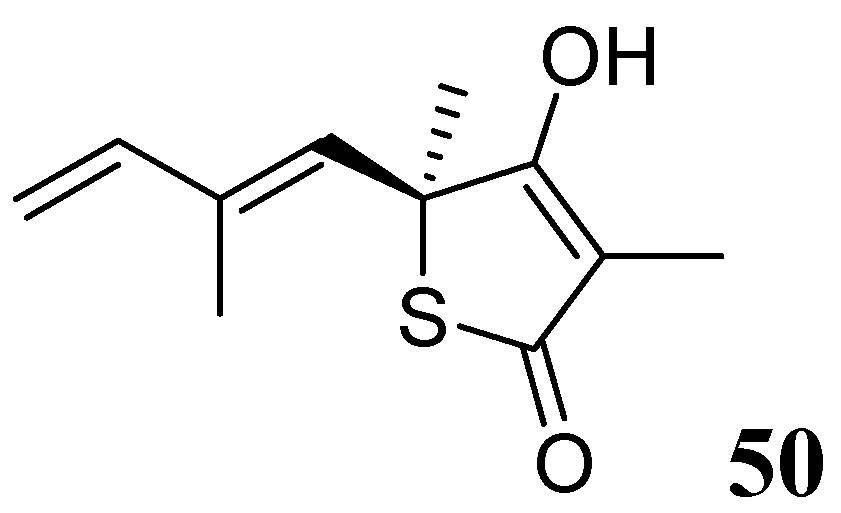
Figure 18.
Structure of thiolactomycin.
Figure 18.
Structure of thiolactomycin.
3.2. Molecules in Development
The following are compounds at various stages of preclinical and clinical development [91,92].
3.2.1. Molecules at Pre-Clinical Stage
3.2.1.1. CPZEN-45
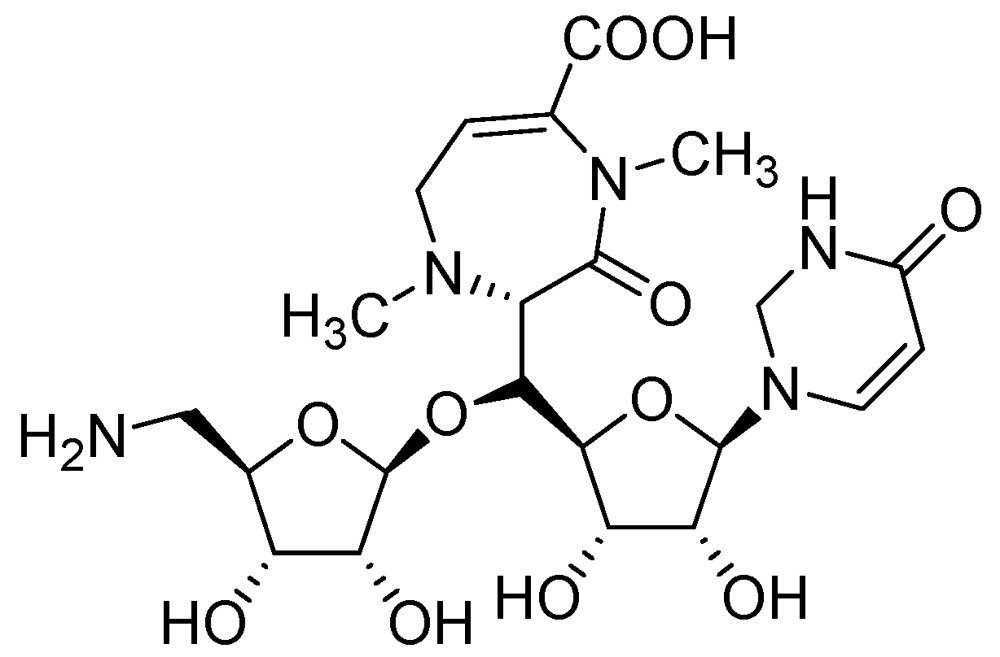
CPZEN-45 (MIC of 1.56 μg/mL, Mtb H37Rv and 6.25 μg/mL, MDR strain of Mtb) is a nucleoside antibiotic produced by Streptomyces spp. CPZEN-45 is active against both replicating and on-replicating Mtb in vitro. It is also effective against both drug sensitive and extremely drug resistant (XDR) Mtb in a mouse model of acute tuberculosis with 1–1.5 log CFU reduction in the lungs. Its mode of action is not specified [93] (Figure 19).
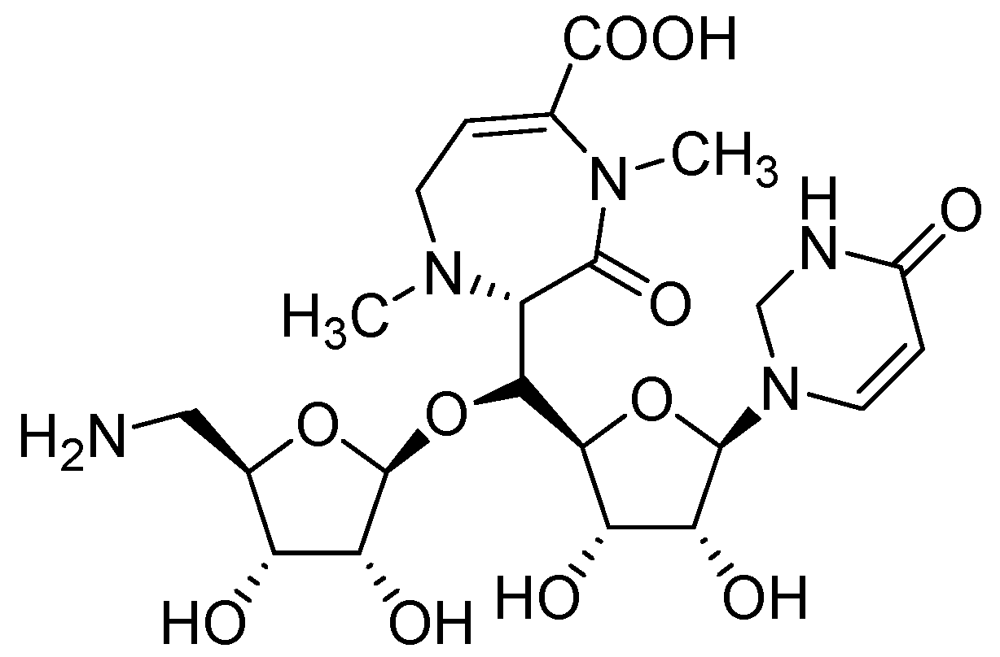
Figure 19.
Structure of CPZEN-45.
Figure 19.
Structure of CPZEN-45.
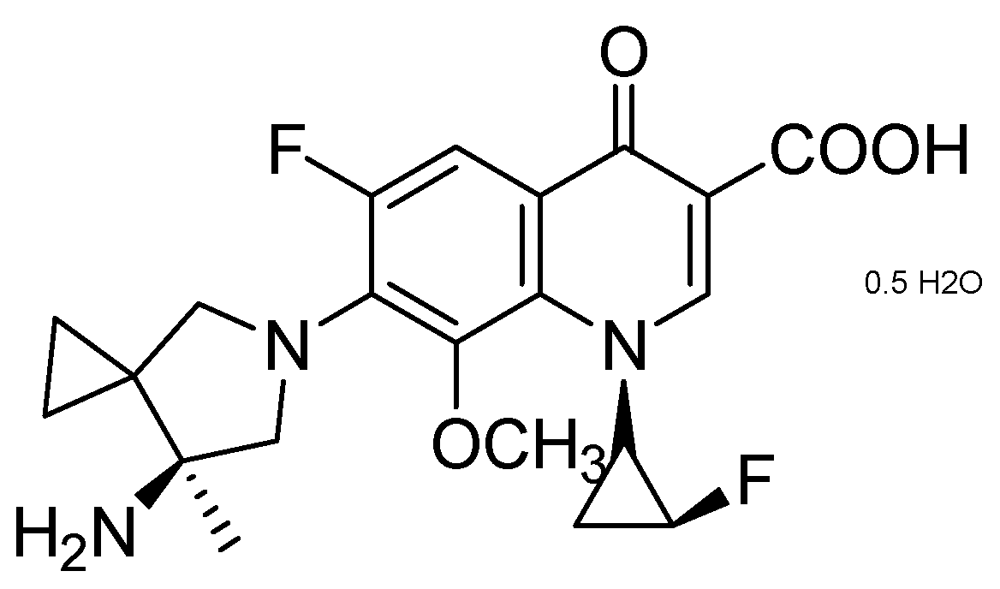
3.2.1.2. Quinolone DC-159a
DC-159a (MIC = 0.03 μg/mL) exhibited better early bactericidal activity (EBA) and higher log reduction of CFU in lungs against drug-susceptible, and quinolone-resistant (QR) MDR-TB, compared to that of moxifloxacin, gatifloxacin, levofloxacin and rifampicin. It acts by inhibiting DNA gyrase of wild-type and MDR-Mtb. In the QR MDR-TB infection model, it showed 2–3 times longer “mean survival days” which was superior to moxifloxacin, levofloxacin, INH and RMP (Figure 20).
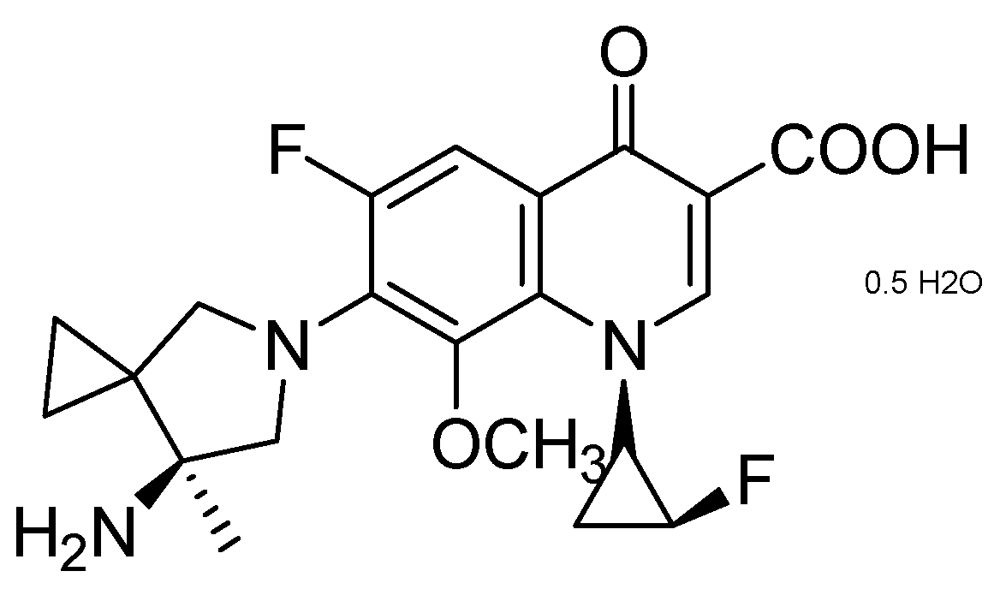
Figure 20.
Structure of DC-159a.
Figure 20.
Structure of DC-159a.
3.2.1.3. SQ-609
Sequella identified a promising candidate, SQ609, as the most potent among a new series of potential cell-wall inhibiting dipiperidines (MIC = 4 μg/mL). The precise mode of action of SQ 609 is unknown [94,95] (Figure 21).
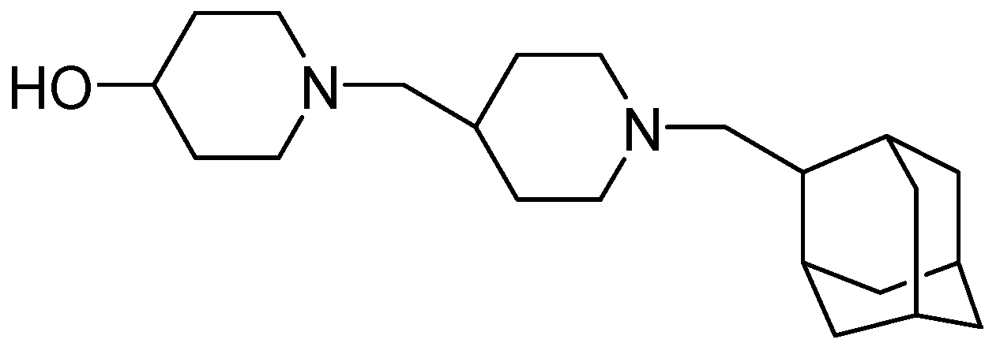
Figure 21.
Structure of SQ609.
Figure 21.
Structure of SQ609.
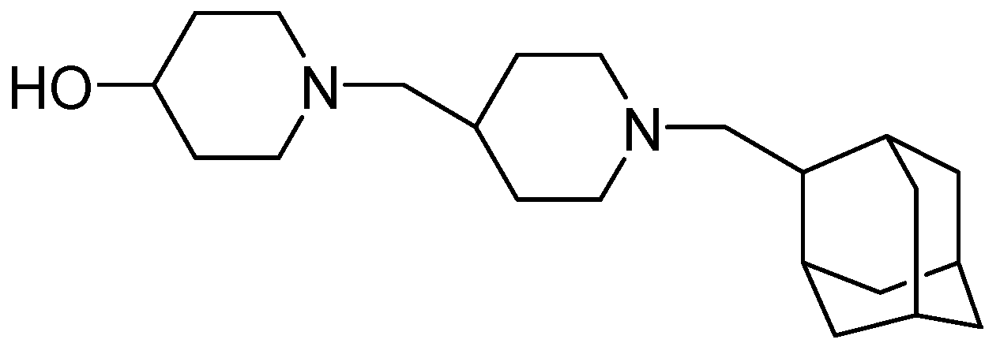
3.2.1.4. SQ-641
The enzyme translocase 1 (TL1), which is absent in eukaryotic cells, is an essential enzyme in bacteria for the biosynthesis peptidoglycan in the cell wall. SQ-641, which targets TL1, possesses activity against MDR clinical strains of Mtb (MIC = 0.5 μg/mL). It has shown efficacy in a mouse model of chronic TB by reducing the CFU in lungs of infected mice by 1.0 to 1.5 log [91,94] (Figure 22).
Figure 22.
Structure of SQ-641.
Figure 22.
Structure of SQ-641.
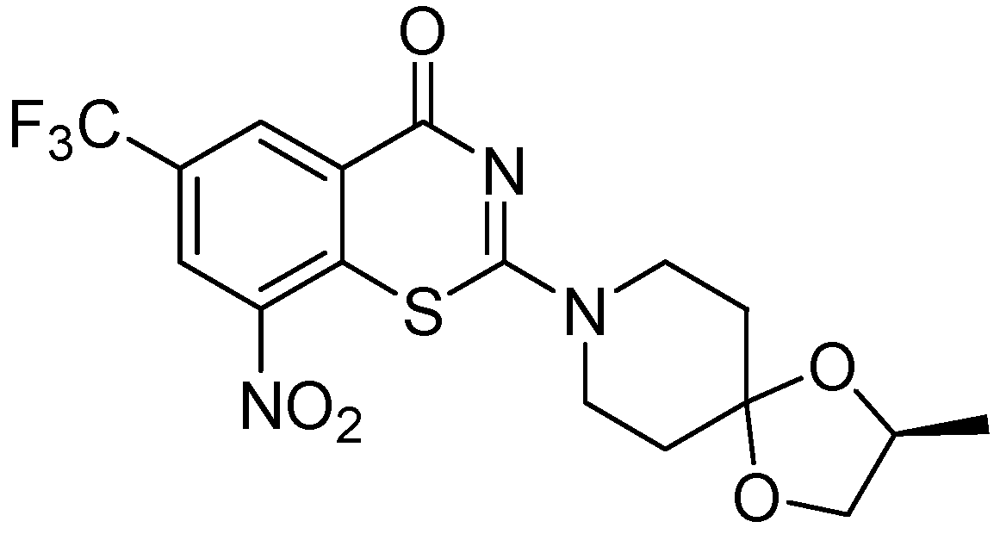
3.2.1.5. Benzothiazinone (BTZ-043)
BTZ-043 is highly active against Mtb (MIC = 1–10 ng/mL) and other actinobacteria. BTZ-043 also possesses activity against MDR and XDR strains. It inhibits cell wall biosynthesis, and targets the DprE1 (Rv3790) subunit of the enzyme decaprenylphosphoryl-beta-D-ribose 2'-epimerase. BTZ-043 has good oral bioavailability (Figure 23).
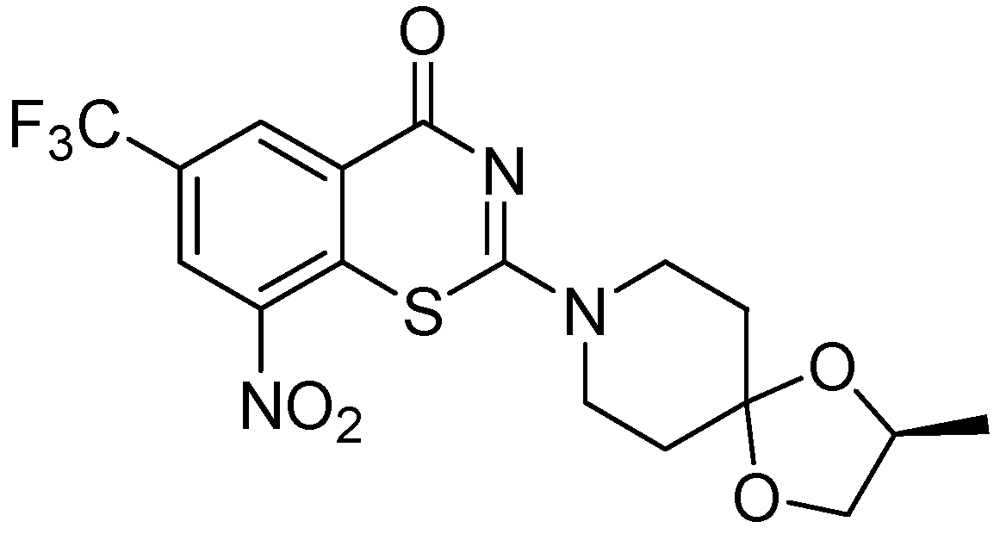
Figure 23.
Structure of BTZ-043.
Figure 23.
Structure of BTZ-043.
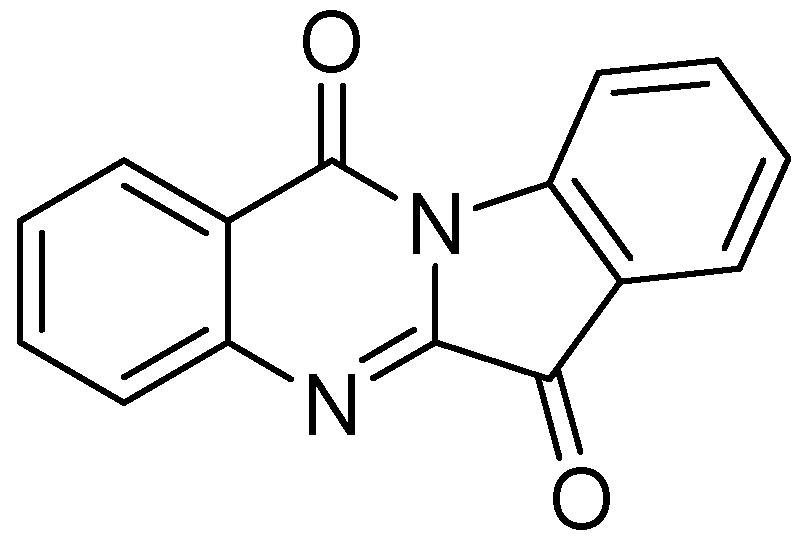
3.2.1.6. Tryptanthrin
Tryptanthrin (indolo [2,1-b]quinazolin-6,12-dione), is a natural product that was obtained from a Chinese plant, Strobilanthes cusia. It has broad-spectrum biological activities including anti-tuberculosis property. Tryptanthrin demonstrated MIC of 1 μg/mL against Mtb in BACTEC assay. It showed MIC values of 0.5–1.0 μg/mL against MDR-TB strains [95]. Preclinical evaluation of tryptanthrin has been conducted [96] (Figure 24).
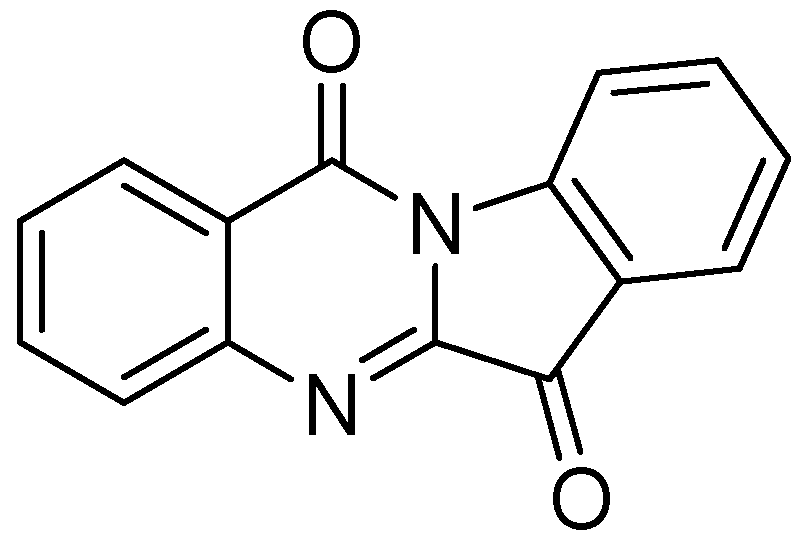
Figure 24.
Structure of tryptanthrin.
Figure 24.
Structure of tryptanthrin.
3.2.2. Molecules in Phase I Clinical Trials
AZD-5847
AZD-5847 is an oxazolidinone antibiotic (structure is not disclosed), originally developed for staphylococcal infections. It possesses an MIC90 of 1 μg/mL against laboratory Mtb strains and clinical isolates resistant to INH, RMP, streptomycin, EMB or OFX [97].
3.2.3. Molecules in Phase II Clinical Trials
3.2.3.1. PNU-100480
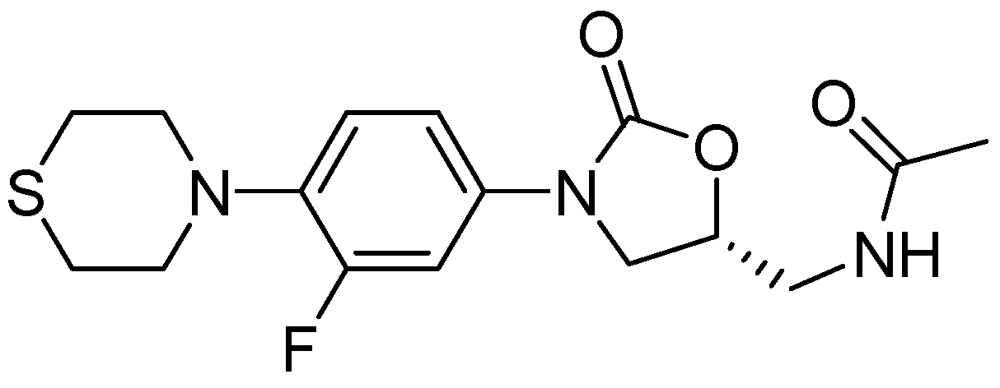
PNU-100480 is a linezolid derivative and is more active (MIC = 0.0625–0.5 μg/mL) than the parent compound and with similar efficacy to that of INH and RMP [98] (Figure 25).
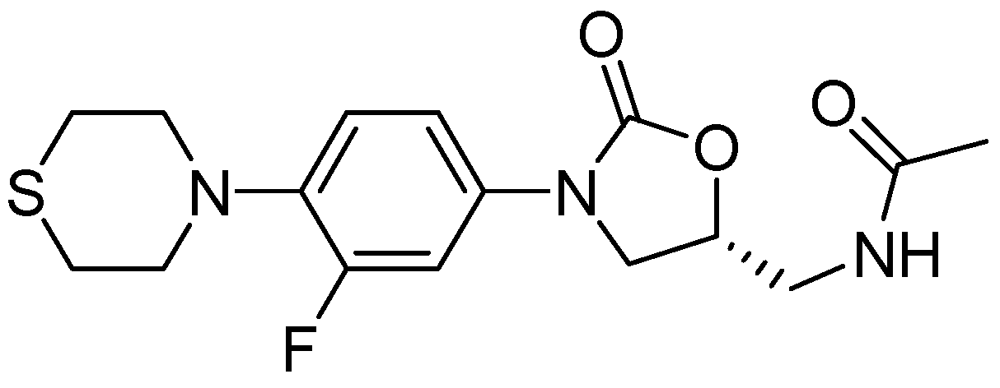
Figure 25.
Structure of PNU-100480.
Figure 25.
Structure of PNU-100480.
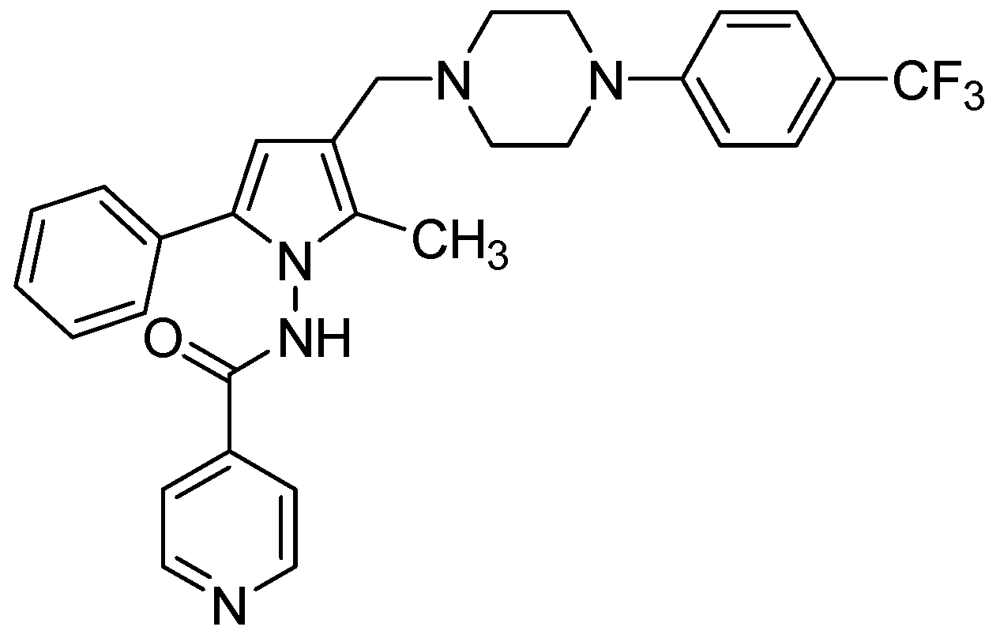
3.2.3.2. LL-3858 or Sudoterb
LL3858 (MIC90 0.25 μg/mL) in combination with current anti-TB drugs, is reported to clear Mtb from the lungs and spleens in less time than conventional therapy [99]. The mechanism of action for this class of compounds has not yet been established (Figure 26).
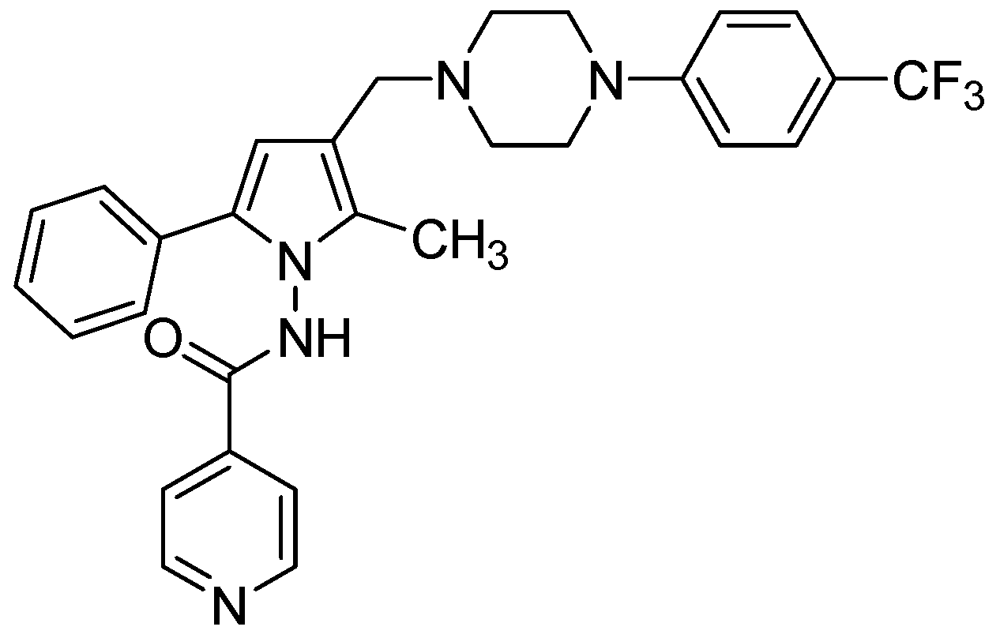
Figure 26.
Structure of LL3858.
Figure 26.
Structure of LL3858.
3.2.3.3. SQ-109
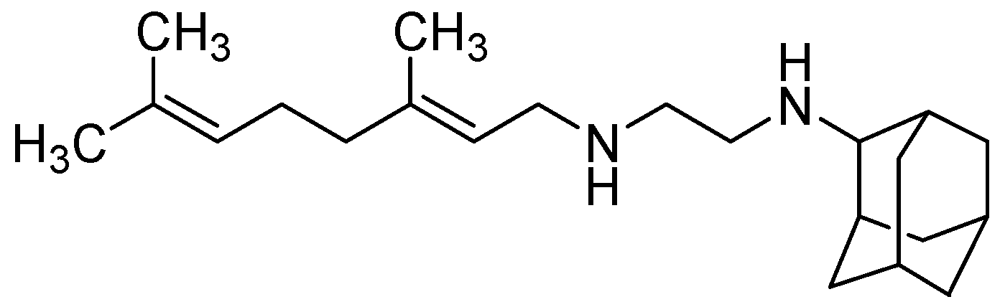
SQ109 has an MIC = 0.1–0.63 μg/mL [100]. In vivo it exhibited 1 to 2.0-log reduction in CFU counts in the lungs and spleens at 25 mg/kg. However, its oral bioavailability was found to be poor (only 4%) [101] (Figure 27).
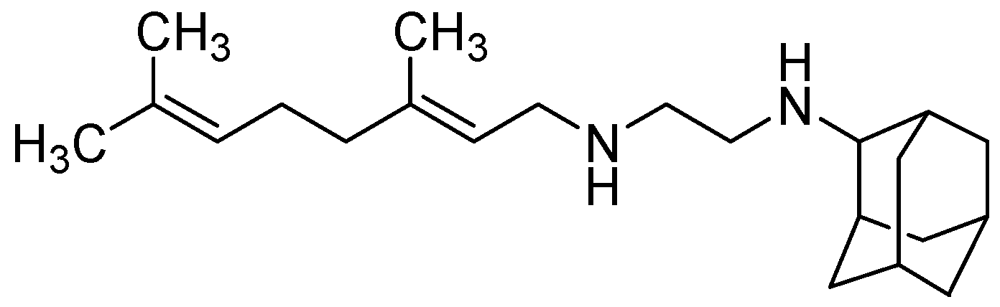
Figure 27.
Structure of SQ109.
Figure 27.
Structure of SQ109.
3.2.3.4. Nitroimidazoles
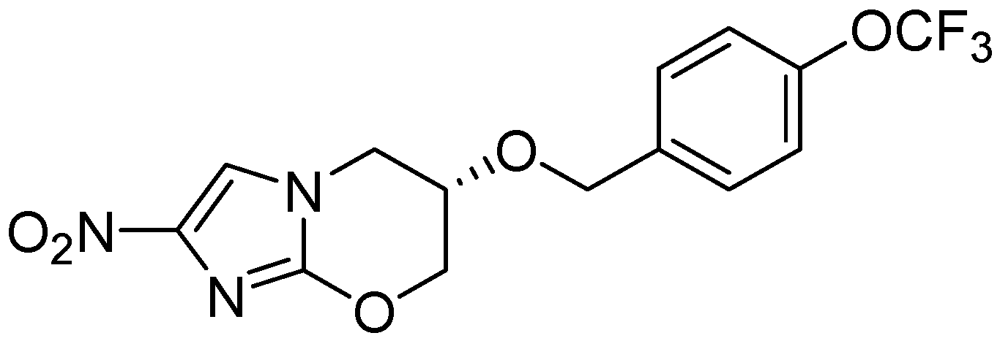
PA-824
PA-824 possesses an MIC in the range of 0.015 to 0.25 μg/ml and also retains activity against resistant isolates. It acts by inhibiting the synthesis of protein and cell wall lipids [102]. In a mouse model PA-824 was highly active for latent TB in combination with moxifloxacin [103]. Its minimum bactericidal dose (to reduce the lung CFU count by 99%) was found to be 100 mg/kg/day in murine studies. It is also effective against MDR strains and Mtb grown under oxygen depletion [104,105] (Figure 28).
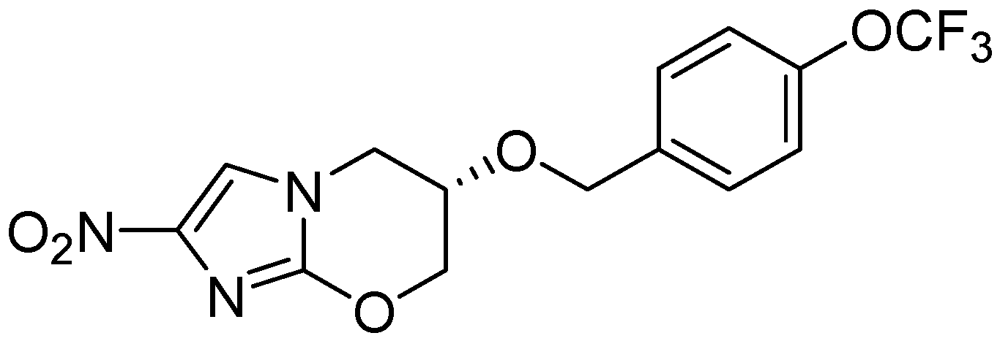
Figure 28.
Structure of PA-824.
Figure 28.
Structure of PA-824.
3.2.3.5. OPC-67683 (Delamanid)
OPC-67683 exhibited an MIC of 0.006 μg/mL [106]. In a mouse model, its efficacy was reported to be superior to existing anti-tuberculosis drugs without any evidence of cross-resistance. The mechanism of action of OPC-67683 is suggested to be similar to PA-824 [107] (Figure 29).
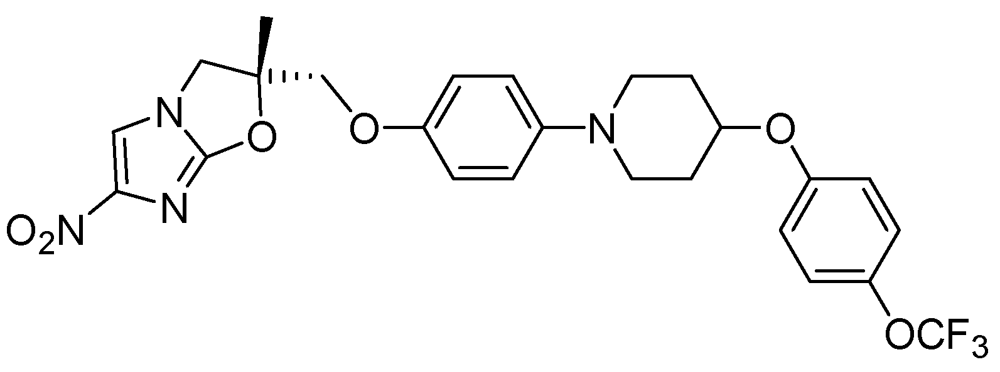
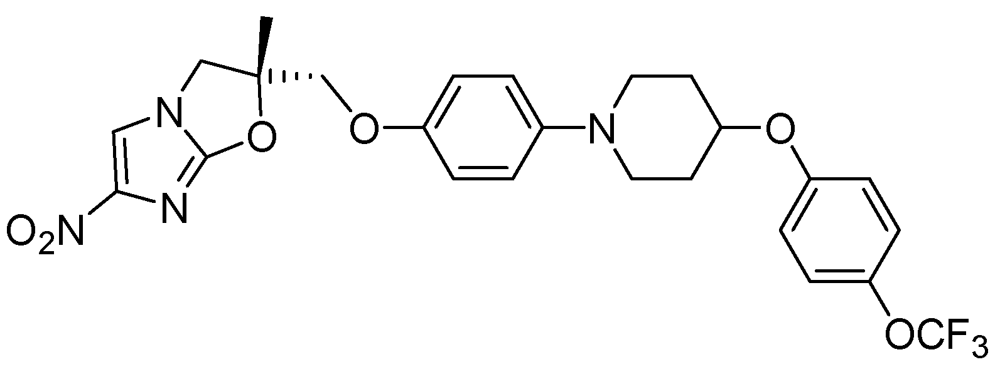
Figure 29.
Structure of OPC-67683.
Figure 29.
Structure of OPC-67683.
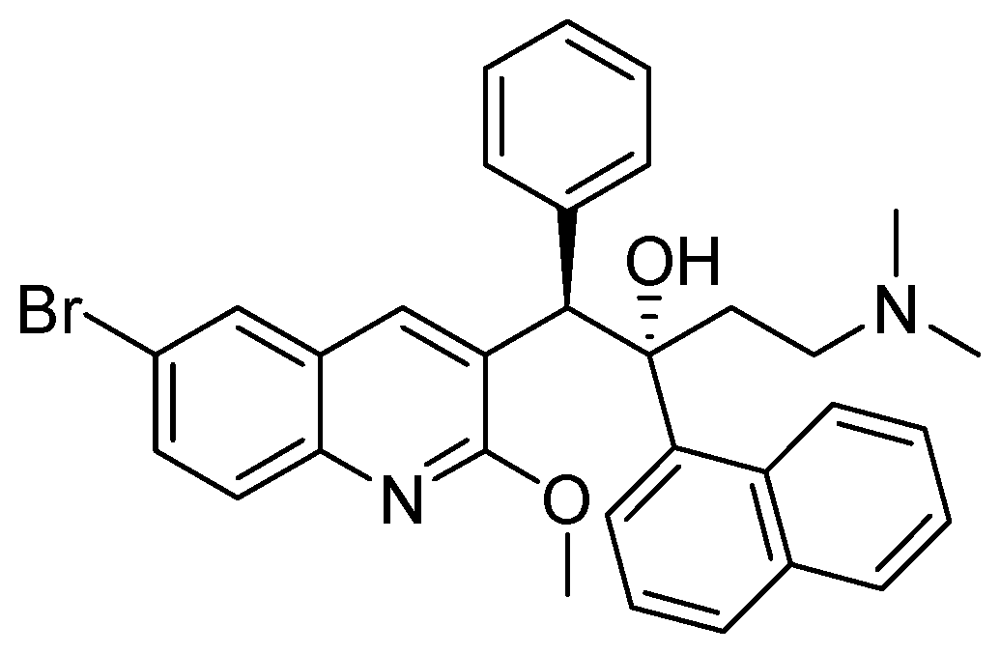
3.2.3.6. TMC-207 or R-207910 or Bedaquiline
The MIC value of TMC-207 ranges from 0.002 to 0.06 µg/mL for drug susceptible and drug resistant (INH, RMP, streptomycin, EMB, PZA and moxifloxacin) strains. It works on the proton pump of ATP synthase [108,109]. In mice, a single dose had bactericidal potency for about eight days. When used as monotherapy, a single dose of TMC-207 was as potent as the triple combination of RMP, INH, and PZA and was more active than RMP alone (Figure 30).
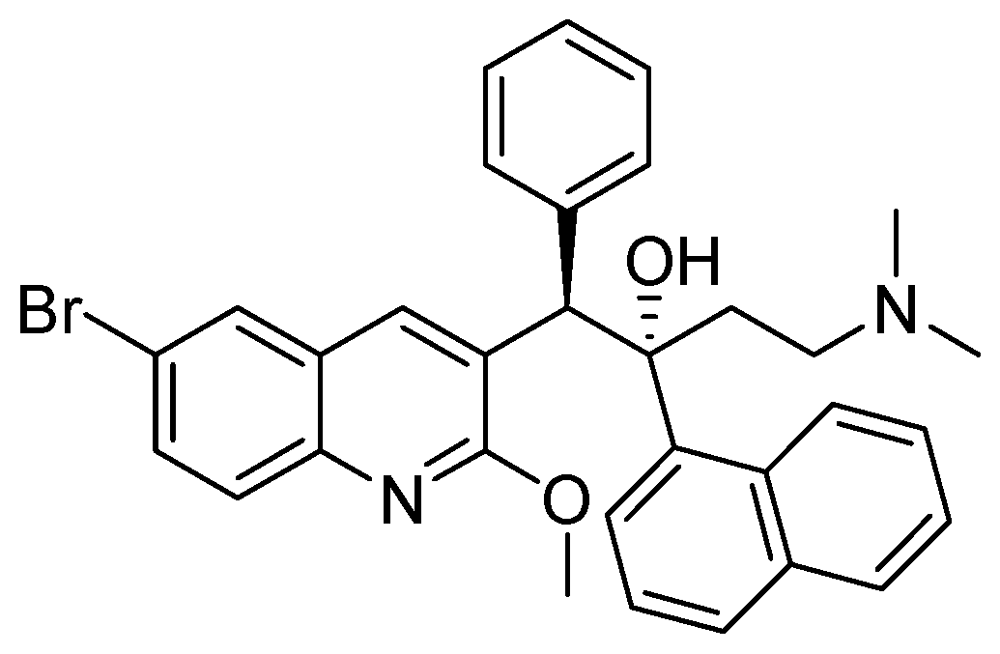
Figure 30.
Structure of TMC-207.
Figure 30.
Structure of TMC-207.
3.2.3.7. Linezolid for the Treatment of MDR-Tuberculosis
Linezolid is an approved antibacterial drug (MIC90 1–2 μg/mL) but has not been approved for TB [110]. One of the major concerns for its use as an anti-TB drug is the lack of information on its efficacy [111]. Its long-term use indicated thrombocytopenia, neuropathy and haematopoietic suppression [112] (Figure 31).
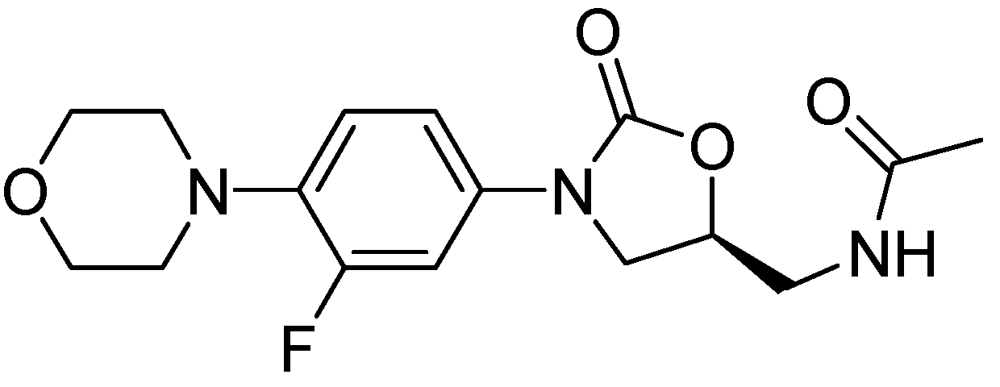
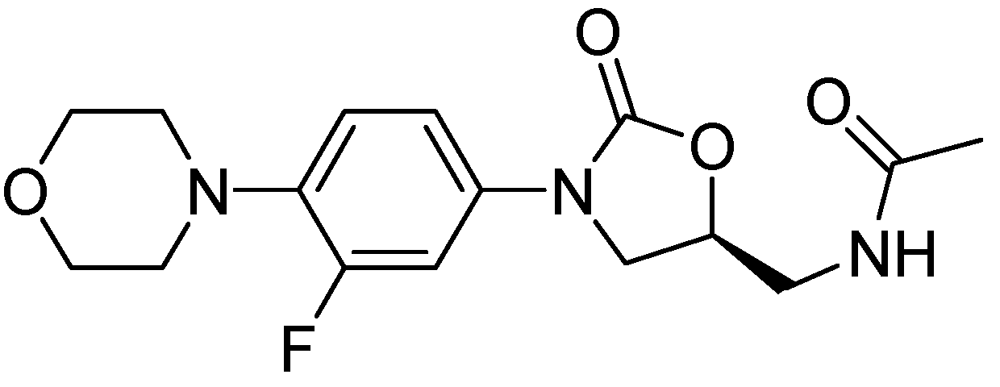
Figure 31.
Structure of linezolid.
Figure 31.
Structure of linezolid.
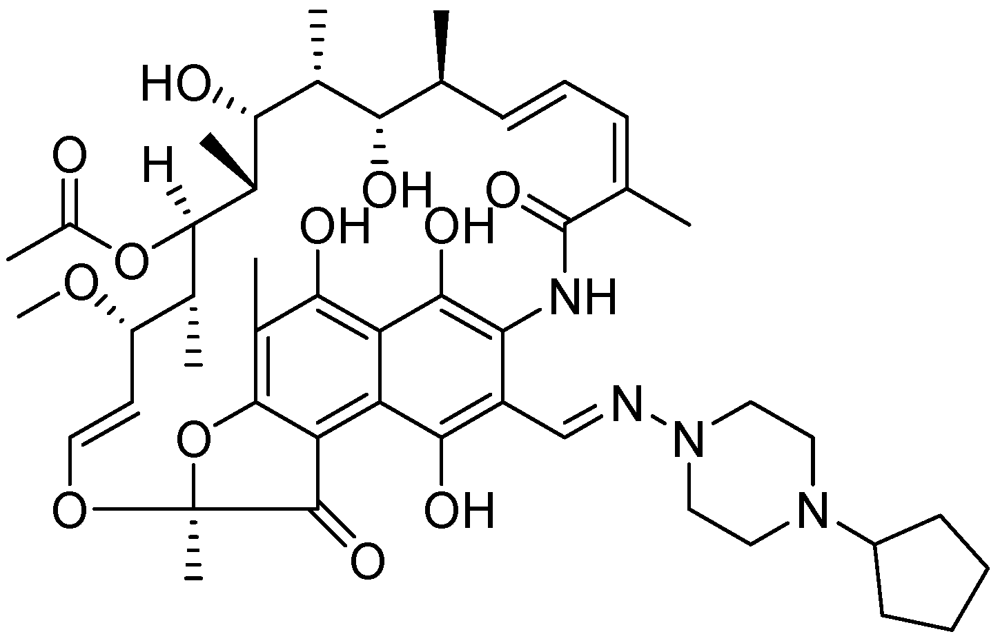
3.2.3.8. Rifapentine (TBTC Study)
Rifapentine is a derivative of rifampicin with an MIC of 0.03 μg/mL [113]. Its mode of action is similar to that of rifampicin [114]. Rifapentine can be used to treat latent TB in combination with either moxifloxacin or INH [103] (Figure 32).
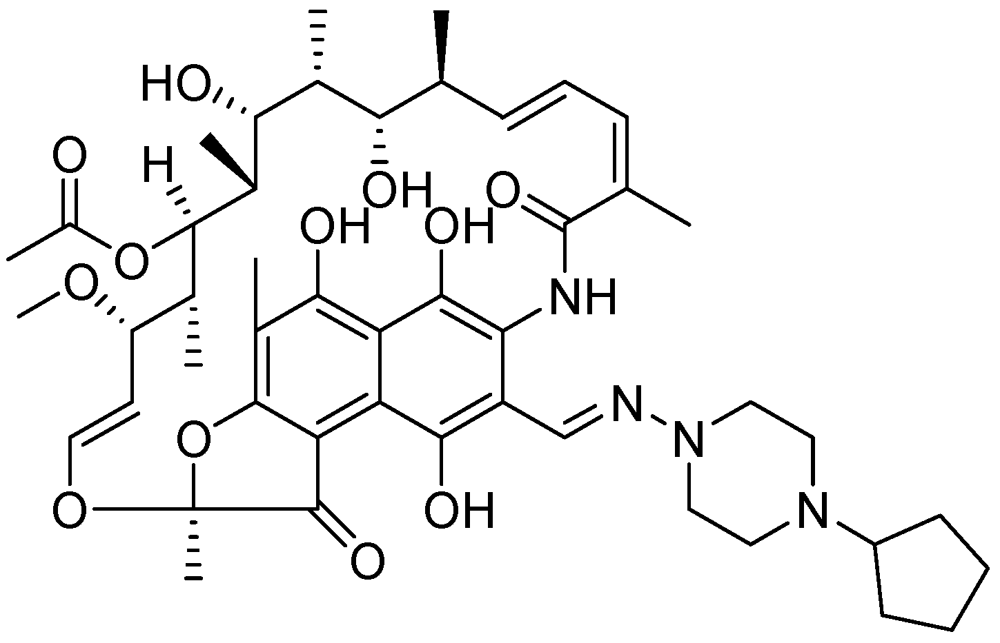
Figure 32.
Structure of rifapentine.
Figure 32.
Structure of rifapentine.
3.2.4. Molecules in Phase III Clincal Trials
Fluoroquinolones (Moxifloxacin and Gatifloxacin)
Moxifloxacin, a broad-spectrum antibiotic (400 mg/day dose, MIC of 0.5 μg/mL), is active against both gram-positive and gram-negative bacteria. It displayed early bactericidal activity comparable to INH and rifampin in humans [115,116]. It binds to DNA gyrase and topisomerase IV, which are involved in bacterial replication. Moxifloxacin has no cross-resistance to other antituberculosis drug classes and has been shown to display good activity against MDR strains [117]. However, it has CNS side effects and drug interactions with other fluoroquinolones. Moxifloxacin has not been reported to be safe or effective in children younger than 18 year or in pregnant or lactating women [118]. Nuermberger et al. found that substituting moxifloxacin for INH shortens the duration of therapy for active disease much better than does substituting moxifloxacin for EMB [119] (Figure 33).
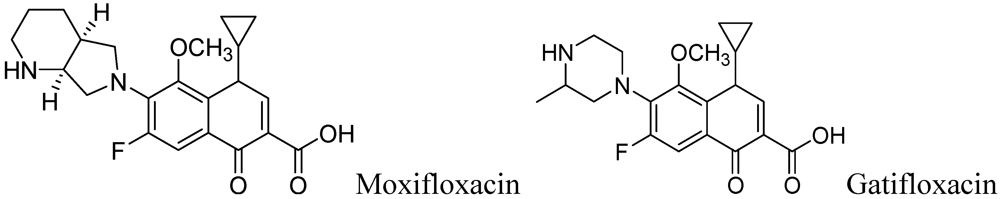
Figure 33.
Structures of moxifloxacin and gatifloxacin.
Figure 33.
Structures of moxifloxacin and gatifloxacin.
Gatifloxacin is also a broad-spectrum antibiotic (dosage of 400 mg/day) and has the same mechanism of action as moxifloxacin. It is active against occasionally dividing Mtb, but not for dormant bacteria [120]. However, it can cause CNS toxicity and has been associated with increases in insulin levels among diabetics. Like moxifloxacin, it has also not been shown to be safe or effective in children younger than 18 years or in pregnant or lactating women.
4. Conclusions
Drug resistance is a critical issue in the treatment of TB. Combined and intensive efforts are required to discover new classes of anti-tuberculosis drugs, otherwise TB could become untreatable in the near future. Currently, several groups/institutions are working together to achieve this goal. These efforts should be continued and intensified to fight this ancient but re-emerging disease. To augment and bolster the development of new drugs for TB, government, private and public authorities need to enhance financial support for research at all levels, and modify regulations to ease the process of evaluation, validation and approval of new drugs. In addition, education and awareness by government, public and private agencies must contribute to preventing the spread of TB and drug resistant MDR or XDR TB.
References
- World Health Organisation (WHO). WHO Global Task Force outlines measures to combat XDR-TB worldwide (17 October 2006). Available online: http://www.who.int/mediacentre/news/notes/2006/np29/en/index.html/ (accessed on 4 April 2012).
- Rowland, K. Totally drug-resistant TB emerges in India, Nature News. Available online: http://www.nature.com/news/totally-drug-resistant-tb-emerges-in-india-1.9797/ (accessed on 9 June 2012).
- Kaufmann, S.H.E. Tuberculosis vaccines—A new kid on the block. Nat. Med. 2011, 17, 159–160. [Google Scholar] [CrossRef]
- Nau, R.; Prange, H.W.; Menck, S.; Kolenda, H.; Visser, K.; Seydel, J.K. Penetration of rifampicin into the cerebrospinal fluid of adults with uninflamed meninges. J. Antimicrob. Chemother. 1992, 29, 719–724. [Google Scholar] [CrossRef]
- Tomioka, H. Current status of some antituberculosis drugs and the development of new antituberculous agents with special reference to their in vitro and in vivo antimicrobial activities. Curr. Pharm. Des. 2006, 12, 4047–4070. [Google Scholar] [CrossRef]
- Aristoff, P.A.; Garcia, G.A.; Kirchhoff, P.D.; Hollis Showalter, H.D. Rifamycins-obstacles and opportunities. Tuberculosis 2010, 90, 94–118. [Google Scholar] [CrossRef]
- The American Thoracic Society [ATS]. Available online: http://www.thoracic.org/assemblies/mtpi/resources/istc-report.pdf (accessed on 4 April 2012).
- Telenti, A.; Imboden, P.; Marchesi, F.; Schmidheini, T.; Bodmer, T. Direct, automated detection of rifampin-resistant mycobacterium tuberculosis by polymerase chain reaction and single-strand conformation polymorphism analysis. Antimicrob. Agents Chemother. 1993, 37, 2054–2058. [Google Scholar] [CrossRef]
- International Programme on Chemical Safety [INCHEM]. Available online: http://www.inchem.org/documents/pims/pharm/rifam.htm#SectionTitle:2.1%20Main%20—risks%20and%20target%20organs/ and reference therein/ (accessed on 4 April 2012).
- Drobac, P.C.; del Castillo, H.; Sweetland, A.; Anca, G.; Joseph, J.K.; Furin, J.; Shin, S. Treatment of multidrug-resistant tuberculosis during pregnancy: Long-term follow-up of 6 children with intrauterine exposure to second-line agents. Clin. Infect. Dis. 2005, 40, 1689–1692. [Google Scholar] [CrossRef]
- Peters, C.; Nienhaus, A. Case report-tuberculosis in a health care worker during pregnancy. Pneumologie 2008, 62, 695–698. [Google Scholar] [CrossRef]
- Baciewicz, A.M.; Chrisman, C.R.; Finch, C.K.; Self, T.H. Update on rifampin and rifabutin drug interactions. Am. J. Med. Sci. 2008, 335, 126–136. [Google Scholar] [CrossRef]
- Singh, B.; Mitchison, D.A. Bactericidal activity of streptomycin and isoniazid against tubercle bacilli. Br. Med. J. 1954, 130, 130–132. [Google Scholar]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Suarez, J.; Ranguelova, K.; Jarzecki, A.A.; Manzerova, J.; Krymov, V.; Zhao, X.; Yu, S.; Metlitsky, L.; Gerfen, G.J.; Magliozzo, R.S. An oxyferrous heme/protein-based radical intermediate is catalytically competent in the catalase reaction of Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 2009, 284, 7017–7029. [Google Scholar]
- McIlleron, H.; Willemse, M.; Werely, C.J.; Hussey, G.D.; Schaaf, H.S.; Smith, P.J.; Donald, P.R. Isoniazid plasma concentrations in a cohort of South African children with tuberculosis: Implications for international pediatric dosing guidelines. Clin. Infect. Dis. 2009, 48, 1547–1553. [Google Scholar] [CrossRef]
- Ellard, G.A.; Gammon, P.T. Pharmacokinetics of Isoniazid Metabolism in Man. J. Pharmacokinet. Biopharm. 1976, 4, 83–113. [Google Scholar] [CrossRef]
- International Programme on Chemical Safety [INCHEM]. Available online: http://www.inchem.org/documents/pims/pharm/pim288.htm#SectionTitle:2.1%20%20Main%20risks%20and%20target%20organs/ (accessed on 4 April 2012).
- Heifets, L. Antimycobacterial agents: Pyrazinamide. In Antimicrobial Therapy and Vaccines; Yu, V.L., Merigan, T.C., Barriere, S.L., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1999. [Google Scholar]
- Hong Kong Chest Service, Medical Research Council. Controlled trial of four thrice weekly regimens and a daily regimen given for 6 months for pulmonary tuberculosis. Lancet 1981, 1, 171–174.
- Donald, P.R.; Seifart, H. Cerebrospinal fluid pyrazinamide concentrations in children with tuberculous meningitis. Pediatr. Infect. Dis. J. 1988, 7, 469–471. [Google Scholar] [CrossRef]
- Zhang, Y.; Mitchison, D. The Curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar]
- Zimhony, O.; Vilcheze, C.; Arai, M.; Welch, J.; Jacobs, W.R. Pyrazinoic acid and its n’Propyl Ester Inhibit Fatty Acid Synthase I in Replicating Tubercle Bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef]
- Mitchison, D.; Davies, G. The chemotherapy of tuberculosis: Past, present and future. Int. J. Tuberc. Lung Dis. 2012, 16, 724–732. [Google Scholar] [CrossRef]
- Scorpio, A.; Zhang, Y. Mutations In pnca, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Scorpio, A.; Lindholm-Levy, P.; Heifets, L.; Gilman, R.; Siddiqi, S.; Cynamon, M.; Zhang, Y. Characterization of pncA Mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1997, 41, 540–543. [Google Scholar]
- Jureen, P.; Werngren, J.; Toro, J.C.; Hoffner, S. Pyrazinamide resistance and PNCA gene mutations in Mycobacterium tuberculosis. Antimicrob. Agents. Chemother. 2008, 52, 1852–1854. [Google Scholar]
- Lacroix, C.; Phan Hoang, T.; Nouveau, J.; Guyonnaud, C.; Laine, G.; Duwoos, H.; Lafont, O. Pharmacokinetics of pyrazinamide and its metabolites in healthy subjects. Eur. J. Clin. Pharmacol. 1989, 36, 395–400. [Google Scholar] [CrossRef]
- Forget, E.J.; Menzies, D. Adverse reactions to first-line antituberculosis drugs. Expert Opin. Drug Saf. 2006, 5, 231–249. [Google Scholar] [CrossRef]
- Telenti, A.; Philipp, W.J.; Sreevatsan, S.; Bernasconi, C.; Stockbauer, K.E.; Wieles, B.; Musser, J.M.; Jacobs W.R., Jr. The Emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat. Med. 1997, 3, 567–570. [Google Scholar] [CrossRef]
- Belanger, A.E.; Besra, G.S.; Ford, M.E.; Mikusova, K.; Belisle, J.T.; Brennan, P.J.; Inamine, J.M. The Embab genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. USA 1996, 93, 11919–11924. [Google Scholar]
- Peloquin, C.A.; Bulpitt, A.E.; Jaresko, G.S.; Jelliffe, R.W.; Childs, J.M.; Nix, D.E. Pharmacokinetics of ethambutol under fasting conditions, with food, and with antacids. Antimicrob. Agents Chemother. 1999, 43, 568–572. [Google Scholar]
- Ethambutol (Ethambutol Hydrochloride)-Indications and Dosage. Available online: http://www.druglib.com/druginfo/ethambutol/indications_doses/ (accessed on 4 April 2012).
- Lim, S.A. Ethambutol-associated optic neuropathy. Ann. Acad. Med. Singapore 2006, 35, 274–278. [Google Scholar]
- Source partly from: North Dakota Department of Health. 2011. Available online: http://www.ndhealth.gov/disease/tb/Documents/Second line TB drugs.pdf (accessed on 4 April 2012).
- Heifets, L. MIC as a quantitative measurement of susceptibility of M. Avium to seven antituberculosis drugs. Antimicrob. Agents Chemother. 1988, 32, 1131–1136. [Google Scholar] [CrossRef]
- Heifets, L.; Lindholm-Levy, P.A. Comparison of bactericidal activities of streptomycin, amikacin, kanamycin, and capreomycin against M. Avium and M. tuberculosis. Antimicrob. Agents Chemother. 1989, 33, 1298–1303. [Google Scholar] [CrossRef]
- Johansen, S.K.; Maus, C.E.; Plikaytis, B.B.; Douthwaite, S. Douthwaite S. Capreomycin binds across the ribosomal subunit interface using Tlya-encoded 2'-O-methylations in 16S and 23S rRNAs. Mol. Cell 2006, 23, 173–182. [Google Scholar] [CrossRef]
- Centre for Disease Control and Prevention [CDC] (1994). Available online: http://wonder.cdc.gov/wonder/prevguid/p0000413/p0000413.asp#head006001000000000/ (accessed on 4 April 2012).
- Lu, Y.; Zheng, M.Q.; Wang, B.; Zhao, W.J.; Li, P.; Chu, N.H.; Liang, B.W. Activities of clofazimine against Mycobacterium tuberculosis in vitro and in vivo. Zhonghua Jie He He Hu Xi Za Zhi 2008, 31, 752–755. [Google Scholar]
- Rastogi, N.; Goh, K.S.; Bryskier, A.; Devallois, A. In vitro activities of levofloxacin used alone and in combination with first- and second-line antituberculous drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1996, 40, 1610–1616. [Google Scholar]
- Vacher, S.; Pellegrin, J.L.; Leblanc, F.; Fourche, J.; Maugein, J. Comparative antimycobacterial activities of ofloxacin, ciprofloxacin and grepafloxacin. J. Antimicrob. Chemother. 1999, 44, 647–652. [Google Scholar] [CrossRef]
- Trimble, K.A.; Clark, R.B.; Sanders, W.E., Jr.; Frankel, J.W.; Cacciatore, R.; Valdez, H. Activity of ciprofloxacin against Mycobacteria in vitro: Comparison of BACTEC and macrobroth dilution methods. J. Antimicrob. Chemother. 1987, 19, 617–622. [Google Scholar] [CrossRef]
- Inderlied, C.B.; Salfinger, M. Antimycobacterial agents and susceptibility tests. In Manual of Clinical Microbiology, III; Murray, P.R., Baron, E.J., Pfaller, M.A., Tenover, F.C., Yolken, R.H., Eds.; ASM Press: Washington, DC, USA, 1999; pp. 1601–1623. [Google Scholar]
- Vanheusden, V.; Munier-Lehmann Froeyen, M.; Busson, R.; Rozenski, J.; Herdewijn, P.; van Calenbergh, S. Discovery of bicyclic thymidine analogues as selective and high-affinity inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J. Med. Chem. 2004, 47, 6187–6194. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar]
- Johar, M.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Synthesis and in vitro anti-mycobacterial activity of 5-substituted pyrimidine nucleosides. Bioorg. Med. Chem. 2005, 13, 6663–6671. [Google Scholar] [CrossRef]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis Isolates by using the microplate alamar blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar]
- Vanheusden, V.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; van Calenbergh, S. Synthesis and evaluation of thymidine-5O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg. Med. Chem. Lett. 2002, 12, 2695–2698. [Google Scholar] [CrossRef]
- Vanheusden, V.; van Rompaey, P.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; van Calenbergh, S. Thymidine and thymidine-5'O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg. Med. Chem. Lett. 2003, 13, 3045–3048. [Google Scholar]
- Pochet, S.; Dugue, L.; Labesse, G.; Delepierre, M.; Munier-Lehmann, H. Comparative study of purine and pyrimidine nucleoside analogues acting on the thymidylate kinases of Mycobacterium tuberculosis and of humans. ChemBioChem 2003, 4, 742–747. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Design and studies of novel 5-substituted alkynylpyrimidine nucleosides as potent inhibitors of mycobacteria. J. Med. Chem. 2005, 48, 7012–7017. [Google Scholar]
- Johar, M.; Manning, T.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, Y.; Kumar, R. Growth inhibition of Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium in vitro: Effect of 1-b-D-2'-b-D-arabinofuranosyl and 1-(2-deoxy-2-fluoro-b-D-2'-ribofuranosyl) pyrimidine nucleoside. J. Med. Chem. 2007, 50, 3696–3705. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Srivastav, N.C.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Inhibition of Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium avium by novel dideoxy nucleosides. J. Med. Chem. 2007, 50, 4766–4774. [Google Scholar]
- Srivastav, N.C.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Studies on acyclic pyrimidines as inhibitors of mycobacteria. Bioorg. Med. Chem. 2007, 15, 2045–2053. [Google Scholar] [CrossRef]
- Shakya, N.; Srivastav, N.C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. 3-Bromo analogues of pyrimidine nucleosides as a new class of potent inhibitors of Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4130–4140. [Google Scholar]
- Bermudez, L.E.; Inderlied, C.B.; Kolonoski, P.; Wu, M.; Aralar, P.; Young, L.S. Telithromycin is active against Mycobacterium avium in mice despite lacking significant activity in standard in vitro and macrophage assays and is associated with low frequency of resistance during treatment. Antimicrob. Agents Chemother. 2001, 45, 2210–2214. [Google Scholar] [CrossRef]
- Srivastav, N.C.; Rai, D.; Tse, C.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Inhibition of mycobacterial replication by pyrimidines possessing various c-5 functionalities and related 20-deoxynucleoside analogues using in vitro and in vivo models. J. Med. Chem. 2010, 53, 6180–6187. [Google Scholar] [CrossRef]
- Kogler, M.; Vanderhoydonck, B.; de Jonghe, S.; Rozenski, J.; van Belle, K.; Herman, J.; Louat, T.; Parchina, A.; Sibley, C.; Lescrinier, E.; et al. Synthesis and evaluation of 5-substituted 2'-deoxyuridine monophosphate analogues as inhibitors of flavin-dependent thymidylate synthase in mycobacterium tuberculosis. J. Med. Chem. 2011, 54, 4847–4862. [Google Scholar]
- Somu, R.V.; Boshoff, H.; Qiao, C.; Bennett, E.; Barry, C.E.; Aldrich, C.C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 31–34. [Google Scholar]
- Gupte, A.; Boshoff, H.I.; Wilson, D.J.; Neres, J.; Labello, N.P.; Somu, R.; Xing, C.; Barry, C.E.; Aldrich, C.C. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5'-O-[N-(Salicyl)sulfamoyl]adenosine: Antibacterial nucleosides effective against Mycobacterium tuberculosis. J. Med. Chem. 2008, 51, 7495–7507. [Google Scholar]
- Long, M.C.; Parker, W.B. Structure-activity relationship for nucleoside analogs as inhibitors or substrates of adenosine kinase from Mycobacterium tuberculosis I. Modifications to the adenine moiety. Biochem. Pharmacol. 2006, 71, 1671–1682. [Google Scholar]
- Tripathi, R.P.; Tiwari, V.K.; Tewari, N.; Katiyar, D.; Saxena, N.; Sinha, S.; Gaikwad, A.; Srivastava, A.; Chaturvedi, V.; Manju, Y.K.; et al. Synthesis and antitubercular activities of Bis-glycosylated diamino alcohols. Bioorg. Med. Chem. 2005, 13, 5668–5679. [Google Scholar]
- Chiba, T.; Takii, T.; Nishimura, K.; Yamamoto, Y.; Morikawa, H.; Abec, C.; Onozaki, K. Synthesis of new sugar derivatives from stachys sieboldi miq and antibacterial evaluation against Mycobacterium tuberculosis, Mycobacterium avium, and Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2007, 17, 2487–2491. [Google Scholar]
- Horita, Y.; Takii, T.; Kuroishi, R.; Chiba, T.; Ogawa, K.; Kremer, L.; Sato, Y.; Lee, Y.; Hasegawa, T.; Onozaki, K. Synthesis and evaluation of anti-tubercular activity of new dithiocarbamate sugar derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 899–903. [Google Scholar]
- Sriram, D.; Aubry, A.; Yogeeswari, P.; Fisher, L.M. Gatifloxacin derivatives: Synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 2006, 16, 2982–2985. [Google Scholar]
- Dinakaran, M.; Senthilkumar, P.; Yogeeswari, P.; China, A.; Nagaraja, V.; Sriram, D. Novel ofloxacin derivatives: Synthesis, antimycobacterial and toxicological evaluation. Bioorg. Med. Chem. Lett. 2008, 18, 1229–1236. [Google Scholar]
- Dinakaran, M.; Senthilkumar, P.; Yogeeswari, P.; China, A.; Nagaraja, V.; Sriram, D. Antimycobacterial activities of novel 2-(sub)-3-fluoro/nitro-5, 12-dihydro-5-oxobenzothiazolo[3,2-A]quinoline-6-carboxylic acid. Bioorg. Med. Chem. 2008, 16, 3408–3418. [Google Scholar]
- Senthilkumar, P.; Dinakaran, M.; Yogeeswari, P.; Sriram, D.; China, A.; Nagaraja, V. Synthesis and antimycobacterial activities of novel 6-nitroquinolone-3-carboxylic acids. Eur. J. Med. Chem. 2009, 44, 345–358. [Google Scholar]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef]
- Vicente, E.; Pérez-Silanes, S.; Lima, L.M.; Ancizu, S.; Burguete, A.; Solano, B.; Villar, R.; Aldana, I.; Monge, A. Selective activity against Mycobacterium tuberculosis of new quinoxaline 1,4-di-N-oxides. Bioorg. Med. Chem. 2009, 17, 385–389. [Google Scholar]
- Ancizu, S.; Moreno, E.; Solano, B.; Burguete, A.; Torres, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A. New 3-methylquinoxaline-2-carboxamide 1,4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2010, 18, 2713–2719. [Google Scholar]
- Carta, A.; Palomba, M.; Paglietti, G.; Molicotti, P.; Paglietti, B.; Cannas, S.; Zanetti, S. [1,2,3]Triazolo[4,5-h]quinolones. A new class of potent antitubercular agents against multidrug resistantnt Mycobacterium tuberculosis strains. Bioorg. Med. Chem. Lett. 2007, 17, 4791–4794. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Shinde, P.; Kadam, S.A.; Bawane, A.N.; Sayyed, A.Y.; Kardile, R.A.; Gitay, P.N.; Lahore, S.V.; Dixit, S.S.; Földesi, A.; et al. Synthesis and antimycobacterial activity of prodrugs of indeno[2,1-c]quinoline derivatives. Eur. J. Med. Chem. 2011, 46, 1306–1324. [Google Scholar]
- Lilienkampf, A.; Mao, J.; Wan, B.; Wang, Y.; Franzblau, S.G.; Kozikowski, A. Structure-activity relationships for a series of quinoline-based compounds active against replicating and nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 2109–2118. [Google Scholar]
- Khoje, A.D.; Kulendrn, A.; Charnock, C.; Wan, B.; Franzblau, S.; Gundersen, L.L. Synthesis of non-purine analogs of 6-aryl-9-benzylpurines, and their antimycobacterial activities. Compounds modified in the imidazole ring. Bioorg. Med. Chem. 2010, 18, 7274–7282. [Google Scholar]
- Trivedi, A.R.; Bhuva, V.R.; Dholariya, B.P.; Dodiya, D.K.; Kataria, V.B.; Shah, V.H. Novel dihydropyrimidines as a potential new class of antitubercular agents. Bioorg. Med. Chem. Lett. 2010, 20, 6100–6102. [Google Scholar]
- Shiradkar, M.R.; Murahari, K.K.; Gangadasu, H.R.; Suresh, T.; Kalyan, C.A.; Panchal, D.; Kaur, R.; Burange, P.; Ghogare, J.; Mokale, V.; et al. Synthesis of new S-derivatives of clubbed triazolyl thiazole as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2007, 15, 3997–4008. [Google Scholar]
- Velaparthi, S.; Brunsteiner, R.; Petukhov, P.A. 5-tert-Butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors of Mycobacterium tuberculosis pantothenate synthetase: Initiating a quest for new antitubercular drugs. J. Med. Chem. 2008, 51, 1999–2002. [Google Scholar] [CrossRef]
- Walczak, K.; Gondela, A.; Suwinski, J. Synthesis and anti-tuberculosis activity of N-Aryl-C-nitroazoles. Eur. J. Med. Chem. 2004, 39, 849–853. [Google Scholar] [CrossRef]
- Huang, Q.; Mao, J.; Wan, B.; Wang, Y.; Brun, R.; Franzblau, S.G.; Kozikowski, A.P. Searching for new cures for tuberculosis: design, synthesis, and biological evaluation of 2-methylbenzothiazoles. J. Med. Chem. 2009, 52, 6757–6767. [Google Scholar] [CrossRef]
- Palmer, B.D.; Thompson, A.M.; Sutherland, H.S.; Blaser, A.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A. Synthesis and structure-activity studies of biphenyl analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2010, 53, 282–294. [Google Scholar]
- Hearn, M.J.; Cynamon, M.H.; Chen, M.F.; Coppins, R.; Davis, J.; Joo-On Kang, H.; Noble, A.; Tu-Sekine, B.; Terrot, M.S.; Trombino, D.; et al. Preparation and antitubercular activities in vitro and in vivo of novel schiff bases of isoniazid. Eur. J. Med. Chem. 2009, 44, 4169–4178. [Google Scholar]
- Lourenço, M.C.; de Lima Ferreira, M.; de Souza, M.V.N.; Peralta, M.A.; Vasconcelos, T.R.A.; Henriques, M.G.M.O. Synthesis and anti-mycobacterial activity of (E)-N'-(monosubstituted-benzylidene) isonicotinohydrazide derivatives. Eur. J. Med. Chem. 2008, 43, 1344–1347. [Google Scholar]
- Miller, M.J.; Walz, A.J.; Zhu, H.; Wu, C.; Moraski, G.; Mcollmann, U.; Tristani, E.M.; Crumbliss, A.L.; Ferdig, M.T.; Checkley, L.; et al. Design, synthesis, and study of a mycobactin-artemisinin conjugat that has selective and potent activity against tuberculosis and malaria. J. Am. Chem. Soc. 2011, 133, 2076–2079. [Google Scholar]
- Falzari, K.; Zhu, Z.; Pan, D.; Liu, H.; Hongmanee, P.; Franzblau, S.G. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 1447–1454. [Google Scholar] [CrossRef]
- Kim, P.; Zhang, Y.M.; Shenoy, G.; Nguyen, Q.A.; Boshoff, H.I.; Manjunatha, U.H.; Goodwin, M.B.; Lonsdale, J.; Price, A.C.; Miller, D.J.; et al. Structure-activity relationships at the 5-position of thiolactomycin: An intact (5R)-isoprene unit is required for activity against the condensing enzymes from Mycobacterium tuberculosis and Escherichia coli. J. Med. Chem. 2006, 49, 159–171. [Google Scholar]
- Slayden, R.A.; Lee, R.E.; Armour, J.W.; Cooper, A.M.; Orme, I.M.; Brennan, P.J.; Besra, G.S. Antimycobacterial action of thiolactomycin: An inhibitor of fatty acid and mycolic acid synthesis. Antimicrob. Agents Chemother. 1996, 40, 2813–2819. [Google Scholar]
- McFadden, J.M.; Medghalchi, S.M.; Thupari, J.N.; Pinn, M.L.; Vadlamudi, A.; Miller, K.I.; Kuhajda, F.P.; Townsend, C.A. Application of a flexible synthesis of (5R)-thiolactomycin to develop new inhibitors of type I fatty acid synthase. J. Med.Chem. 2005, 48, 946–961. [Google Scholar]
- Miyakawa, S.; Suzuki, K.; Noto, T.; Harada, Y.; Okazaki, H. Thiolactomycin, a new antibiotic. IV. Biological properties and chemotherapeutic activity in mice. J. Antibiot. 1982, 35, 411–419. [Google Scholar]
- Working Group on New Drugs. (WGND) (8 December 2011). Available online: http://www.newtbdrugs.org/ (accessed on 4 April 2012).
- TB Alliance, and Tuberculosis Trial Consortium [TBTC]). Available online: http://www.cdc.gov/tb/topic/research/tbtc/introduction.htm/ (accessed on 26 June 2012).
- Hirano, S.; Ichikawa, S.; Matsuda, A. Structure-activity relationship of truncated analogs of caprazamycins as potential anti-tuberculosis agents. Bioorg. Med. Chem. 2008, 16, 5123–5133. [Google Scholar] [CrossRef]
- Bogatcheva, E.; Hanrahan, C.; Nikonenko, B.; de los Santos, G.; Reddy, V.; Chen, C.; Barbosa, F.; Einck, L.; Nacy, C.; Protopopova, M. Identification of SQ609 as a lead compound from a library of dipiperidines. Bioorg. Med. Chem. Lett. 2011, 21, 5353–5357. [Google Scholar]
- Mitscher, L.A.; Baker, W. Tuberculosis: A search for novel therapy starting with natural products. Med. Res. Rev. 1998, 18, 363–374. [Google Scholar] [CrossRef]
- Global Alliance for TB Drug Development. “Background”. Available online: http://www.tballiance.org/new/portfolio/html-portfolio-item.php?id=29/ (accessed on 9 June 2012).
- Balasubramanian, V.; Gaonkar, S.; Solapure, S.; Sambandamurthy, V.; Shandil, R.; Mahesh, K.N.; Sharma, S.; Kaur, P.; Deepthi, R.; Subbulakshmi, V.; et al. (Scheduled Presentation on Monday, 19 September 2011) AZD5847, an Oxazolidinone for the Treatment of Tuberculosis: Pre-clinical Studies. [Presentation no. F1-1364]. American Society for Microbiology, 1752 N Street NW Washington, DC. USA. Available online: http://www.abstractsonline.com/plan/ViewAbstract.aspx?mID=2789&sKey=5f3fa01a-9c86-4ebd-8cab-50756d6faa6f&cKey=c393491b-4fdf-42a8-8857-3756dce2a517&mKey={0C918954-D607-46A7-8073-44F4B537A439}/ (accessed on 4 April 2012).
- Alffenaar, J.W.C.; van der Laan, T.; Simons, S.; van der Werf, T.S.; van de Kasteele, P.J.; de Neeling, H.; van Soolingen, D. Susceptibility of Clinical Mycobacterium tuberculosis isolates to a potentially less toxic derivate of linezolid, PNU-100480. Antimicrob. Agents Chemother. 2011, 55, 1287–1289. [Google Scholar]
- Sinha, R.K.; Arora, S.K.; Sinha, N.; Modak, V.M. In vivo activity of LL4858 against Mycobacterium tuberculosis [abstract F-1116]. In Program and Abstracts of the 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (Washington, DC); American Society for Microbiology: Washington, DC, USA, 2004; p. 212. [Google Scholar]
- Lee, R.; Protopopova, M.; Crooks, E.; Slayden, R.A.; Terrot, M.; Barry, C.E. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 2003, 5, 172–187. [Google Scholar] [CrossRef]
- Jia, L.; Tomaszewski, J.E.; Hanrahan, C.; Coward, L.; Noker, P.; Gorman, G.; Nikonenko, B.; Protopopova, M. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005, 144, 80–87. [Google Scholar]
- Stover, C.K.; Warrener, P.; van Devanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar]
- Nuermberger, E.; Tyagi, S.; Williams, K.N.; Rosenthal, I.; Bishai, W.R.; Grosset, J.H. Rifapentine, moxifloxacin, or dna vaccine improves treatment of latent tuberculosis in a mouse model. Am. J. Respir. Crit. Care Med. 2005, 172, 1452–1456. [Google Scholar] [CrossRef]
- Lenaerts, A.J.; Gruppo, V.; Marietta, K.S.; Johnson, C.M.; Driscoll, D.K.; Tompkins, N.M.; Rose, J.D.; Reynolds, R.C.; Orme, I.M. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 2005, 49, 2294–2301. [Google Scholar]
- Tyagi, S.; Nuermberger, E.; Yoshimatsu, T.; Williams, K.; Rosenthal, I.; Bishai, W.; Grosset, J. Bactericidal activity of the nitroimidazopyran PA-824 in the murine model of tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 2289–2293. [Google Scholar]
- Matsumoto, M.; Hshizume, H.; Tomishige, T.; Kawasaki, M. In vitro and in vivo efficacy of novel antituberculous candidate OPC-67683 [abstract F-1462]. In Program and abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy (Washington, DC); American Society for Microbiology: Washington, DC, USA, 2005. [Google Scholar]
- Kawasaki, M.; Yamamoto, K.; Matusmoto, M. Mechanism of action of OPC-67683 against Mycobacterium tuberculosis [abstract F-1463]. In Program and abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy (Washington, DC); American Society for Microbiology: Washington, DC, USA, 2005. [Google Scholar]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.M.; Winkler, H.; Gestel, J.V.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline atp-synthase inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef]
- Alcalá, L.; Ruiz-Serrano, M.J.; Turégano, C.P.F.; de Viedma, D.G.; Díaz-Infantes, M.; Marín-Arriaza, M.; Bouza, E. In vitro activities of linezolid against clinical isolates of Mycobacterium tuberculosis that are susceptible or resistant to first-line antituberculous drugs. Antimicrob. Agents Chemother. 2003, 47, 416–417. [Google Scholar]
- Migliori, G.B.; Eker, B.; Richardson, M.D.; Sotgiu, G.; Zellweger, J.P.; Skrahina, A.; Ortmann, J.; Girardi, E.; Hosffmann, H.; Besozzi, G.; Bevilacqua, N.; et al. A retrospective TBNET assessment of linezolid, safety, tolerability and efficacy in multidrug resistant tuberculosis. Eur. Respir. J. 2009, 34, 387–393. [Google Scholar]
- Gerson, S.L.; Kaplan, S.L.; Bruss, J.B.; Le, V.; Arellano, F.M.; Hafkin, B.; Kuter, D.J. Hematologic effects of linezolid: Summary of clinical experience. Antimicrob. Agents Chemother. 2002, 46, 2723–2726. [Google Scholar]
- Heifets, L.; Sanchez, T.; Vanderkolk, J.; Pham, V. Development of rifapentine susceptibility tests for Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1999, 43, 25–28. [Google Scholar]
- Williams, D.L.; Spring, L.; Collins, L.; Miller, L.P.; Heifets, L.B.; Gangadharam, P.R.J.; Gillis, T.P. Contribution of rpoB mutations to development of rifamycin cross-resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998, 42, 1853–1857. [Google Scholar]
- Gosling, R.D.; Uiso, L.O.; Sam, N.E.; Bongard, E.; Kanduma, E.G.; Nyindo, M.; Morris, R.W.; Gillespie, S.H. The bactericidal activity of moxifloxacin in patients with pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 2003, 168, 1342–1345. [Google Scholar] [CrossRef]
- Pletz, M.W.R.; de Roux, A.; Roth, A.; Neumann, K.H.; Mauch, H.; Lode, H. Early bactericidal activity of moxifloxacin in treatment of pulmonary tuberculosis: A prospective, randomized study. Antimicrob. Agents Chemother. 2004, 48, 780–782. [Google Scholar]
- Tortoli, E.; Dionisio, D.; Fabbri, C. Evaluation of moxifloxacin activity in vitro against Mycobacterium tuberculosis, including resistant and multidrug-resistant strains. J. Chemother. 2004, 16, 334–336. [Google Scholar]
- Bayer. Avelox® (moxifloxacin hydrochloride) Product Safety. Available online: http://www.avelox.com/en/physician/product_information/avelox_safety/index.php (accessed on 4 April 2012).
- Nuermberger, E.L.; Yoshimatsu, T.; Tyagi, S.; Williams, K.; Rosenthal, I.; O’brien, R.J.; Vernon, A.A.; Chaisson, R.E.; Bishai, W.R.; Grosset, J.H. Moxifloxacin-containing regimens of reduced duration produce a stable cure in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2004, 170, 1131–1134. [Google Scholar] [CrossRef]
- Paramasivan, C.N.; Sulochana, S.; Kubendiran, G.; Venkatesan, P.; Mitchison, D.A. Bactericidal action of gatifloxacin, rifampin, and isoniazid on logarithmic- and stationary-phase cultures of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 627–631. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).