
On the Quest of Cellular Functions of PEA-15 and the Therapeutic Opportunities

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Protective Roles of PEA-15 and Modulations of PEA-15 Functions
2.1. PEA-15 Protects Mature Neurons from Fas/TNFα-Induced Apoptosis
2.2. PEA-15 Protects Tissues from Malignant Cell Growth
2.3. PEA-15 Prevents Tissue Damage from Excessive Inflammation
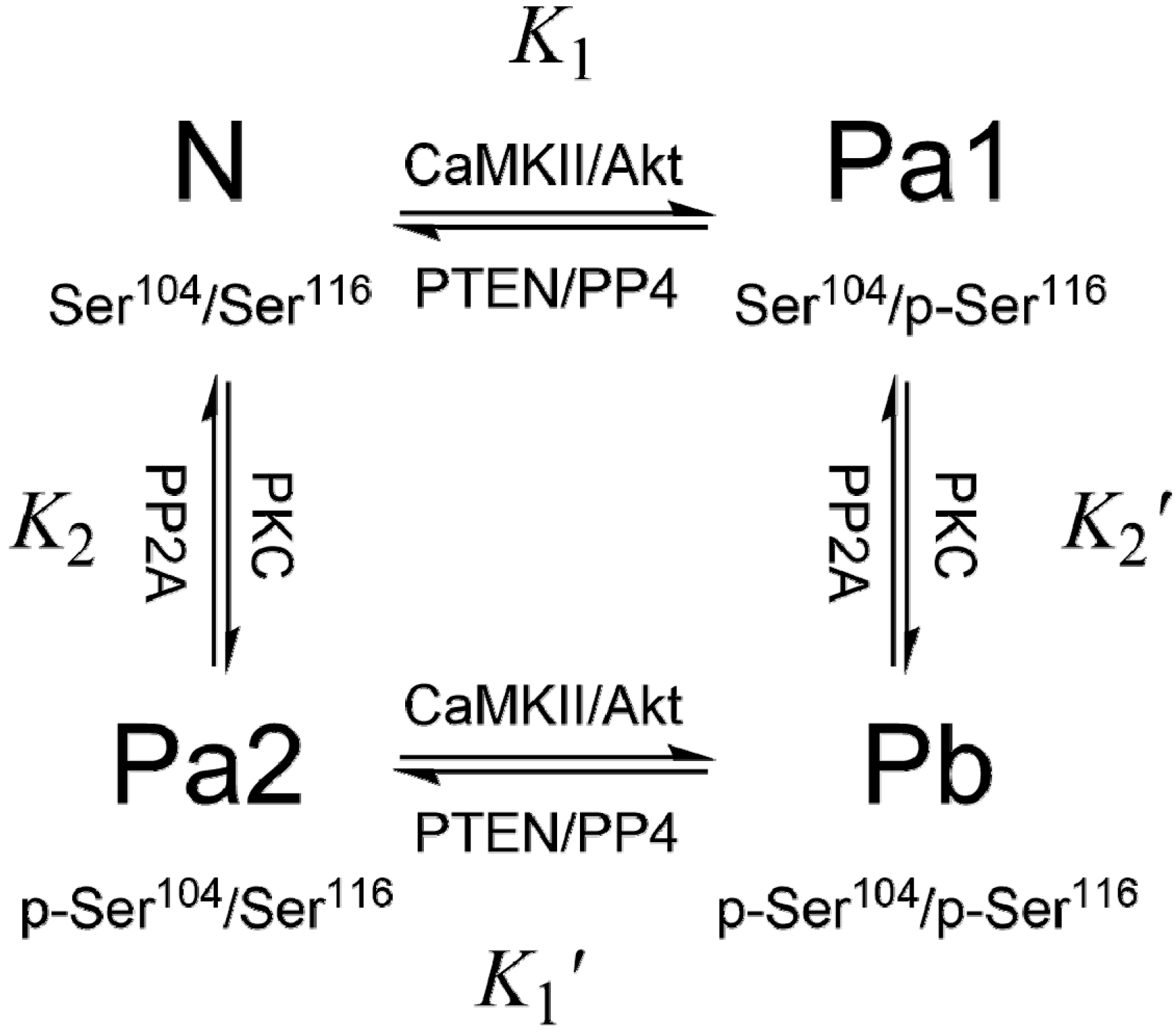
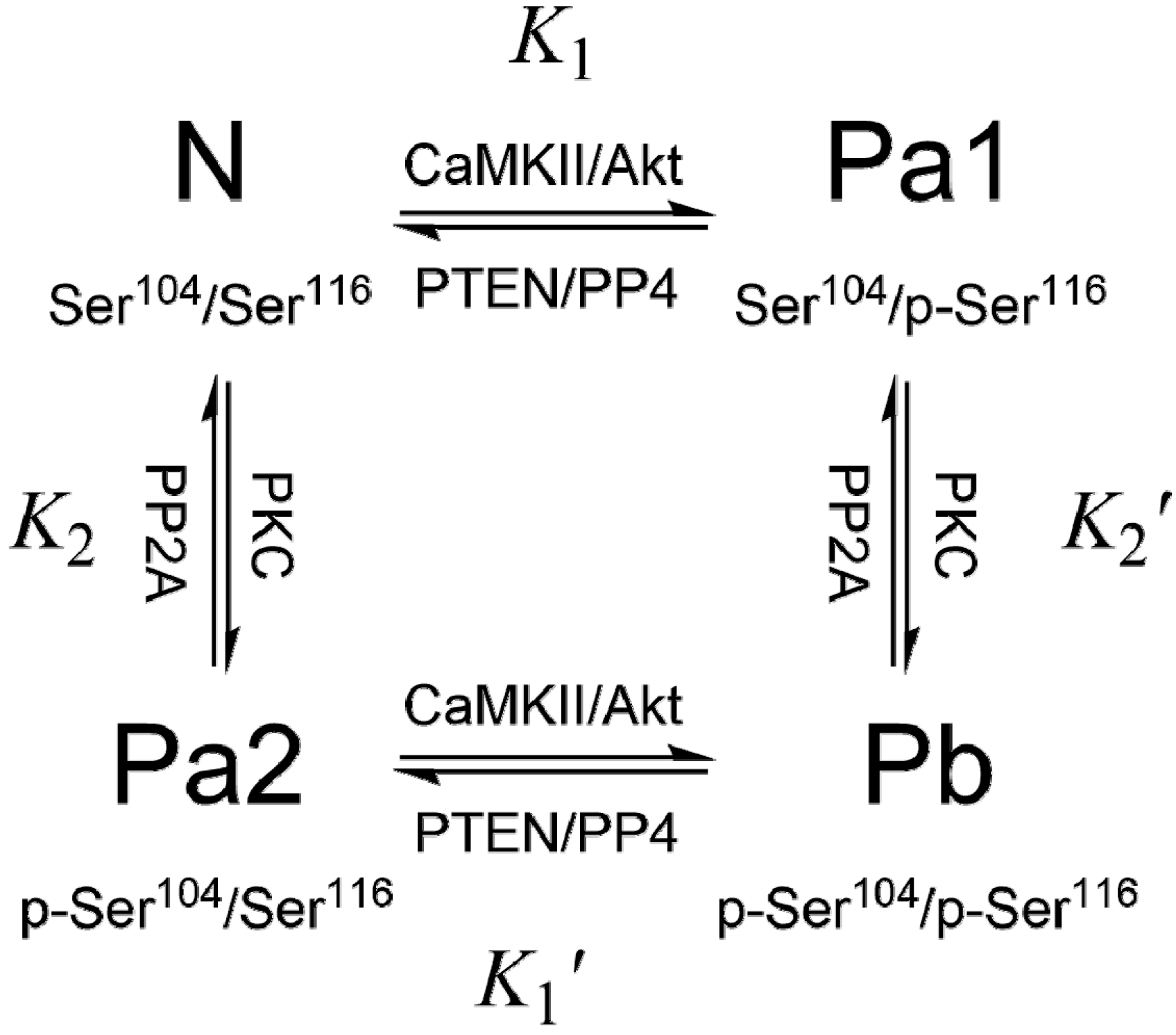
3. PEA-15 Phosphorylation Homeostasis
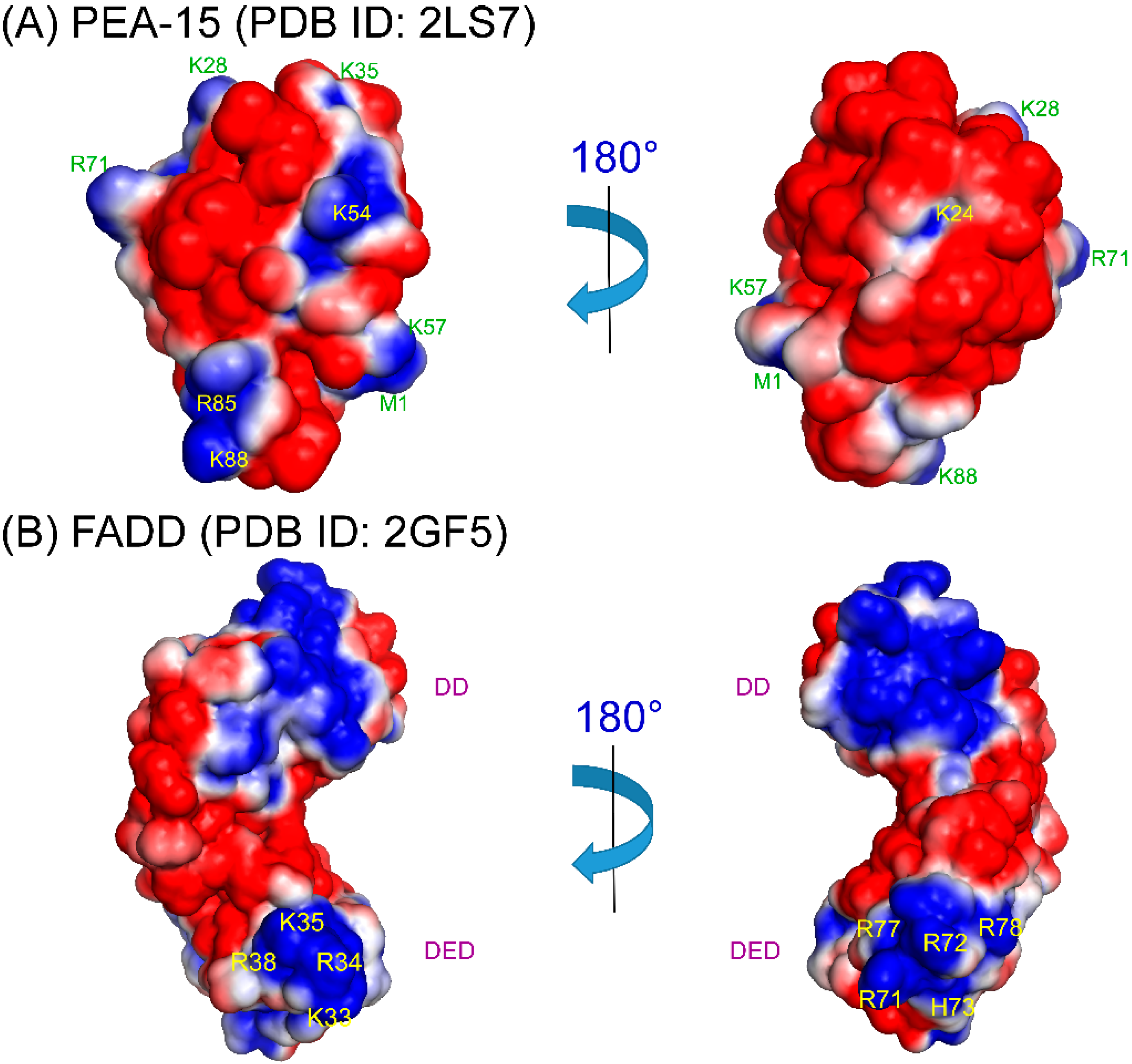
4. Structural Basis of PEA-15 Interactions
5. Therapeutic Interventions Involving PEA-15

Acknowledgments
Conflicts of Interest
References
- Araujo, H.; Danziger, N.; Cordier, J.; Glowinski, J.; Chneiweiss, H. Characterization of pea-15, a major substrate for protein kinase c in astrocytes. J. Biol. Chem. 1993, 268, 5911–5920. [Google Scholar] [PubMed]
- Danziger, N.; Yokoyama, M.; Jay, T.; Cordier, J.; Glowinski, J.; Chneiweiss, H. Cellular expression, developmental regulation, and phylogenic conservation of pea-15, the astrocytic major phosphoprotein and protein kinase c substrate. J. Neurochem. 1995, 64, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Estellés, A.; Yokoyama, M.; Nothias, F.; Vincent, J.D.; Glowinski, J.; Vernier, P.; Chneiweiss, H. The major astrocytic phosphoprotein pea-15 is encoded by two mrnas conserved on their full length in mouse and human. J. Biol. Chem. 1996, 271, 14800–14806. [Google Scholar] [PubMed]
- Kubes, M.; Cordier, J.; Glowinski, J.; Girault, J.A.; Chneiweiss, H. Endothelin induces a calcium-dependent phosphorylation of pea-15 in intact astrocytes: Identification of ser104 and ser116 phosphorylated, respectively, by protein kinase c and calcium/calmodulin kinase ii in vitro. J. Neurochem. 1998, 71, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Trencia, A.; Perfetti, A.; Cassese, A.; Vigliotta, G.; Miele, C.; Oriente, F.; Santopietro, S.; Giacco, F.; Condorelli, G.; Formisano, P.; et al. Protein kinase b/akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol. Cell. Biol. 2003, 23, 4511–4521. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Vigliotta, G.; Iavarone, C.; Caruso, M.; Tocchetti, C.G.; Andreozzi, F.; Cafieri, A.; Tecce, M.F.; Formisano, P.; Beguinot, L.; et al. PED/PEA-15 gene controls glucose transport and is overexpressed in type 2 diabetes mellitus. EMBO J. 1998, 17, 3858–3866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Redina, O.; Altshuller, Y.M.; Yamazaki, M.; Ramos, J.; Chneiweiss, H.; Kanaho, Y.; Frohman, M.A. Regulation of expression of phospholipase d1 and d2 by PEA-15, a novel protein that interacts with them. J. Biol. Chem. 2000, 275, 35224–35232. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Vigliotta, G.; Trencia, A.; Maitan, M.A.; Caruso, M.; Miele, C.; Oriente, F.; Santopietro, S.; Formisano, P.; Beguinot, F. Protein kinase c (pkc)-α activation inhibits pkc-ζ and mediates the action of PED/PEA-15 on glucose transport in the l6 skeletal muscle cells. Diabetes 2001, 50, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.; Lupoli, G.; Raciti, G.; Oriente, F.; Farinaro, E.; Della Valle, E.; Salomone, M.; Riccardi, G.; Vaccaro, O.; Donnarumma, G.; et al. The PEA15 gene is overexpressed and related to insulin resistance in healthy first-degree relatives of patients with type 2 diabetes. Diabetologia 2006, 49, 3058–3066. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Vigliotta, G.; Cafieri, A.; Trencia, A.; Andalo, P.; Oriente, F.; Miele, C.; Caruso, M.; Formisano, P.; Beguinot, F. PED/PEA-15: An anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. Oncogene 1999, 18, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Kitsberg, D.; Formstecher, E.; Fauquet, M.; Kubes, M.; Cordier, J.; Canton, B.; Pan, G.; Rolli, M.; Glowinski, J.; Chneiweiss, H. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from tnfα-induced apoptosis. J. Neurosci. 1999, 19, 8244–8251. [Google Scholar] [PubMed]
- Hao, C.; Beguinot, F.; Condorelli, G.; Trencia, A.; Van Meir, E.G.; Yong, V.W.; Parney, I.F.; Roa, W.H.; Petruk, K.C. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (trail) mediated apotosis in human malignant glioma cells. Cancer Res. 2001, 61, 1162–1170. [Google Scholar] [PubMed]
- Ramos, J.W.; Hughes, P.E.; Renshaw, M.W.; Schwartz, M.A.; Formstecher, E.; Chneiweiss, H.; Ginsberg, M.H. Death effector domain protein PEA-15 potentiates ras activation of extracellular signal receptor-activated kinase by an adhesion-independent mechanism. Mol. Biol. Cell 2000, 11, 2863–2872. [Google Scholar] [CrossRef] [PubMed]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Haling, J.R.; Wang, F.; Ginsberg, M.H. Phosphoprotein enriched in astrocytes 15 kDa (PEA-15) reprograms growth factor signaling by inhibiting threonine phosphorylation of fibroblast receptor substrate 2α. Mol. Biol. Cell 2010, 21, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, H.; Ramos, J.W. RSK2 activity is regulated by its interaction with PEA-15. J. Biol. Chem. 2003, 278, 32367–32372. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, H.; Opoku-Ansah, J.; Pastorino, S.; Renganathan, H.; Matter, M.L.; Ramos, J.W. ERK MAP kinase is targeted to RSK2 by the phosphoprotein PEA-15. Proc. Natl. Acad. Sci. USA 2007, 104, 19837–19842. [Google Scholar] [CrossRef] [PubMed]
- Greig, F.H.; Nixon, G.F. Phosphoprotein enriched in astrocytes (PEA)-15: A potential therapeutic target in multiple disease states. Pharmacol. Therap. 2014, 143, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Vaidyanathan, H.; Ramos, J.W.; Ginsberg, M.H.; Werner, M.H. Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. EMBO J. 2002, 21, 6494–6504. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Lo, Y.C.; Lin, S.C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Ann. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Twomey, E.C. NMR Spectroscopic Characterization of Death Domain Superfamily Proteins: Structures, Dynamics and Interactions. In Nuclear Magnetic Resonance (NMR): Theory, Applications and Technology; Rao, D.K., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2014; pp. 83–110. [Google Scholar]
- Twomey, E.C.; Wei, Y. High-definition NMR structure of PED/PEA-15 death effector domain reveals details of key polar side chain interactions. Biochem. Biophys. Res. Commun. 2012, 424, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Fiory, F.; Formisano, P.; Perruolo, G.; Beguinot, F. Frontiers: PED/PEA-15, a multifunctional protein controlling cell survival and glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E592–E601. [Google Scholar] [CrossRef] [PubMed]
- Valmiki, M.; Ramos, J. Death effector domain-containing proteins. Cell. Mol. Life Sci. 2009, 66, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Mace, P.D.; Wallez, Y.; Egger, M.F.; Dobaczewska, M.K.; Robinson, H.; Pasquale, E.B.; Riedl, S.J. Structure of ERK2 bound to PEA-15 reveals a mechanism for rapid release of activated mapk. Nat. Commun. 2013, 4, 1681. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Cordasco, D.F.; Wei, Y. Profound conformational changes of PED/PEA-15 in ERK2 complex revealed by NMR backbone dynamics. Biochim. Biophys. Acta 2012, 1824, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Cordasco, D.F.; Kozuch, S.D.; Wei, Y. Substantial conformational change mediated by charge-triad residues of the death effector domain in protein-protein interactions. PLoS ONE 2013, 8, e83421. [Google Scholar] [CrossRef] [PubMed]
- Viparelli, F.; Cassese, A.; Doti, N.; Paturzo, F.; Marasco, D.; Dathan, N.A.; Monti, S.M.; Basile, G.; Ungaro, P.; Sabatella, M.; et al. Targeting of PED/PEA-15 molecular interaction with phospholipase d1 enhances insulin sensitivity in skeletal muscle cells. J. Biol. Chem. 2008, 283, 21769–21778. [Google Scholar] [CrossRef] [PubMed]
- Farina, B.; Doti, N.; Pirone, L.; Malgieri, G.; Pedone, E.M.; Ruvo, M.; Fattorusso, R. Molecular basis of the PED/PEA15 interaction with the c-terminal fragment of phospholipase d1 revealed by NMR spectroscopy. Biochim. Biophys. Acta 2013, 1834, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Sharif, A.; Renault, F.; Beuvon, F.; Castellanos, R.; Canton, B.; Barbeito, L.; Junier, M.P.; Chneiweiss, H. The expression of PEA-15 (phosphoprotein enriched in astrocytes of 15 kDa) defines subpopulations of astrocytes and neurons throughout the adult mouse brain. Neuroscience 2004, 126, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Thomason, L.A.; Smithson, L.J.; Hazrati, L.N.; McLaurin, J.; Kawaja, M.D. Reactive astrocytes associated with plaques in tgcrnd8 mouse brain and in human alzheimer brain express phosphoprotein enriched in astrocytes (PEA-15). FEBS Lett. 2013, 587, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Kim, D.W.; Shin, M.J.; Kim, H.R.; Kim, S.M.; Woo, S.J.; Eom, S.A.; Jo, H.S.; Kim, D.S.; Cho, S.W.; et al. Pep-1-PEA-15 protects against toxin-induced neuronal damage in a mouse model of parkinson’s disease. BBA 2014, 1840, 1686–1700. [Google Scholar] [PubMed]
- Xiao, C.; Yang, B.F.; Asadi, N.; Beguinot, F.; Hao, C. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-flip and PED/PEA-15 in glioma cells. J. Biol. Chem. 2002, 277, 25020–25025. [Google Scholar] [CrossRef] [PubMed]
- Stassi, G.; Garofalo, M.; Zerilli, M.; Ricci-Vitiani, L.; Zanca, C.; Todaro, M.; Aragona, F.; Limite, G.; Petrella, G.; Condorelli, G. Ped mediates AKT-dependent chemoresistance in human breast cancer cells. Cancer Res. 2005, 65, 6668–6675. [Google Scholar] [CrossRef] [PubMed]
- Zanca, C.; Garofalo, M.; Quintavalle, C.; Romano, G.; Acunzo, M.; Ragno, P.; Montuori, N.; Incoronato, M.; Tornillo, L.; Baumhoer, D.; et al. PED is overexpressed and mediates trail resistance in human non-small cell lung cancer. J. Cell. Mol. Med. 2008, 12, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Torii, S.; Nishida, E. Control of map kinase signaling to the nucleus. Chromosoma 2005, 114, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Glading, A.; Koziol, J.A.; Krueger, J.; Ginsberg, M.H. PEA-15 inhibits tumor cell invasion by binding to extracellular signal-regulated kinase 1/2. Cancer Res. 2007, 67, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M.; Kazansky, A.; Krishnamurthy, S.; Liu, P.; Yuan, L.X.H.; Yamasaki, F.; Liu, S.; Hayashi, N.; Zhang, D.; et al. PEA-15 inhibits tumorigenesis in an mda-mb-468 triple-negative breast cancer xenograft model through increased cytoplasmic localization of activated extracellular signal-regulated kinase. Clin. Cancer Res. 2010, 16, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Rosen, D.; Wei, C.; Kazansky, A.; Yamasaki, F.; Takahashi, T.; Itamochi, H.; Kondo, S.; Liu, J.; Ueno, N.T. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer Res. 2008, 68, 9302–9310. [Google Scholar] [CrossRef] [PubMed]
- Buonomo, R.; Giacco, F.; Vasaturo, A.; Caserta, S.; Guido, S.; Pagliara, V.; Garbi, C.; Mansueto, G.; Cassese, A.; Perruolo, G.; et al. PED/PEA-15 controls fibroblast motility and wound closure by ERK1/2-dependent mechanisms. J. Cell. Physiol. 2012, 227, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Böck, B.C.; Tagscherer, K.E.; Fassl, A.; Krämer, A.; Oehme, I.; Zentgraf, H.W.; Keith, M.; Roth, W. The PEA-15 protein regulates autophagy via activation of JNK. J. Biol. Chem. 2010, 285, 21644–21654. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Itamochi, H.; Nitta, M.; Saya, H.; Ginsberg, M.H.; Ueno, N.T. Antitumor effect of e1a in ovarian cancer by cytoplasmic sequestration of activated ERK by PEA15. Oncogene 2006, 25, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Botta, G.; Perruolo, G.; Libertini, S.; Cassese, A.; Abagnale, A.; Beguinot, F.; Formisano, P.; Portella, G. PED/PEA-15 modulates coxsackievirus–adenovirus receptor expression and adenoviral infectivity via ERK-mediated signals in glioma cells. Hum. Gene Ther. 2010, 21, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Yamasaki, F.; Kajiwara, Y.; Saito, T.; Nishimoto, T.; Bartholomeusz, C.; Ueno, N.; Sugiyama, K.; Kurisu, K. Expression of phosphoprotein enriched in astrocytes 15 kDa (PEA-15) in astrocytic tumors: A novel approach of correlating malignancy grade and prognosis. J. Neurooncol. 2010, 100, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Funke, V.; Lehmann-Koch, J.; Bickeboller, M.; Benner, A.; Tagscherer, K.E.; Grund, K.; Pfeifer, M.; Herpel, E.; Schirmacher, P.; Chang-Claude, J.; et al. The PEA-15/PED protein regulates cellular survival and invasiveness in colorectal carcinomas. Cancer Lett. 2013, 335, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Dogra, S.K.; Liu, A.Y.; Green, M.R.; Wajapeyee, N. PEA15 regulates the DNA damage-induced cell cycle checkpoint and oncogene-directed transformation. Mol. Cell. Biol. 2014, 34, 2264–2282. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by mdm2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Kim, H.S.; Lim, I.K. Downregulation of PEA-15 reverses g1 arrest, and nuclear and chromatin changes of senescence phenotype via perk1/2 translocation to nuclei. Cell Signal 2015, 27, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, S.; Renganathan, H.; Caliva, M.J.; Filbert, E.L.; Opoku-Ansah, J.; Sulzmaier, F.J.; Gawecka, J.E.; Werlen, G.; Shaw, A.S.; Ramos, J.W. The death effector domain protein PEA-15 negatively regulates t-cell receptor signaling. FASEB J. 2010, 24, 2818–2828. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, P.; Teperino, R.; Mirra, P.; Cassese, A.; Fiory, F.; Perruolo, G.; Miele, C.; Laakso, M.; Formisano, P.; Beguinot, F. Molecular cloning and characterization of the human PED/PEA-15 gene promoter reveal antagonistic regulation by hepatocyte nuclear factor 4α and chicken ovalbumin upstream promoter transcription factor ii. J. Biol. Chem. 2008, 283, 30970–30979. [Google Scholar] [CrossRef] [PubMed]
- Renault, F.; Formstecher, E.; Callebaut, I.; Junier, M.P.; Chneiweiss, H. The multifunctional protein PEA-15 is involved in the control of apoptosis and cell cycle in astrocytes. Biochem. Pharmacol. 2003, 66, 1581–1588. [Google Scholar] [CrossRef]
- Ramos-Miguel, A.; García-Fuster, M.J.; Callado, L.F.; La Harpe, R.; Meana, J.J.; García-Sevilla, J.A. Phosphorylation of Fadd (Fas-associated death domain protein) at serine 194 is increased in the prefrontal cortex of opiate abusers: Relation to mitogen activated protein kinase, phosphoprotein enriched in astrocytes of 15 kDa, and AKT signaling pathways involved in neuroplasticity. Neuroscience 2009, 161, 23–38. [Google Scholar] [PubMed]
- Ramos-Miguel, A.; Esteban, S.; García-Sevilla, J.A. The time course of unconditioned morphine-induced psychomotor sensitization mirrors the phosphorylation of fadd and MEK/ERK in rat striatum: Role of PEA-15 as a fadd-ERK binding partner in striatal plasticity. Eur. Neuropsychopharmacol. 2010, 20, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Álvaro-Bartolomé, M.; La Harpe, R.; Callado, L.F.; Meana, J.J.; García-Sevilla, J.A. Molecular adaptations of apoptotic pathways and signaling partners in the cerebral cortex of human cocaine addicts and cocaine-treated rats. Neuroscience 2011, 196, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Fujikura-Ouchi, Y.; Ito, C.; Matsuoka, H.; Shimoda, K.; Akiyama, K. An association study on polymorphisms in the PEA15, entpd4, and gas2l1 genes and schizophrenia. Psychiatry Res. 2011, 185, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.; Chou, F.L.; Glading, A.; Schaefer, E.; Ginsberg, M.H. Phosphorylation of phosphoprotein enriched in astrocytes (PEA-15) regulates extracellular signal-regulated kinase-dependent transcription and cell proliferation. Mol. Biol. Cell 2005, 16, 3552–3561. [Google Scholar] [CrossRef] [PubMed]
- Renganathan, H.; Vaidyanathan, H.; Knapinska, A.; Ramos, J.W. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to fadd. Biochem. J. 2005, 390, 729–735. [Google Scholar] [PubMed]
- Sulzmaier, F.; Opoku-Ansah, J.; Ramos, J.W. Phosphorylation is the switch that turns PEA-15 from tumor suppressor to tumor promoter. Small GTPases 2012, 3, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Gervais, M.; Dugourd, C.; Muller, L.; Ardidie, C.; Canton, B.; Loviconi, L.; Corvol, P.; Chneiweiss, H.; Monnot, C. Akt down-regulates ERK1/2 nuclear localization and angiotensin ii-induced cell proliferation through PEA-15. Mol. Biol. Cell 2006, 17, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Bock, B.C.; Tagscherer, K.E.; Haas, T.L.; Grund, K.; Sykora, J.; Herold-Mende, C.; Ehemann, V.; Hollstein, M.; Chneiweiss, H.; et al. The PEA-15/PED protein protects glioblastoma cells from glucose deprivation-induced apoptosis via the ERK/MAP kinase pathway. Oncogene 2007, 27, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E. Ampk in health and disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.K.; Balaji, S.A.; Saxena, M.; Pandey, S.; Sravan, G.S.; Heda, N.; Kumar, M.V.; Mukherjee, G.; Dey, D.; Rangarajan, A. Identification of a novel ampk-PEA15 axis in the anoikis-resistant growth of mammary cells. Breast Cancer Res. 2014, 16, 420. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; Belotti, D.; Giavazzi, R.; Sobel, M.E.; Castronovo, V. Enhancement of metastatic potential of murine and human melanoma cells by laminin receptor peptide g: Attachment of cancer cells to subendothelial matrix as a pathway for hematogenous metastasis. J. Natl. Cancer Inst. 1993, 85, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Formisano, P.; Ragno, P.; Pesapane, A.; Alfano, D.; Alberobello, A.T.; Rea, V.E.; Giusto, R.; Rossi, F.W.; Beguinot, F.; Rossi, G.; et al. Ped/PEA-15 interacts with the 67 kd laminin receptor and regulates cell adhesion, migration, proliferation and apoptosis. J. Cell. Mol. Med. 2012, 16, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Di Costanzo, S.; Zanca, C.; Tasset, I.; Fraldi, A.; Incoronato, M.; Mirabelli, P.; Monti, M.; Ballabio, A.; Pucci, P.; et al. Phosphorylation-regulated degradation of the tumor-suppressor form of ped by chaperone-mediated autophagy in lung cancer cells. J. Cell. Physiol. 2014, 229, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.H.; Cho, E.H.; Min, W.; Kim, M.J.; Kim, M.O.; Jung, E.J.; Koh, P.O. Identification of proteins regulated by estradiol in focal cerebral ischemic injury—A proteomics approach. Neurosci. Lett. 2010, 477, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.C.; Tarnawski, A.S. Pten regulatory functions in tumor suppression and cell biology. Med. Sci. Monit. 2004, 10, Ra235–241. [Google Scholar] [PubMed]
- Whang, Y.E.; Yuan, X.J.; Liu, Y.; Majumder, S.; Lewis, T.D. Regulation of sensitivity to trail by the PTEN tumor suppressor. Vitam. Horm. 2004, 67, 409–426. [Google Scholar] [PubMed]
- Peacock, J.W.; Palmer, J.; Fink, D.; Ip, S.; Pietras, E.M.; Mui, A.L.; Chung, S.W.; Gleave, M.E.; Cox, M.E.; Parsons, R.; et al. Pten loss promotes mitochondrially dependent type II FAS-induced apoptosis via PEA-15. Mol. Cell. Biol. 2009, 29, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Peacock, J.W.; Beraldi, E.; Zoubeidi, A.; Gleave, M.E.; Ong, C.J. Hsp27 silencing coordinately inhibits proliferation and promotes FAS-induced apoptosis by regulating the PEA-15 molecular switch. Cell Death Differ. 2012, 19, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T.; Philp, A.; Vazquez-Martin, C. Protein phosphatase 4—From obscurity to vital functions. FEBS Lett. 2005, 579, 3278–3286. [Google Scholar] [CrossRef] [PubMed]
- Mourtada-Maarabouni, M.; Williams, G.T. Protein phosphatase 4 regulates apoptosis, proliferation and mutation rate of human cells. Biochim Biophys Acta 2008, 1783, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Mourtada-Maarabouni, M.; Williams, G.T. Protein phosphatase 4 regulates apoptosis in leukemic and primary human t-cells. Leuk. Res. 2009, 33, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Valmiki, M.K.; Nelson, D.A.; Caliva, M.J.; Geerts, D.; Matter, M.L.; White, E.P.; Ramos, J.W. PEA-15 potentiates h-ras-mediated epithelial cell transformation through phospholipase D. Oncogene 2012, 31, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Callaway, K.; Abramczyk, O.; Martin, L.; Dalby, K.N. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the d-recruitment site. Biochemistry 2007, 46, 9187–9198. [Google Scholar] [CrossRef] [PubMed]
- Kaoud, T.S.; Devkota, A.K.; Harris, R.; Rana, M.S.; Abramczyk, O.; Warthaka, M.; Lee, S.; Girvin, M.E.; Riggs, A.F.; Dalby, K.N. Activated ERK2 is a monomer in vitro with or without divalent cations and when complexed to the cytoplasmic scaffold PEA-15. Biochemistry 2011, 50, 4568–4578. [Google Scholar] [CrossRef] [PubMed]
- Vigliotta, G.; Miele, C.; Santopietro, S.; Portella, G.; Perfetti, A.; Maitan, M.A.; Cassese, A.; Oriente, F.; Trencia, A.; Fiory, F.; et al. Overexpression of the ped/PEA-15 gene causes diabetes by impairing glucose-stimulated insulin secretion in addition to insulin action. Mol. Cell. Biol. 2004, 24, 5005–5015. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Cassese, A.; Marasco, D.; Paturzo, F.; Sabatella, M.; Viparelli, F.; Dathan, N.; Monti, S.M.; Miele, C.; Formisano, P.; et al. Residues 762–801 of PLD1 mediate the interaction with ped/PEA15. Mol. Biosyst. 2010, 6, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Wang, L.; Zheng, L.; Wan, F.; Ahmed, M.; Lenardo, M.J.; Wu, H. Crystal structure of MC159 reveals molecular mechanism of disc assembly and flip inhibition. Mol. Cell 2005, 20, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Jeffrey, P.D.; Yu, J.W.; Shi, Y. Crystal structure of a viral flip. J. Biol. Chem. 2006, 281, 2960–2968. [Google Scholar] [CrossRef] [PubMed]
- Carrington, P.E.; Sandu, C.; Wei, Y.; Hill, J.M.; Morisawa, G.; Huang, T.; Gavathiotis, E.; Wei, Y.; Werner, M.H. The structure of fadd and its mode of interaction with procaspase-8. Mol. Cell 2006, 22, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Park, H. Pyrin domains and their interactions in the apoptosis and inflammation signaling pathway. Apoptosis 2012, 17, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Bartholomeusz, C.; Krishnamurthy, S.; Liu, P.; Saso, H.; Lafortune, T.A.; Hortobagyi, G.N.; Ueno, N.T. PEA-15 unphosphorylated at both serine 104 and serine 116 inhibits ovarian cancer cell tumorigenicity and progression through blocking beta-catenin. Oncogenesis 2012, 1, e22. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Bartholomeusz, C.; Ahmed, A.A.; Kazansky, A.; Diao, L.; Baggerly, K.A.; Hortobagyi, G.N.; Ueno, N.T. Bisphosphorylated PEA-15 sensitizes ovarian cancer cells to paclitaxel by impairing the microtubule-destabilizing effect of sclip. Mol. Cancer Ther. 2013, 12, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Oishi, T.; Saso, H.; Akar, U.; Liu, P.; Kondo, K.; Kazansky, A.; Krishnamurthy, S.; Lee, J.; Esteva, F.J.; et al. Mek1/2 inhibitor selumetinib (azd6244) inhibits growth of ovarian clear cell carcinoma in a PEA-15-dependent manner in a mouse xenograft model. Mol. Cancer Ther. 2012, 11, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Lee, K.E.; Yang, E.G.; Jeon, H.; Song, H.K. PEA-15 facilitates egfr dephosphorylation via ERK sequestration at increased ER-PM contacts in TNBC cells. FEBS Lett. 2015, 589, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Roccaro, A.; Hong, F.; Weller, E.; Rubin, N.; Leduc, R.; Rourke, M.; Chuma, S.; Sacco, A.; Jia, X.; et al. Clinical and translational studies of a phase ii trial of the novel oral AKT inhibitor perifosine in relapsed or relapsed/refractory waldenstrom’s macroglobulinemia. Clin. Cancer Res. 2010, 16, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Hilger, R.A.; Scheulen, M.E.; Strumberg, D. The RAS-RAF-MEK-ERK pathway in the treatment of cancer. Onkologie 2002, 25, 511–518. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the RAF/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Das, T.K.; Shokat, K.M.; Cagan, R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, L.; Xiao, X.; Guo, J.; Kong, Y.; Wu, M.; Liu, W.; Gao, G.; Hsu, J.L.; Wei, W.; et al. Targeted expression of bikdd eliminates breast cancer with virtually no toxicity in noninvasive imaging models. Mol. Cancer Ther. 2012, 11, 1915–1924. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Tang, H.; Liu, P.; Kong, Y.; Wu, M.; Xiao, X.; Yang, L.; Gao, J.; Wei, W.; Lee, J.; et al. Development of PEA-15 using a potent non-viral vector for therapeutic application in breast cancer. Cancer Lett. 2015, 356, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci U S A 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The pymol molecular graphics system, version 1.7.2. Available online: http://www.pymol.org/ (accessed on 15 February 2015).
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y. On the Quest of Cellular Functions of PEA-15 and the Therapeutic Opportunities. Pharmaceuticals 2015, 8, 455-473. https://doi.org/10.3390/ph8030455
Wei Y. On the Quest of Cellular Functions of PEA-15 and the Therapeutic Opportunities. Pharmaceuticals. 2015; 8(3):455-473. https://doi.org/10.3390/ph8030455
Chicago/Turabian StyleWei, Yufeng. 2015. "On the Quest of Cellular Functions of PEA-15 and the Therapeutic Opportunities" Pharmaceuticals 8, no. 3: 455-473. https://doi.org/10.3390/ph8030455
APA StyleWei, Y. (2015). On the Quest of Cellular Functions of PEA-15 and the Therapeutic Opportunities. Pharmaceuticals, 8(3), 455-473. https://doi.org/10.3390/ph8030455