
Therapeutic Potential of Interferon-? and Its Antagonists in Autoinflammation: Lessons from Murine Models of Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome

Abstract
:1. Introduction
2. Pro- and Anti-Inflammatory Aspects of Interferon-γ

{kind=link}
{kind=link}
| Proinflammatory | Anti-Inflammatory |
|---|---|
| Macrophage activation | Inhibition of Th2 and Th17 polarization |
| Upregulation of antigen presentation pathways | Induction of IDO |
| NK cell activation | Induction of T cell apoptosis |
| Induction of Th1 polarization | Inhibition of neutrophil specific chemokines |
| Stimulation of monocyte chemoattractants | Inhibition of tissue damage |
| Upregulation of cell adhesion molecules | Inhibition of osteoclastogenesis |
| B cell maturation | Induction of IL-18 binding protein |
2.1. Macrophage Activation and Enhancement of Immune Pathways
2.2. Modulation of T Helper and Regulatory T Cell Responses
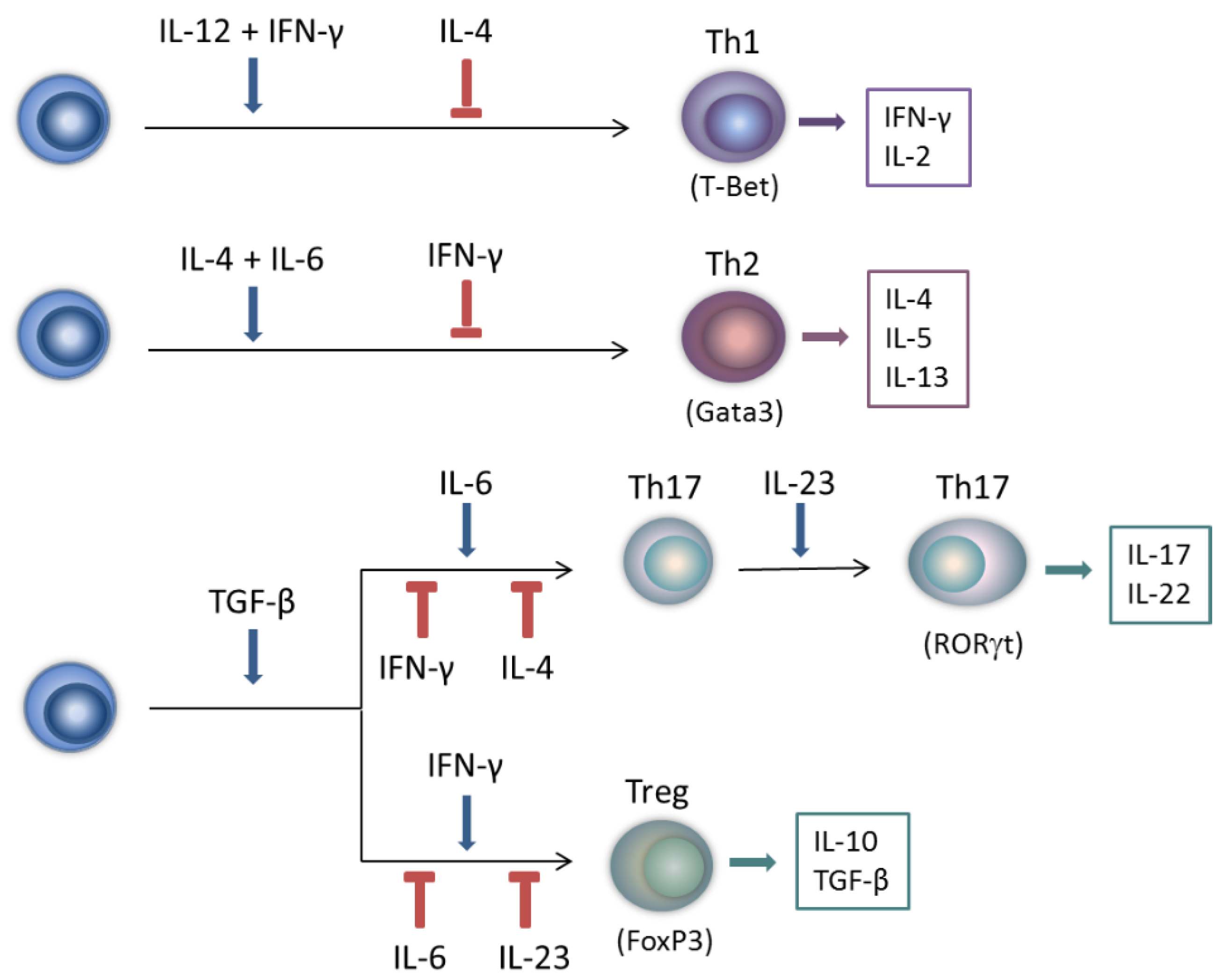
2.3. Influence on Chemokine Production and Attenuation of Tissue Destruction
2.4. Recombinant IFN-γ as Therapy in Mycobacterial Infections
3. Interferon-γ in Autoinflammation: Focus on Systemic Juvenile Idiopathic Arthritis
3.1. Pathogenesis of Systemic Juvenile Idiopathic Arthritis and Current Treatment Recommendations
| Feature | sJIA | MAS | |
|---|---|---|---|
| Incidence | ~1/100,000 | ~10% of sJIA patients | |
| Clinical Features | Fever | Quotidian | Persistent |
| Rash | Evanescent | Petechial/macular | |
| Hepatomegaly | + | + | |
| Splenomegaly | + | + | |
| Lymphadenopathy | + | + | |
| Arthritis | + | - | |
| Serositis | + | - | |
| CNS dysfunction | −/+ | + | |
| Laboratory Characteristics | Neutrophil count | ↑↑ | ↓ |
| Platelet count | ↑↑ | ↓ | |
| Anemia | + | + | |
| ESR | ↑↑ | Normal or ↓ | |
| CRP | ↑ | ↑ | |
| ALT/AST | Normal or ↑ | ↑↑ | |
| Fibrinogen | ↑ | ↓ | |
| Ferritin | Normal or ↑ | ↑↑ | |
| D-dimers | ↑ | ↑↑ | |
| sCD25 | Normal or ↑ | ↑↑ | |
| sCD163 | Normal or ↑ | ↑↑ | |
| Histopathological Features | NK cell dysfunction | Possible | Frequent |
| Hemophagocytic macrophages | Possible | Frequent |
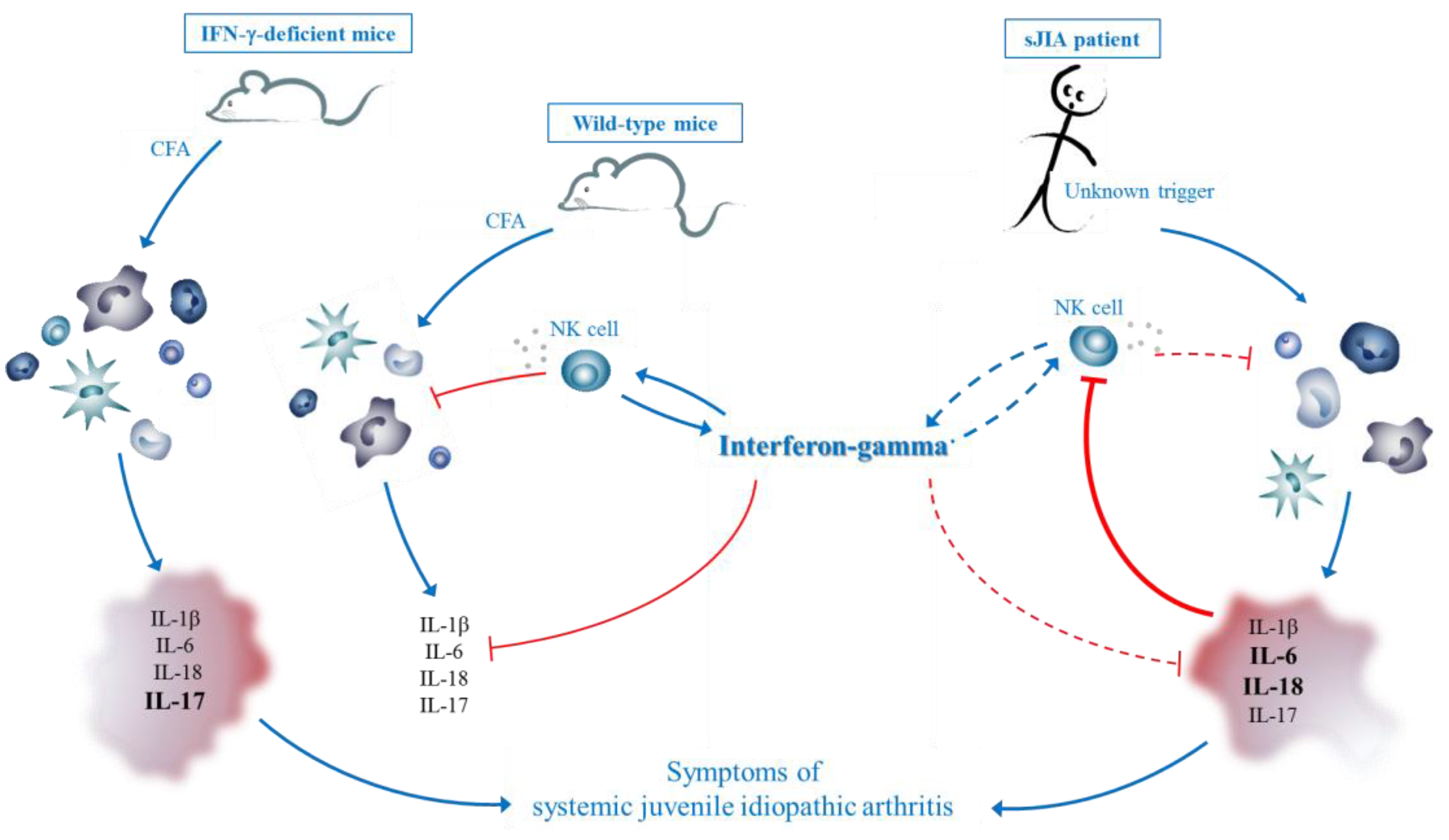
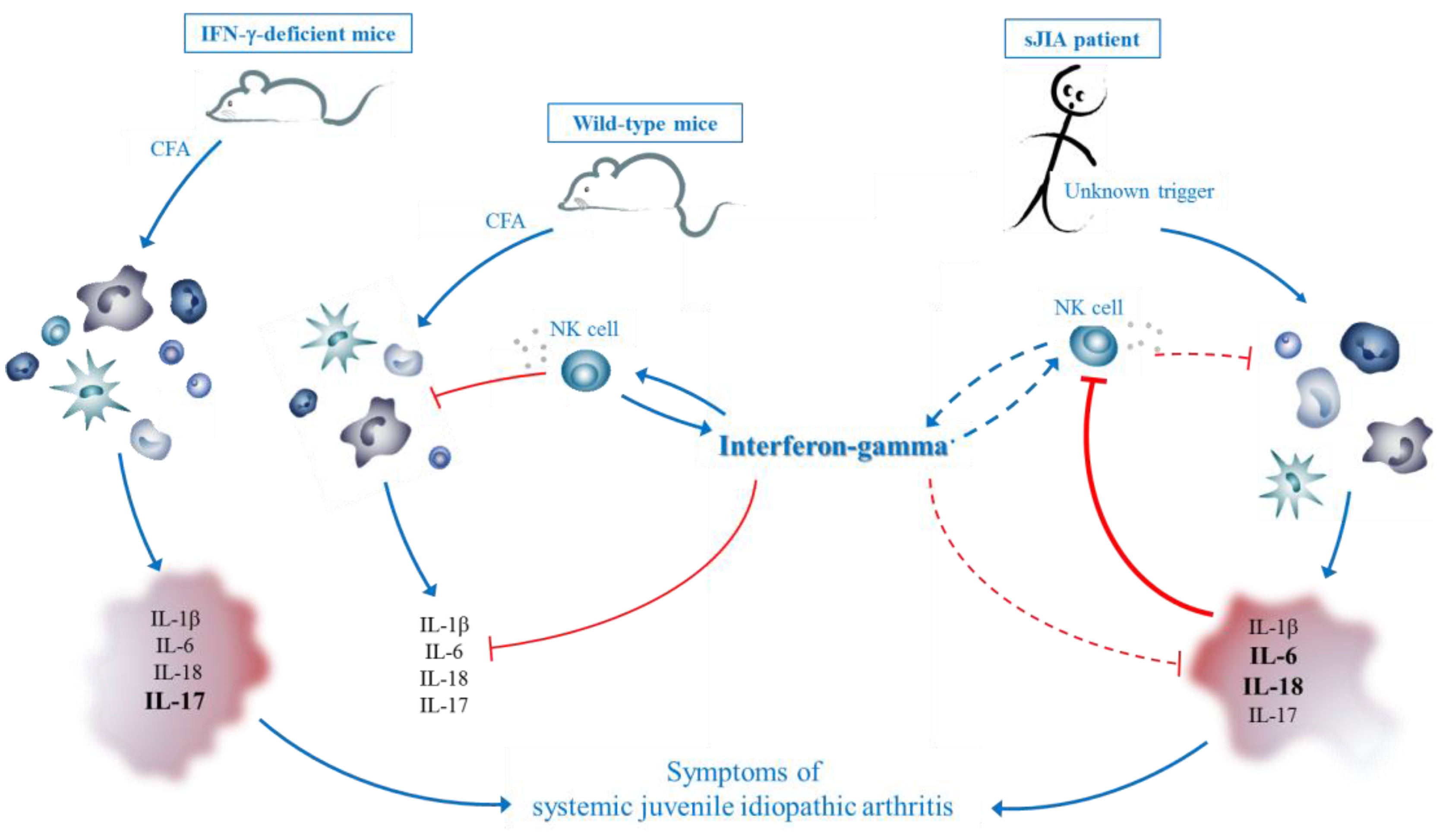
3.2. IFN-γ in sJIA: Lessons from a New Mouse Model
3.3. Comparison with CFA-Induced Experimental Autoimmune Diseases
3.4. Comparison with Mouse Models of MAS
3.5. Hypothesis
4. Therapeutic Potential of IFN-γ in Autoinflammatory Disorders
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Young, H.A.; Hardy, K.J. Role of interferon-γ in immune cell regulation. J. Leukoc. Biol. 1995, 58, 373–381. [Google Scholar] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Boisson-Dupuis, S.; Chapgier, A.; Yang, K.; Bustamante, J.; Puel, A.; Picard, C.; Abel, L.; Jouanguy, E.; Casanova, J.L. Inborn errors of interferon (IFN)-mediated immunity in humans: Insights into the respective roles of IFN-α/β, IFN-γ, and IFN-lambda in host defense. Immunol. Rev. 2008, 226, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Yin and yang interplay of IFN-γ in inflammation and autoimmune disease. J. Clin. Investig. 2007, 117, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Kelchtermans, H.; Billiau, A.; Matthys, P. How interferon-γ keeps autoimmune diseases in check. Trends Immunol. 2008, 29, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.; Matthys, P. Interferon-γ: A historical perspective. Cytokine Growth Factor Rev. 2009, 20, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Herrero, C.; Li, W.P.; Antoniv, T.T.; Falck-Pedersen, E.; Koch, A.E.; Woods, J.M.; Haines, G.K., III; Ivashkiv, L.B. Sensitization of IFN-γ Jak-STAT signaling during macrophage activation. Nat. Immunol. 2002, 3, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of signaling pathways by interferon-γ: Implications for immune responses and autoimmune diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar]
- Basler, M.; Kirk, C.J.; Groettrup, M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.; Carotta, S.; Thong, R.P.; Chan, C.J.; Hayakawa, Y.; Smyth, M.J.; Nutt, S.L. The interactions of multiple cytokines control NK cell maturation. J. Immunol. 2010, 185, 6679–6688. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Weigent, D.A.; Langford, M.P.; Fleischmann, W.R., Jr.; Stanton, G.J. Potentiation of lymphocyte natural killing by mixtures of α or β interferon with recombinant γ interferon. Infect. Immun. 1983, 40, 35–38. [Google Scholar] [PubMed]
- Senik, A.; Stefanos, S.; Kolb, J.P.; Lucero, M.; Falcoff, E. Enhancement of mouse natural killer cell activity by type II interferon. Ann. Immunol. 1980, 131C, 349–361. [Google Scholar]
- Catalona, W.J.; Ratliff, T.L.; McCool, R.E. γ-Interferon induced by S. aureus protein A augments natural killing and ADCC. Nature 1981, 291, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Suzuki, R.; Matsui, H.; Shimizu, Y.; Kumagai, K. Natural killer (NK) cells as a responder to interleukin 2 (IL 2). II. IL 2-induced interferon γ production. J. Immunol. 1983, 130, 988–992. [Google Scholar] [PubMed]
- Dalton, D.K.; Pitts-Meek, S.; Keshav, S.; Figari, I.S.; Bradley, A.; Stewart, T.A. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science 1993, 259, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Avau, A.; Mitera, T.; Put, S.; Put, K.; Brisse, E.; Filtjens, J.; Uyttenhove, C.; Van Snick, J.; Liston, A.; Leclercq, G.; et al. Systemic Juvenile Idiopathic Arthritis-like Syndrome in Mice Following Stimulation of the Immune System With Freund’s Complete Adjuvant: Regulation by Interferon-γ. Arthritis Rheumatol. 2014, 66, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, K.G.; Jonak, Z.L.; Trulli, S.; Gollob, J.A. IL-12 induces monocyte IL-18 binding protein expression via IFN-γ. J. Immunol. 2002, 168, 2282–2287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Mangan, P.R.; Weaver, C.T. Expanding the effector CD4 T-cell repertoire: The Th17 lineage. Curr. Opin. Immunol. 2006, 18, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Oukka, M.; Kuchroo, V.; Bettelli, E. Th17 cells: Effector T cells with inflammatory properties. Semin. Immunol. 2007, 19, 362–371. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Chen, Y.; Tato, C.M.; Laurence, A.; Joyce-Shaikh, B.; Blumenschein, W.M.; McClanahan, T.K.; O’Shea, J.J.; Cua, D.J. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 2009, 10, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Blumenschein, W.M.; Brovont-Porth, K.; McGeachy, M.J.; Basham, B.; Desai, B.; Pierce, R.; McClanahan, T.K.; Sadekova, S.; de Waal Malefyt, R. Human Th17 cells comprise heterogeneous subsets including IFN-γ-producing cells with distinct properties from the Th1 lineage. J. Immunol. 2010, 185, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Kelchtermans, H.; De, K.B.; Mitera, T.; Van, B.M.; Bullens, D.; Billiau, A.; Leclercq, G.; Matthys, P. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: An important factor in pathogenesis, counter-regulated by endogenous IFN-γ. Arthritis Res. Ther. 2005, 7, R402–R415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, A.A.; Hunt, N.H.; Ball, H.J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: Characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Refaeli, Y.; van Parijs, L.; Alexander, S.I.; Abbas, A.K. Interferon γ is required for activation-induced death of T lymphocytes. J. Exp. Med. 2002, 196, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Dalton, D.K.; Haynes, L.; Chu, C.Q.; Swain, S.L.; Wittmer, S. Interferon γ eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J. Exp. Med. 2000, 192, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Reljic, R. IFN-γ therapy of tuberculosis and related infections. J. Interferon Cytokine Res. 2007, 27, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Kastner, D.L. Familial autoinflammatory diseases: Genetics, pathogenesis and treatment. Curr. Opin. Rheumatol. 2005, 17, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Mellins, E.D.; Macaubas, C.; Grom, A.A. Pathogenesis of systemic juvenile idiopathic arthritis: Some answers, more questions. Nat. Rev. Rheumatol. 2011, 7, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Still, G.F. On a Form of Chronic Joint Disease in Children. Med. Chir. Trans. 1897, 80, 47–60. [Google Scholar] [PubMed]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar] [PubMed]
- Prakken, B.; Albani, S.; Martini, A. Juvenile idiopathic arthritis. Lancet 2011, 377, 2138–2149. [Google Scholar] [CrossRef]
- Ravelli, A.; Martini, A. Juvenile idiopathic arthritis. Lancet 2007, 369, 767–778. [Google Scholar] [CrossRef]
- Gowdie, P.J.; Tse, S.M. Juvenile idiopathic arthritis. Pediatr. Clin. N. Am. 2012, 59, 301–327. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Beukelman, T.; Gallo, L.; Spangler, J.; Rosenkranz, M.; Arkachaisri, T.; Ayala, R.; Groh, B.; Finkel, T.H.; Cron, R.Q. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: Data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J. Rheumatol. 2008, 35, 343–348. [Google Scholar] [PubMed]
- De Benedetti, F.; Schneider, R. Systemic juvenile idiopathic arthritis. In Textbook of Pediatric Rheumatology; Cassidy, J.T., Laxer, R.M., Petty, R.E., Lindsey, C.B., Eds.; Saunders: Philadelphia, PA, USA, 2011; pp. 236–248. [Google Scholar]
- Woo, P. Systemic juvenile idiopathic arthritis: Diagnosis, management, and outcome. Nat. Clin. Pract. Rheumatol. 2006, 2, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.A.; Grom, A.A. Update on the pathogenesis and treatment of systemic idiopathic arthritis. Curr. Opin. Pediatr. 2011, 23, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.; Remmers, E.F.; Grom, A.A.; Thomson, W.; Martini, A.; Gattorno, M.; Ozen, S.; Gul, A.; Bohnsack, J.F.; Zeft, A.S.; et al. Systemic juvenile idiopathic arthritis is associated with HLA-DRB1 in Europeans and Americans of European descent. Pediatr. Rheumatol. 2012, 10, A6. [Google Scholar] [CrossRef]
- Ombrello, M.; Remmers, E.F.; Grom, A.A.; Thomson, W.; Martini, A.; Gattorno, M.; Ozen, S. Genome-Wide Association Meta-Analysis of Eight Independent Systemic Juvenile Idiopathic Arthritis Collections Reveals Regional Association Spanning the Major Histocompatibility Complex Class II and III Gene Cluster. In Proceedings of the ACR/ARHP Annual Meeting, Washington, DC, USA, 9–14 November 2012; pp. S1126–S1126.
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J. Clin. Investig. 1998, 102, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Seki, N.; Kamizono, S.; Higuchi, T.; Hirata, T.; Miyata, K.; Ohkuni, M.; Tatsuzawa, O.; Yokota, S.; Joo, K.; et al. Identification of a genetic risk factor for systemic juvenile rheumatoid arthritis in the 5ʹ-flanking region of the TNFα gene and HLA genes. Arthritis Rheum. 1999, 42, 2577–2582. [Google Scholar] [CrossRef]
- Stock, C.J.; Ogilvie, E.M.; Samuel, J.M.; Fife, M.; Lewis, C.M.; Woo, P. Comprehensive association study of genetic variants in the IL-1 gene family in systemic juvenile idiopathic arthritis. Genes Immun. 2008, 9, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, E.M.; Fife, M.S.; Thompson, S.D.; Twine, N.; Tsoras, M.; Moroldo, M.; Fisher, S.A.; Lewis, C.M.; Prieur, A.-M.; Glass, D.N.; et al. The -174G allele of the interleukin-6 gene confers susceptibility to systemic arthritis in children: A multicenter study using simplex and multiplex juvenile idiopathic arthritis families. Arthritis Rheum. 2003, 48, 3202–3206. [Google Scholar] [CrossRef] [PubMed]
- Fife, M.S.; Gutierrez, A.; Ogilvie, E.M.; Stock, C.J.; Samuel, J.M.; Thomson, W.; Mack, L.F.; Lewis, C.M.; Woo, P. Novel IL10 gene family associations with systemic juvenile idiopathic arthritis. Arthritis Res. Ther. 2006, 8, R148. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.D.; Miller, J.J., III; Bernstein, B.; Shafai, T. Consumption coagulopathy associated with systemic juvenile rheumatoid arthritis. J. Pediatr. 1983, 103, 872–876. [Google Scholar] [CrossRef]
- Grom, A.A.; Mellins, E.D. Macrophage activation syndrome: Advances towards understanding pathogenesis. Curr. Opin. Rheumatol. 2010, 22, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A. Macrophage activation syndrome. Curr. Opin. Rheumatol. 2002, 14, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; Behrens, E.M. Not all hemophagocytes are created equally: Appreciating the heterogeneity of the hemophagocytic syndromes. Curr. Opin. Rheumatol. 2012, 24, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Vastert, S.J.; Kuis, W.; Grom, A.A. Systemic JIA: New developments in the understanding of the pathophysiology and therapy. Best Pract. Res. Clin. Rheumatol. 2009, 23, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.L.; Zeller, J.; Hubert, P.; Herbelin, C.; Dayer, J.M.; Prieur, A.M. Macrophage activation syndrome and rheumatic disease in childhood: A report of four new cases. Clin. Exp. Rheumatol. 1993, 11, 451–456. [Google Scholar] [PubMed]
- Ramanan, A.V.; Grom, A.A. Does systemic-onset juvenile idiopathic arthritis belong under juvenile idiopathic arthritis? Rheumatology 2005, 44, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Grom, A.A. Macrophage activation syndrome and reactive hemophagocytic lymphohistiocytosis: The same entities? Curr. Opin. Rheumatol. 2003, 15, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M. Macrophage activation syndrome in rheumatic disease: What is the role of the antigen presenting cell? Autoimmun. Rev. 2008, 7, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Beukelman, T.; Paessler, M.; Cron, R.Q. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J. Rheumatol. 2007, 34, 1133–1138. [Google Scholar] [PubMed]
- Grom, A.A. Natural killer cell dysfunction: A common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis? Arthritis Rheum. 2004, 50, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Allen, C.E.; Weitzman, S.; Filipovich, A.H.; McClain, K.L. How I treat hemophagocytic lymphohistiocytosis. Blood 2011, 118, 4041–4052. [Google Scholar] [CrossRef] [PubMed]
- Janka, G.E. Familial and acquired hemophagocytic lymphohistiocytosis. Annu. Rev. Med. 2012, 63, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Grom, A.A.; Behrens, E.M.; Cron, R.Q. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: Diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012, 13, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S.; Woo, P.; Murray, K.J. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch. Dis. Child. 2001, 85, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lehmberg, K.; Pink, I.; Eulenburg, C.; Beutel, K.; Maul-Pavicic, A.; Janka, G. Differentiating macrophage activation syndrome in systemic juvenile idiopathic arthritis from other forms of hemophagocytic lymphohistiocytosis. J. Pediat. 2013, 162, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Quartier, P.; Taupin, P.; Bourdeaut, F.; Lemelle, I.; Pillet, P.; Bost, M.; Sibilia, J.; Koné-Paut, I.; Gandon-Laloum, S.; LeBideau, M.; et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 2003, 48, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Ponchio, L.; de, B.F.; Ravelli, A.; Rosti, V.; Beguin, Y.; Invernizzi, R.; Barosi, G.; Martini, A. Defective iron supply for erythropoiesis and adequate endogenous erythropoietin production in the anemia associated with systemic-onset juvenile chronic arthritis. Blood 1996, 87, 4824–4830. [Google Scholar] [PubMed]
- De Benedetti, F.; Meazza, C.; Oliveri, M.; Pignatti, P.; Vivarelli, M.; Alonzi, T.; Fattori, E.; Garrone, S.; Barreca, A.; Martini, A. Effect of IL-6 on IGF binding protein-3: A study in IL-6 transgenic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology 2001, 142, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Avau, A.; Put, K.; Wouters, C.H.; Matthys, P. Cytokine balance and cytokine-driven natural killer cell dysfunction in systemic juvenile idiopathic arthritis. Cytokine Growth Factor Rev. 2015, 26, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.; Horneff, G.; Brik, R.; McCann, L.; Kasapcopur, O.; Rutkowska-Sak, L.; et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Quartier, P.; Allantaz, F.; Cimaz, R.; Pillet, P.; Messiaen, C.; Bardin, C.; Bossuyt, X.; Boutten, A.; Bienvenu, J.; Duquesne, A.; et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann. Rheum. Dis. 2011, 70, 747–754. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Brunner, H.I.; Ruperto, N.; Kenwright, A.; Wright, S.; Calvo, I.; Cuttica, R.; Ravelli, A.; Schneider, R.; Woo, P.; et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2385–2395. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.; Wilkinson, N.; Prieur, A.M.; Southwood, T.; Leone, V.; Livermore, P.; Wythe, H.; Thomson, D.; Kishimoto, T. Open label phase II trial of single, ascending doses of MRA in Caucasian children with severe systemic juvenile idiopathic arthritis: Proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged clinical improvement. Arthritis Res. Ther. 2005, 7, R1281–R1288. [Google Scholar] [PubMed]
- Canna, S.W. Editorial: Interferon-γ: Friend or foe in systemic juvenile idiopathic arthritis and adult-onset Still’s Disease? Arthritis Rheumatol. 2014, 66, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Yokoyama, T.; Yamada, K.; Kaneda, H.; Wada, H.; Wada, T.; Toma, T.; Ohta, K.; Kasahara, Y.; Yachie, A. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology 2010, 49, 1645–1653. [Google Scholar]
- Shimizu, M.; Nakagishi, Y.; Yachie, A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine 2013, 61, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Gattorno, M.; Piccini, A.; Lasiglie, D.; Tassi, S.; Brisca, G.; Carta, S.; Delfino, L.; Ferlito, F.; Pelagatti, M.A.; Caroli, F.; et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- De Kleer, I.M.; Brinkman, D.M.; Ferster, A.; Abinun, M.; Quartier, P.; van Der Net, J.; Ten Cate, R.; Wedderburn, L.R.; Horneff, G.; Oppermann, J.; et al. Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: Analysis of clinical effects, mortality, and transplant related morbidity. Ann. Rheum. Dis. 2004, 63, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Wulffraat, N.M.; Brinkman, D.; Ferster, A.; Opperman, J.; ten, C.R.; Wedderburn, L.; Foster, H.; Abinun, M.; Prieur, A.M.; Horneff, G.; et al. Long-term follow-up of autologous stem cell transplantation for refractory juvenile idiopathic arthritis. Bone Marrow Transplant. 2003, 32, S61–S64. [Google Scholar] [CrossRef] [PubMed]
- Sandborg, C.; Mellins, E.D. A new era in the treatment of systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2439–2440. [Google Scholar] [CrossRef] [PubMed]
- Put, K.; Avau, A.; Brisse, E.; Mitera, T.; Put, S.; Proost, P.; Bader-Meunier, B.; Westhovens, R.; van den Eynde, B.J.; Orabona, C.; et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: Tipping the balance between interleukin-18 and interferon-γ. Rheumatology 2015, 54, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Osugi, Y.; Hara, J.; Tagawa, S.; Takai, K.; Hosoi, G.; Matsuda, Y.; Ohta, H.; Fujisaki, H.; Kobayashi, M.; Sakata, N.; et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 1997, 89, 4100–4103. [Google Scholar] [PubMed]
- Akashi, K.; Hayashi, S.; Gondo, H.; Mizuno, S.; Harada, M.; Tamura, K.; Yamasaki, K.; Shibuya, T.; Uike, N.; Okamura, T. Involvement of interferon-γ and macrophage colony-stimulating factor in pathogenesis of haemophagocytic lymphohistiocytosis in adults. Br. J. Haematol. 1994, 87, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.D.; Roskams, T.; van Damme-Lombaerts, R.; Matthys, P.; Wouters, C. Macrophage activation syndrome: Characteristic findings on liver biopsy illustrating the key role of activated, IFN-γ-producing lymphocytes and IL-6- and TNF-α-producing macrophages. Blood 2005, 105, 1648–1651. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Hildeman, D.; Kappler, J.; Marrack, P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon γ are essential for the disorder. Blood 2004, 104, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Pachlopnik, S.J.; Ho, C.H.; Chretien, F.; Lefebvre, J.M.; Pivert, G.; Kosco-Vilbois, M.; Ferlin, W.; Geissmann, F.; Fischer, A.; de Saint Basile, G. Neutralization of IFNγ defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol. Med. 2009, 1, 112–124. [Google Scholar]
- De Jager, W.; Vastert, S.J.; Beekman, J.M.; Wulffraat, N.M.; Kuis, W.; Coffer, P.J.; Prakken, B.J. Defective phosphorylation of interleukin-18 receptor β causes impaired natural killer cell function in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009, 60, 2782–2793. [Google Scholar] [CrossRef] [PubMed]
- Fall, N.; Barnes, M.; Thornton, S.; Luyrink, L.; Olson, J.; Ilowite, N.T.; Gottlieb, B.S.; Griffin, T.; Sherry, D.D.; Thompson, S.; et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007, 56, 3793–3804. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, E.M.; Khan, A.; Hubank, M.; Kellam, P.; Woo, P. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007, 56, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.A.; Fall, N.; Thornton, S.; Grom, A.A. The limited role of interferon-γ in systemic juvenile idiopathic arthritis cannot be explained by cellular hyporesponsiveness. Arthritis Rheum. 2012, 64, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Yoshikai, Y.; Matsuzaki, G.; Inoue, T.; Nomoto, K. An increase in number of T-cell receptor γ/δ-bearing T cells in athymic nude mice treated with complete Freund’s adjuvants. Immunology 1990, 70, 61–65. [Google Scholar] [PubMed]
- Billiau, A.; Matthys, P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar] [PubMed]
- Janis, E.M.; Kaufmann, S.H.; Schwartz, R.H.; Pardoll, D.M. Activation of γ δ T cells in the primary immune response to Mycobacterium tuberculosis. Science 1989, 244, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Matthys, P.; Vermeire, K.; Mitera, T.; Heremans, H.; Huang, S.; Schols, D.; de Wolf-Peeters, C.; Billiau, A. Enhanced autoimmune arthritis in IFN-γ receptor-deficient mice is conditioned by mycobacteria in Freund’s adjuvant and by increased expansion of Mac-1+ myeloid cells. J. Immunol. 1999, 163, 3503–3510. [Google Scholar] [PubMed]
- Murray, P.J.; Young, R.A.; Daley, G.Q. Hematopoietic remodeling in interferon-γ-deficient mice infected with mycobacteria. Blood 1998, 91, 2914–2924. [Google Scholar] [PubMed]
- Vermeire, K.; Heremans, H.; Vandeputte, M.; Huang, S.; Billiau, A.; Matthys, P. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J. Immunol. 1997, 158, 5507–5513. [Google Scholar] [PubMed]
- Manoury-Schwartz, B.; Chiocchia, G.; Bessis, N.; Abehsira-Amar, O.; Batteux, F.; Muller, S.; Huang, S.; Boissier, M.C.; Fournier, C. High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J. Immunol. 1997, 158, 5501–5506. [Google Scholar] [PubMed]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 1996, 156, 5–7. [Google Scholar] [PubMed]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 2009, 361, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Oukka, M.; Kuchroo, V.K. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007, 8, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Berghmans, N.; Nuyts, A.; Uyttenhove, C.; van Snick, J.; Opdenakker, G.; Heremans, H. Interferon-γ orchestrates the number and function of Th17 cells in experimental autoimmune encephalomyelitis. J. Interferon Cytokine Res. 2011, 31, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Kelchtermans, H.; Schurgers, E.; Geboes, L.; Mitera, T.; van Damme, J.; van Snick, J.; Uyttenhove, C.; Matthys, P. Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-γ and counteraction by interferon-γ. Arthritis Res. Ther. 2009, 11, R122. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Q.; Swart, D.; Alcorn, D.; Tocker, J.; Elkon, K.B. Interferon-γ regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007, 56, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Doodes, P.D.; Cao, Y.; Hamel, K.M.; Wang, Y.; Rodeghero, R.L.; Mikecz, K.; Glant, T.T.; Iwakura, Y.; Finnegan, A. IFN-γ regulates the requirement for IL-17 in proteoglycan-induced arthritis. J. Immunol. 2010, 184, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Matthys, P.; Lories, R.J.; De, K.B.; Heremans, H.; Luyten, F.P.; Billiau, A. Dependence on interferon-γ for the spontaneous occurrence of arthritis in DBA/1 mice. Arthritis Rheum. 2003, 48, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.; Matthys, P. Collagen-induced arthritis and related animal models: How much of their pathogenesis is auto-immune, how much is auto-inflammatory? Cytokine Growth Factor Rev. 2011, 22, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Marcovecchio, M.L.; Mohn, A.; Chiarelli, F. Inflammatory cytokines and growth in childhood. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 57–62. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Alonzi, T.; Moretta, A.; Lazzaro, D.; Costa, P.; Poli, V.; Martini, A.; Ciliberto, G.; Fattori, E. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation. J. Clin. Investig. 1997, 99, 643–650. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Meazza, C.; Martini, A. Role of interleukin-6 in growth failure: An animal model. Horm Res. 2002, 58, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, R.; Carvello, F.; Scianaro, R.; Pasquale, L.D.; Vivarelli, M.; Petrini, S.; Bracci-Laudiero, L.; de Benedetti, F. Chronic exposure to interleukin-6 amplifies the response to toll-like receptor ligands: Implication on the pathogenesis of macrophage activation syndrome. Arthritis Rheum. 2011, 9, 210. [Google Scholar] [CrossRef]
- Bracaglia, C.; Caiello, I.; de Graaf, K.; D’Ario, G.; Guilhot, F.; Ferlin, W.; Melli, L.; Prencipe, G.; Davì, S.; Schulert, G.; et al. Interferon-γ (IFNy) in macrophage activation syndrome (MAS) associated with systemic juvenile idiopathic arthritis (sJIA). High levels in patients and a role in a murine mas model. Pediatr. Rheumatol. 2014, 12, O3. [Google Scholar] [CrossRef]
- Behrens, E.M.; Canna, S.W.; Slade, K.; Rao, S.; Kreiger, P.A.; Paessler, M.; Kambayashi, T.; Koretzky, G.A. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J. Clin. Investig. 2011, 121, 2264–2277. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; Wrobel, J.; Chu, N.; Kreiger, P.A.; Paessler, M.; Behrens, E.M. Interferon-γ mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis. Arthritis Rheum. 2013, 65, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Cifaldi, L.; Prencipe, G.; Caiello, I.; Bracaglia, C.; Locatelli, F.; de Benedetti, F.; Strippoli, R. Inhibition of Natural Killer Cell Cytotoxicity by Interleukin-6: Implications for the Pathogenesis of Macrophage Activation Syndrome. Arthritis Rheumatol. 2015, 67, 3037–3046. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.H.; Maher, S.G.; Young, H.A. Clinical Use of Interferon-γ. Ann. N. Y. Acad. Sci. 2009, 1182, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Panitch, H.S.; Hirsch, R.L.; Haley, A.S.; Johnson, K.P. Exacerbations of multiple sclerosis in patients treated with γ interferon. Lancet 1987, 1, 893–895. [Google Scholar] [CrossRef]
- Skurkovich, S.; Boiko, A.; Beliaeva, I.; Buglak, A.; Alekseeva, T.; Smirnova, N.; Kulakova, O.; Tchechonin, V.; Gurova, O.; Deomina, T.; et al. Randomized study of antibodies to IFN-γ and TNF-α in secondary progressive multiple sclerosis. Mult. Scler. 2001, 7, 277–284. [Google Scholar] [PubMed]
- Lemmel, E.M.; Brackertz, D.; Franke, M.; Gaus, W.; Hartl, P.W.; Machalke, K.; Mielke, H.; Obert, H.J.; Peter, H.H.; Sieper, J.; et al. Results of a multicenter placebo-controlled double-blind randomized phase III clinical study of treatment of rheumatoid arthritis with recombinant interferon-γ. Rheumatol. Int. 1988, 8, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, E.; Billiau, A.; Matthys, P. Collagen-induced arthritis as an animal model for rheumatoid arthritis: Focus on interferon-γ. J. Interferon Cytokine Res. 2011, 31, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Sigidin, Y.A.; Loukina, G.V.; Skurkovich, B.; Skurkovich, S. Randomized, double-blind trial of anti-interferon-γ antibodies in rheumatoid arthritis. Scand. J. Rheumatol. 2001, 30, 203–207. [Google Scholar] [PubMed]
- Coto, C.; Varela, G.; Hernandez, V.; Del Rosario, M.; Lopez-Saura, P. Use of recombinant interferon γ in pediatric patients with advanced juvenile chronic arthritis. Biotherapy 1998, 11, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Pernice, W.; Schuchmann, L.; Dippell, J.; Suschke, J.; Vogel, P.; Truckenbrodt, H.; Schindera, F.; Humburg, C.; Brzoska, J. Therapy for systemic juvenile rheumatoid arthritis with γ-interferon: A pilot study of nine patients. Arthritis Rheum. 1989, 32, 643–646. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avau, A.; Matthys, P. Therapeutic Potential of Interferon-? and Its Antagonists in Autoinflammation: Lessons from Murine Models of Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Pharmaceuticals 2015, 8, 793-815. https://doi.org/10.3390/ph8040793
Avau A, Matthys P. Therapeutic Potential of Interferon-? and Its Antagonists in Autoinflammation: Lessons from Murine Models of Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Pharmaceuticals. 2015; 8(4):793-815. https://doi.org/10.3390/ph8040793
Chicago/Turabian StyleAvau, Anneleen, and Patrick Matthys. 2015. "Therapeutic Potential of Interferon-? and Its Antagonists in Autoinflammation: Lessons from Murine Models of Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome" Pharmaceuticals 8, no. 4: 793-815. https://doi.org/10.3390/ph8040793
APA StyleAvau, A., & Matthys, P. (2015). Therapeutic Potential of Interferon-? and Its Antagonists in Autoinflammation: Lessons from Murine Models of Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Pharmaceuticals, 8(4), 793-815. https://doi.org/10.3390/ph8040793