
Targeting TRPM2 in ROS-Coupled Diseases

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TRPM2 Activators and Inhibitors
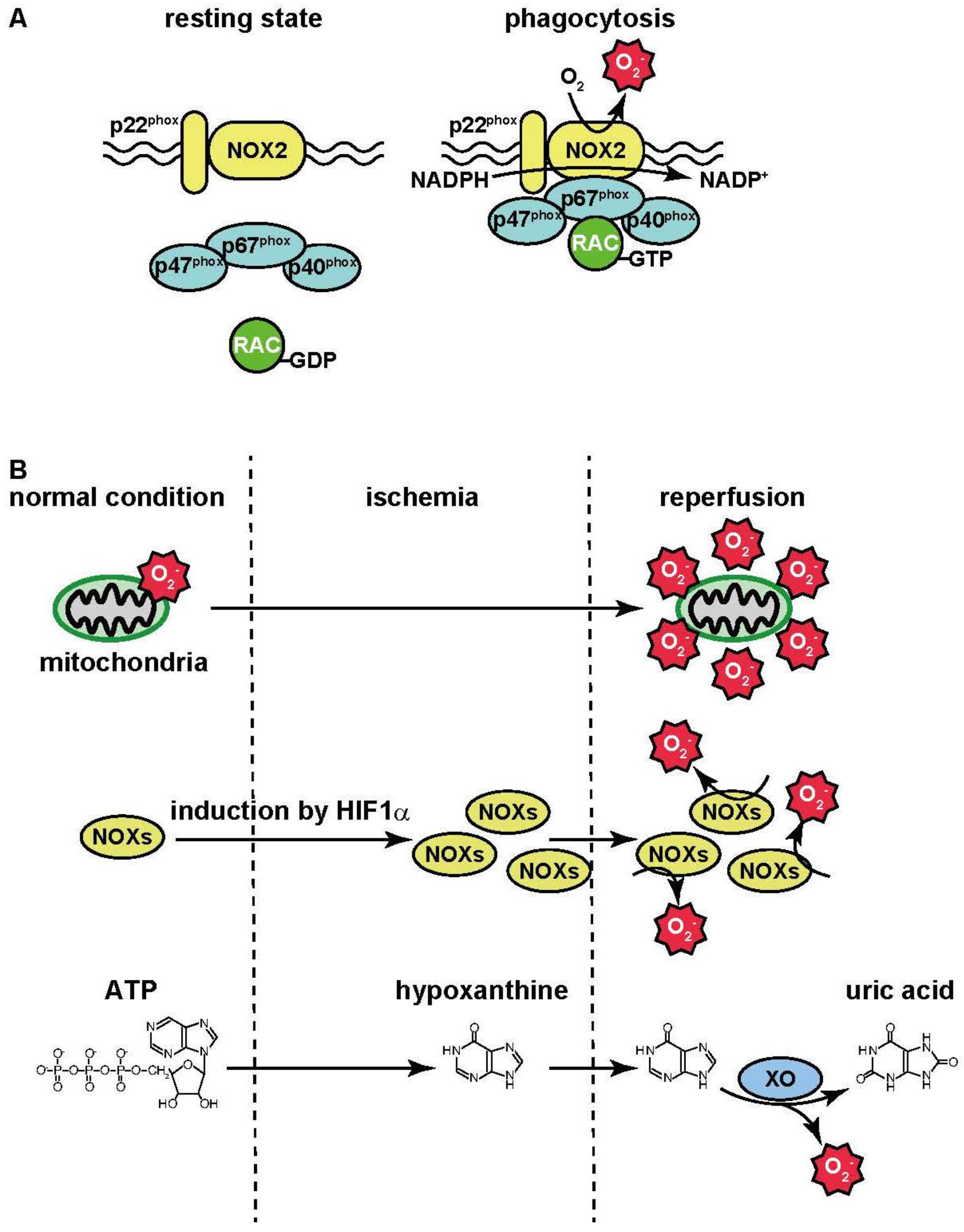
3. ROS Production under Pathological Conditions
3.1. Inflammation
3.2. Ischemia-Reperfusion
4. ROS-Coupled Diseases and TRPM2
4.1. Inflammatory Diseases
4.1.1. TRPM2-Mediated Chemokine Production
4.1.2. LPS-Induced Inflammatory Responses and TRPM2
4.1.3. Functional Roles of TRPM2 during Infection
4.1.4. NLRP3 Inflammasome and TRPM2
4.2. Ischemia-Reperfusion Injury
4.2.1. Brain
4.2.2. Heart
4.2.3. Kidneys
4.3. Other Diseases and Injuries
4.3.1. Acetaminophen-Induced Liver Injury
4.3.2. Radiation-Induced Tissue Damage
4.3.3. Alzheimer’s Disease
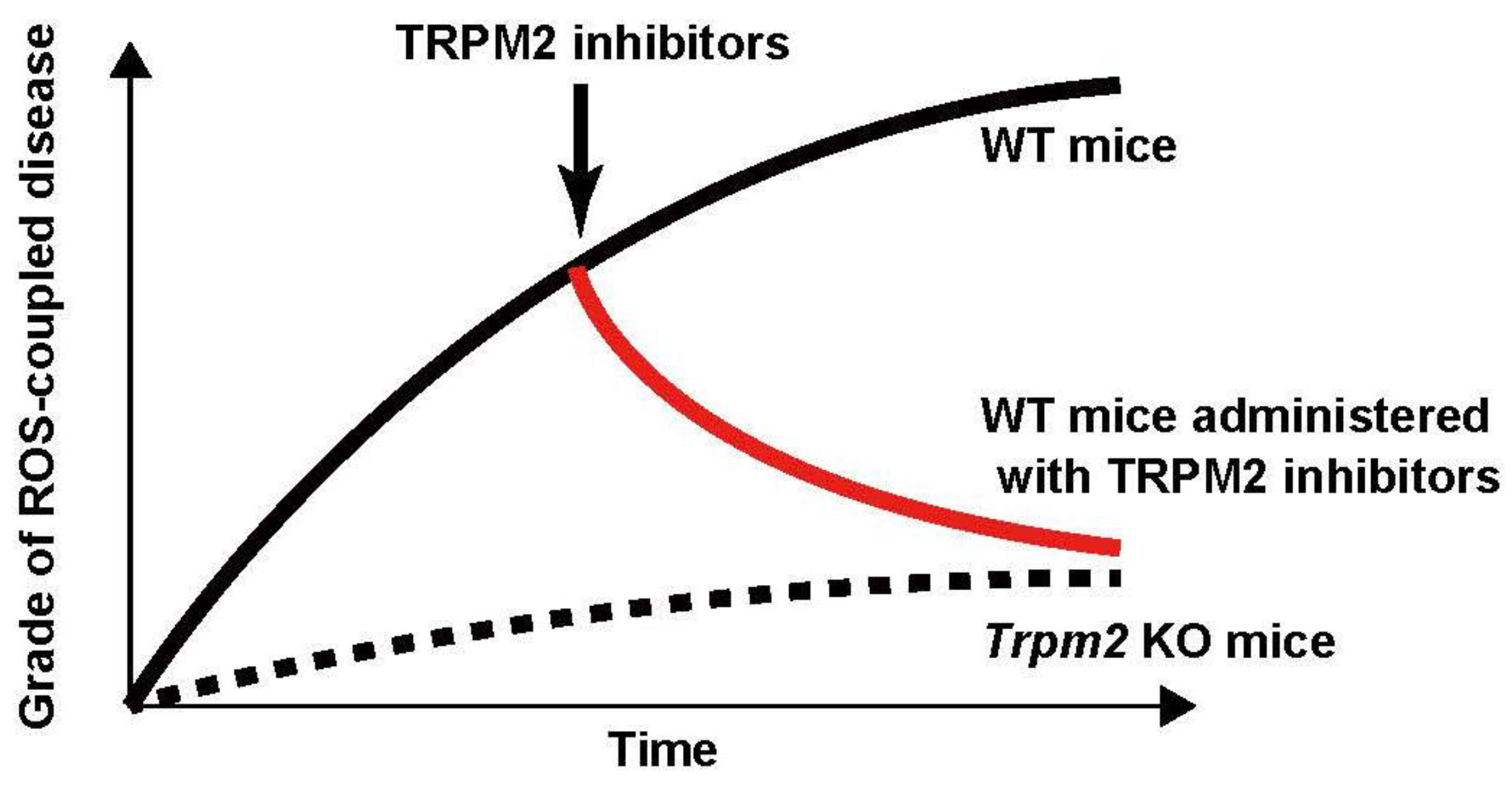
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Clapham, D.E. Calcium signaling. Cell 1995, 80, 259–268. [Google Scholar] [CrossRef]
- Montell, C.; Rubin, G.M. Molecular characterization of the Drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective Association of TRPC Channel Subunits in Rat Brain Synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Sudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [PubMed]
- Okada, T.; Inoue, R.; Yamazaki, K.; Maeda, A.; Kurosaki, T.; Yamakuni, T.; Tanaka, I.; Shimizu, S.; Ikenaka, K.; Imoto, K.; et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. J. Biol. Chem. 1999, 274, 27359–27370. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Okada, T.; Onoue, H.; Hara, Y.; Shimizu, S.; Naitoh, S.; Ito, Y.; Mori, Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ. Res. 2001, 88, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [PubMed]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [PubMed]
- Güler, A.D.; Lee, H.; Iida, T.; Shimizu, I.; Tominaga, M.; Caterina, M. Heat-evoked activation of the ion channel, TRPV4. J. Neurosci. 2002, 22, 6408–6414. [Google Scholar] [PubMed]
- Peier, A.M.; Reeve, A.J.; Andersson, D.A.; Moqrich, A.; Earley, T.J.; Hergarden, A.C.; Story, G.M.; Colley, S.; Hogenesch, J.B.; McIntyre, P.; et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 2002, 296, 2046–2049. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Gunthorpe, M.J.; Kelsell, R.E.; Hayes, P.D.; Reilly, P.; Facer, P.; Wright, J.E.; Jerman, J.C.; Walhin, J.P.; Ooi, L.; et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 2002, 418, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ramsey, I.S.; Kotecha, S.A.; Moran, M.M.; Chong, J.A.; Lawson, D.; Ge, P.; Lilly, J.; Silos-Santiago, I.; Xie, Y.; et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 2002, 418, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Liedtke, W.; Choe, Y.; Marti-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef]
- Strotmann, R.; Harteneck, C.; Nunnenmacher, K.; Schultz, G.; Plant, T.D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000, 2, 695–702. [Google Scholar] [PubMed]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Peier, A.M.; Moqrich, A.; Hergarden, A.C.; Reeve, A.J.; Andersson, D.A.; Story, G.M.; Earley, T.J.; Dragoni, I.; McIntyre, P.; Bevan, S.; et al. A TRP channel that senses cold stimuli and menthol. Cell 2002, 108, 705–715. [Google Scholar] [CrossRef]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Tsuchiya, K.; Nitta, K. Autosomal dominant polycystic kidney disease: Recent advances in pathogenesis and potential therapies. Clin. Exp. Nephrol. 2013, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, H.B.; Cooke, F.T.; Jat, P.; Boucheron, C.; Koizumi, T.; Hayakawa, M.; Kaizawa, H.; Ohishi, T.; Workman, P.; Waterfield, M.D.; et al. A selective PIKfyve inhibitor blocks PtdIns(3,5)P2 production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008, 9, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Mizuno, Y.; Kozai, D.; Yamamoto, S.; Kiyonaka, S.; Shibata, T.; Uchida, K.; Mori, Y. Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels 2008, 2, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, L.J.; Geierstanger, B.H.; Viswanath, V.; Bandell, M.; Eid, S.R.; Hwang, S.; Patapoutian, A. The pungency of garlic: Activation of TRPA1 and TRPV1 in response to allicin. Curr. Biol. 2005, 15, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.E.; Bautista, D.M.; Chuang, H.H.; McKemy, D.D.; Zygmunt, P.M.; Hogestatt, E.D.; Meng, I.D.; Julius, D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kuwaki, T.; Kiyonaka, S.; Numata, T.; Kozai, D.; Mizuno, Y.; Yamamoto, S.; Naito, S.; Knevels, E.; Carmeliet, P.; et al. TRPA1 underlies a sensing mechanism for O2. Nat. Chem. Biol. 2011, 10, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henricks, P.A.; Nijkamp, F.P. Reactive oxygen species as mediators in asthma. Pulm. Pharmacol. Ther. 2001, 14, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Soga, M.; Matsuzawa, A.; Ichijo, H. Oxidative stress-induced diseases via the ASK1 signaling pathway. Int. J. Cell Biol. 2012, 2012. article ID 439587. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Kawakami, S.; Hara, Y.; Wakamori, M.; Itoh, E.; Minami, T.; Takada, Y.; Kume, T.; Katsuki, H.; Mori, Y.; et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J. Pharmacol. Sci. 2006, 101, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Wang, J.; Hirschler-Laszkiewicz, I.; Gao, E.; Song, J.; Zhang, X.; Koch, W.J.; Madesh, M.; Mallilankaraman, K.; Gu, T.; et al. The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1010–H1022. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Yamamoto, S.; Partida-Sánchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 functions as lysosomal Ca2+ release channel in β-cells. Sci. Signal. 2009, 71, ra23. [Google Scholar] [CrossRef] [PubMed]
- Partida-Sánchez, S.; Gasser, A.; Fliegert, R.; Siebrands, C.C.; Dammermann, W.; Shi, G.; Mousseau, B.J.; Sumazo-Toledo, A.; Bhagat, H.; Walseth, T.F.; et al. Chemotaxis of mouse bone marrow neutrophils and dendritic cell is controlled by ADP-ribose, the major product generated by the CD38 enzyme reaction. J. Immunol. 2007, 179, 7827–7839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chu, X.; Tong, Q.; Cheung, J.Y.; Conrad, K.; Masker, K.; Miller, B.A. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J. Biol. Chem. 2003, 278, 16222–16229. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Wehage, E.; Eisfeld, J.; Heiner, I.; Jungling, E.; Zitt, C.; Luckhoff, A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 2002, 277, 23150–23156. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Kühn, F.J.; Lückhoff, A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Starkus, J.; Beck, A.; Fleig, A.; Penner, R. Regulation of TRPM2 by extra- and intracellular calcium. J. Gen. Physiol. 2007, 130, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Penner, R.; Fleig, A.; Beck, A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium 2008, 44, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xie, J.; Yue, L. Intracellular calcium activates TRPM2 and tis alternative spliced isoforms. Proc. Natl. Acad. Sci. USA 2009, 106, 7239–7244. [Google Scholar] [CrossRef] [PubMed]
- Heiner, I.; Eisfeld, J.; Warnstedt, M.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.; Csanády, L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J. Biol. Chem. 2010, 285, 30091–30102. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.; Iordanov, I.; Csanády, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2’-phosphate. J. Gen. Physiol. 2015, 145, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubisha, O.; Rafty, L.A.; Takanishi, C.L.; Xu, X.; Tong, L.; Perraud, A.L.; Scharenberg, A.M.; Denu, J.M. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J. Biol. Chem. 2006, 281, 14057–14065. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed]
- Magnone, M.; Bauer, I.; Poggi, A.; Mannino, E.; Sturla, L.; Brini, M.; Zocchi, E.; De Flora, A.; Nencioni, A.; Bruzzone, S. NAD+ levels control Ca2+ sore replenishment and mitogen-induced increase of cytosolic Ca2+ by cyclic ADP-ribose-dependent TRPM2 channel gating in human T lymphocytes. J. Biol. Chem. 2012, 287, 21067–21081. [Google Scholar] [CrossRef] [PubMed]
- Tanuma, S.; Yagi, T.; Johnson, G.S. Endogenous ADP ribosylation of high mobility group proteins 1 and 2 and histone H1 following DNA damage in intact cells. Arch. Biochem. Biophys. 1985, 237, 38–42. [Google Scholar] [CrossRef]
- De Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Oliver, F.J.; Menissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Virág, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Fonfria, E.; Marshall, I.C.; Benham, C.D.; Boyfield, I.; Brown, J.D.; Hill, K.; Hughes, J.P.; Skaper, S.D.; McNulty, S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 2004, 143, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Buelow, B.; Song, Y.; Andrew, M.; Scharenberg, A.M. The poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Shimizu, S.; Hara, Y.; Hagiwara, T.; Miyazaki, A.; Mori, Y.; Kiuchi, Y. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium 2006, 39, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yonezawa, R.; Negoro, T.; Yamamoto, S.; Numata, T.; Ishii, M.; Mori, Y.; Toda, T. Sensitization of H2O2-induced TRPM2 activation and subsequent interleukin-8 (CXCL8) production by intracellular Fe2+ in human monocytic U937 cells. Int. J. Biochem. Cell Biol. 2015, 68, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tong, Q.; Conrad, K.; Wozney, J.; Cheung, J.Y.; Miller, B.A. Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am. J. Physiol. Cell Physiol. 2007, 292, C1746–C1758. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; McNulty, S.; Randall, A.D. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn-Schmiedeberg's Arch. Pharmacol. 2004, 370, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Benham, C. D.; McNulty, S.; Randall, A.D. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology 2004, 47, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Inada1, H.; Tominaga, M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br. J. Pharmacol. 2008, 153, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.R.; Akbar, S.; Eweida, M.; Kühn, F.J.; Gustafsson, A.J.; Lückhoff, A.; Islam, M.S. H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the beta-cells: Involvement of TRPM2 channels. J. Cell. Mol. Med. 2009, 13, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Kheradpezhouh, E.; Barritt, G.J.; Rychkov, G.Y. Curcumin inhibits activation of TRPM2 channels in rat hepatocytes. Redox Biol. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yonezawa, R.; Hagiwara, T.; Yoshida, T.; Takahashi, N.; Hamano, S.; Negoro, T.; Toda, T.; Wakamori, M.; Mori, Y.; et al. Inhibitory effects of AG490 on H2O2-induced TRPM2-mediated Ca2+ entry. Eur. J. Pharmacol. 2014, 742, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Yamamoto, S.; Yonezawa, R.; Mori, Y.; Shimizu, S. Inhibitory effects of Tyrphostin AG-related compounds on oxidative stress-sensitive transient receptor potential channel activation. Eur. J. Pharmacol. 2016, 786, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Berton, G.; Castaldi, M.A.; Cassatella, M.A.; Nauseef, W.M. Celebrating the 50th anniversary of the seminal discovery that the phagocyte respiratory burst enzyme is an NADPH oxidase. J. Leukoc. Biol. 2015, 97, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receproe signaling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Kohchi, C.; Inagawa, H.; Nishizawa, T.; Soma, G. ROS and innate immunity. Anticancer Res. 2009, 29, 817–822. [Google Scholar] [PubMed]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of liver injury. I. TNF-α-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabó, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Luster, A.D. Chemokines—Chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [PubMed]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, R.; Yamamoto, S.; Takenaka, M.; Kage, Y.; Negoro, T.; Toda, T.; Ohbayashi, M.; Numata, T.; Nakano, Y.; Yamamoto, T.; et al. TRPM2 channels in alveolar epithelial cells mediate bleomycin-induced lung inflammation. Free Radic. Biol. 2016, 90, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wehrhahn, J.; Kraft, R.; Harteneck, C.; Hauschildt, S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J. Immunol. 2010, 184, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Hardaker, L.; Bahra, P.; de Billy, B.C.; Freeman, M.; Kupfer, N.; Wyss, D.; Trifilieff, A. The ion channel transient receptor potential melastatin-2 does not play a role in inflammatory mouse models of chronic obstructive pulmonary diseases. Respir. Res. 2012, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Gao, X.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2012, 13, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, N.P.; Soni, S.; Rajaram, M.V.; Dang, P.M.; Reilly, T.J.; El-Benna, J.; Clay, C.D.; Schlesinger, L.S.; Gunn, J.S. Francisella acid phosphatases inactivate the NADPH oxidase in human phagocytes. J. Imunnol. 2010, 184, 5141–5150. [Google Scholar] [CrossRef] [PubMed]
- Shakerley, N.L.; Chandrasekaran, A.; Trebak, M.; Miller, B.A.; Melendez, J.A. Francisella tularensis catalase restricts immune function by impairing TRPM2 channel activity. J. Imunnol. 2016, 291, 3871–3881. [Google Scholar]
- Knowles, H.; Heizer, J.W.; Li, Y.; Chapman, K.; Ogden, C.A.; Andreasen, K.; Shapland, E.; Kucera, G.; Mogan, J.; Humann, J.; et al. Transient receptor potential melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2011, 108, 11578–11583. [Google Scholar] [CrossRef] [PubMed]
- Partida-Sánchez, S.; Cockayne, D.A.; Monard, S.; Jacobson, E.L.; Oppenheimer, N.; Garvy, B.; Kusser, K.; Goodrich, S.; Howard, M.; Harmsen, A.; et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001, 7, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signaling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Surmeier, D.J.; Reiner, A. NMDA and non-NMDA receptor-mediated excitotoxicity are potentiated in cultured striatal neurons by prior chronic depolarization. Exp. Neurol. 1999, 159, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signaling. Nat. Rev. Neurosci. 2007, 8, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Verma, S.; Nakayama, S.; Quillinan, N.; Grafe, M.R.; Hurn, P.D.; Herson, P.S. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J. Cereb. Blood Flow Metab. 2011, 31, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Quillinan, N.; Yang, Y.; Nakayama, S.; Cheng, J.; Kelley, M.H.; Herson, P.S. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci. Lett. 2012, 530, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Macey, T.A.; Quillinan, N.; Klawitter, J.; Perraud, A.L.; Traystman, R.J.; Herson, P.S. Androgen and PARP-1 regulation of TRPM2 channels after ischemic injury. J. Cereb. Blood Flow Metab. 2013, 33, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Alim, I.; Teves, L.; Li, R.; Mori, Y.; Tymianski, M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability of ischemic cell death. J. Neurosci. 2013, 33, 17264–17277. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Melzer, N.; Schattling, B.; Gob, E.; Hicking, G.; Arunachalam, P.; Bittner, S.; Ufer, F.; Herrmann, A.M.; Bernreuther, C.; et al. Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke 2014, 45, 3395–3402. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Hoffman, N.E.; Merali, S.; Zhang, X.Q.; Wang, J.; Rajan, S.; Shanmughapriya, S.; Gao, E.; Barrero, C.A.; Mallilankaraman, K.; et al. TRPM2 cahnnels protect against cardiac ischemia-reperfusion injury. J. Biol. Chem. 2014, 289, 7615–7629. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Miller, B.A.; Wang, J.; Elrod, J.W.; Rajan, S.; Gao, E.; Song, J.; Zhang, X.; Hirschler-Laszkiewicz, I.; Shanmughapriya, S.; et al. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H637–H650. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, T.; Wajima, T.; Nrgoro, T.; Ishii, M.; Nakano, Y.; Kiuchi, Y.; Mori, Y.; Shimizu, S. Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ishchemia/reperfusion injury. Cardiovasc. Res. 2013, 97, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Wang, W.; Tadagavadi, R.K.; Briley, N.E.; Love, M.I.; Miller, B.A.; Reeves, W.B. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Investig. 2014, 124, 4989–5001. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.G.; Eastham, W.N. Acute liver necrosis following overdose of paracetamol. BMJ 1966, 2, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. role of drug metabolism. J. Pharmacol. Exp. Ther. 1971, 187, 185–194. [Google Scholar]
- Kheradpezhouh, E.; Ma, L.; Morphett, A.; Barritt, G.J.; Rychkov, G.Y. TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. USA 2014, 111, 3176–3181. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cotrim, A.; Teos, L.; Zheng, C.; Swaim, W.; Mitchell, J.; Mori, Y.; Ambudkar, I. Loss of TRPM2 function protects against irradiation-induced salivary gland dysfunction. Nat. Commun. 2013, 4, 1515. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Wajima, T.; Hara, Y.; Nishida, M.; Mori, Y. Transient receptor potential channels in Alzheimer’s disease. Biochim. Biophys. Acta 2007, 1772, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Hemmelgarn, B.T.; Chuang, C.; Besr, T.M. The role of oxidative stress-induced epigenetic alterations in amyloid-β production in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2015, 2015. Article ID 604658. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, S.; Shimizu, S. Targeting TRPM2 in ROS-Coupled Diseases. Pharmaceuticals 2016, 9, 57. https://doi.org/10.3390/ph9030057
Yamamoto S, Shimizu S. Targeting TRPM2 in ROS-Coupled Diseases. Pharmaceuticals. 2016; 9(3):57. https://doi.org/10.3390/ph9030057
Chicago/Turabian StyleYamamoto, Shinichiro, and Shunichi Shimizu. 2016. "Targeting TRPM2 in ROS-Coupled Diseases" Pharmaceuticals 9, no. 3: 57. https://doi.org/10.3390/ph9030057
APA StyleYamamoto, S., & Shimizu, S. (2016). Targeting TRPM2 in ROS-Coupled Diseases. Pharmaceuticals, 9(3), 57. https://doi.org/10.3390/ph9030057