
Aptamers: A New Technological Platform in Cancer Immunotherapy

Abstract
:1. Introduction
2. Oligonucleotide Targeting Aptamers
3. Immune-Checkpoint Antagonizing Aptamers
4. Immunostimulatory Aptamers
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. New Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory hodgkin’s lymphoma. New Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. New Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.F.; Proverbs-Singh, T.A.; Postow, M.A. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: A review. JAMA Oncol. 2016, 2, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Morales-Kastresana, A.; Sanmamed, M.F.; Rodriguez, I.; Palazon, A.; Martinez-Forero, I.; Labiano, S.; Hervas-Stubbs, S.; Sangro, B.; Ochoa, C.; Rouzaut, A.; et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin. Cancer Res. 2013, 19, 6151–6162. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Blind, M.; Blank, M. Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef]
- Tolle, F.; Wilke, J.; Wengel, J.; Mayer, G. By-product formation in repetitive pcr amplification of DNA libraries during selex. PLoS ONE 2014, 9, e114693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, S.; Hirabayashi, N.; Kato, S.; Akitomi, J.; Egashira, H.; Tanaka, T.; Waga, I.; Ohtsu, T. Effective isolation of RNA aptamer through suppression of PCR bias. Biochem. Biophys. Res. Commun. 2009, 386, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.K.; Chang, J.L.; Burke, D.H. Fastaptamer: A bioinformatic toolkit for high-throughput sequence analysis of combinatorial selections. Mol. Ther. Nucleic Acids 2015, 4, e230. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Pastor, F.; Villanueva, H.; Soldevilla, M.M.; Hervas-Stubbs, H.; Oyarzabal, J. In-silico aptamer docking studies. From a retrospective validation to a prospective case study: TIM3 aptamers binding. Mol. Ther. Nucleic Acids 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.E.; Wu, H.; Niu, Y.; Cai, J. Improving the stability of aptamers by chemical modification. Curr. Med. Chem. 2011, 18, 4126–4138. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Yamashige, R.; Matsunaga, K.; Yokoyama, S.; Hirao, I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 2013, 31, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Fecher, L.A.; Agarwala, S.S.; Hodi, F.S.; Weber, J.S. Ipilimumab and its toxicities: A multidisciplinary approach. Oncologist 2013, 18, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Strahotin, S.; Hewes, B.; Zhang, B.; Zhang, Y.; Archer, D.; Spencer, T.; Dillehay, D.; Kwon, B.; Chen, L.; et al. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. J. Immunol. 2007, 178, 4194–4213. [Google Scholar] [CrossRef] [PubMed]
- Oney, S.; Lam, R.T.; Bompiani, K.M.; Blake, C.M.; Quick, G.; Heidel, J.D.; Liu, J.Y.; Mack, B.C.; Davis, M.E.; Leong, K.W.; et al. Development of universal antidotes to control aptamer activity. Nat. Med. 2009, 15, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Rudilla, F.; Arribillaga, L.; Llopiz, D.; Riezu-Boj, J.I.; Lozano, T.; Lopez-Sagaseta, J.; Guembe, L.; Sarobe, P.; Prieto, J.; et al. A peptide inhibitor of Foxp3 impairs regulatory T cell activity and improves vaccine efficacy in mice. J. Immunol. 2010, 185, 5150–5159. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Wu, J.; Riling, C.; Kodrasov, M.; Weinstock, J.; Sokirniy, I.; Mattern, M.; Kambayashi, T.; Kumar, S. Abstract 2228: Cbl-b inhibitors as novel intra-cellular checkpoint inhibitors for cancer immunotherapy. Cancer Res. 2016, 76, 2228–2228. [Google Scholar] [CrossRef]
- Lozano, T.; Villanueva, L.; Durantez, M.; Gorraiz, M.; Ruiz, M.; Belsue, V.; Riezu-Boj, J.I.; Hervas-Stubbs, S.; Oyarzabal, J.; Bandukwala, H.; et al. Inhibition of Foxp3/nfat interaction enhances T cell function after tcr stimulation. J. Immunol. 2015, 195, 3180–3189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hossain, D.M.; Duttagupta, P.; Moreira, D.; Zhao, X.; Won, H.; Buettner, R.; Nechaev, S.; Majka, M.; Zhang, B.; et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood 2016, 127, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Lutz-Nicoladoni, C.; Wolf, D.; Sopper, S. Modulation of immune cell functions by the E3 ligase cbl-b. Front. Oncol. 2015, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Symonds, A.L.; Martin, J.E.; Kioussis, D.; Wraith, D.C.; Li, S.; Wang, P. Early growth response gene 2 (egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J. Exp. Med. 2008, 205, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents t cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Bachmaier, K.; Krawczyk, C.; Kozieradzki, I.; Kong, Y.Y.; Sasaki, T.; Oliveira-dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [PubMed]
- Zheng, Y.; Zha, Y.; Driessens, G.; Locke, F.; Gajewski, T.F. Transcriptional regulator early growth response gene 2 (egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med. 2012, 209, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent rna aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar] [PubMed]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.; Cydzik, M.; Gariepy, J. Targeting the PD-1/PD-l1 immune evasion axis with DNA aptamers as a novel therapeutic strategy for the treatment of disseminated cancers. Mol. Ther. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef] [PubMed]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting TIM-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Hervas-Stubbs, S.; Soldevilla, M.M.; Villanueva, H.; Mancheno, U.; Bendandi, M.; Pastor, F. Identification of TIM3 2′-fluoro oligonucleotide aptamer by HT-SELEX for cancer immunotherapy. Oncotarget 2016, 7, 4522–4530. [Google Scholar] [PubMed]
- Berezhnoy, A.; Stewart, C.A.; McNamara, J.O.; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of sirnas with aptamer-sirna chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.W.; Reebye, V.; Mintz, P.; Tien, Y.W.; Lai, H.S.; Saetrom, P.; Reccia, I.; Swiderski, P.; Armstrong, B.; et al. Targeted delivery of c/ebpalpha-sarna by pancreatic ductal adenocarcinoma-specific RNA aptamers inhibits tumor growth in vivo. Mol. Ther. 2016, 24, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Battig, M.R.; Huang, Y.; Chen, N.; Wang, Y. Aptamer-functionalized superporous hydrogels for sequestration and release of growth factors regulated via molecular recognition. Biomaterials 2014, 35, 8040–8048. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, T.; Zhao, J.; Huang, Y.; Deng, H.; Kumar, A.; Wang, C.; Liang, Z.; Ma, X.; Liang, X.J. Multifunctional aptamer-based nanoparticles for targeted drug delivery to circumvent cancer resistance. Biomaterials 2016, 91, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Lozano, T.; Soldevilla, M.M.; Casares, N.; Villanueva, H.; Bendandi, M.; Lasarte, J.J.; Pastor, F. Targeting inhibition of Foxp3 by a CD28 2′-fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials 2016, 91, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zheng, J.; Song, E.; Donovan, M.; Zhang, K.; Liu, C.; Tan, W. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc. Natl. Acad. Sci. USA 2013, 110, 7998–8003. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Cerchia, L.; Catuogno, S.; de Vita, G.; Dassie, J.P.; Santamaria, G.; Swiderski, P.; Condorelli, G.; Giangrande, P.H.; de Franciscis, V. Multifunctional aptamer-mirna conjugates for targeted cancer therapy. Mol. Ther. 2014, 22, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic administration of optimized aptamer-sirna chimeras promotes regression of psma-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.; Dos Santos, C.; Zhang, Q.; Kozlowska, A.; Liu, H.; Gao, C.; Moreira, D.; Swiderski, P.; Jozwiak, A.; Kline, J.; et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood 2014, 123, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In vivo delivery of sirna to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Priceman, S.J.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; Forman, S.J.; et al. CTLA4 aptamer delivers stat3 sirna to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J. Clin. Investig. 2013, 124, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. 2007, 7, 95–106. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1bb binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Investig. 2008, 118, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Benaduce, A.P.; Brenneman, R.; Schrand, B.; Pollack, A.; Gilboa, E.; Ishkanian, A. 4-1bb aptamer based immunomodulation enhances the therapeutic index of radiotherapy in murine tumor models. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Soldevilla, M.M.; Villanueva, H.; Kolonias, D.; Inoges, S.; de Cerio, A.L.; Kandzia, R.; Klimyuk, V.; Gleba, Y.; Gilboa, E.; et al. CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids 2013, 2, e98. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Villanueva, H.; Bendandi, M.; Inoges, S.; Lopez-Diaz de Cerio, A.; Pastor, F. 2-fluoro-rna oligonucleotide CD40 targeted aptamers for the control of b lymphoma and bone-marrow aplasia. Biomaterials 2015, 67, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Soontornworajit, B.; Wang, Y. A temperature-responsive antibody-like nanostructure. Biomacromolecules 2010, 11, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Salek-Ardakani, S.; Mittler, R.S.; Croft, M. Hypercostimulation through 4-1BB distorts homeostasis of immune cells. J. Immunol. 2009, 182, 6753–6762. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Kolonias, D.; McNamara, J.O., 2nd; Gilboa, E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol. Ther. 2011, 19, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Schrand, B.; Berezhnoy, A.; Brenneman, R.; Williams, A.; Levay, A.; Kong, L.Y.; Rao, G.; Zhou, S.; Heimberger, A.B.; Gilboa, E. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol. Res. 2014, 2, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Villanueva, H.; Casares, N.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; de Cerio, A.L.-D.; Pastor, F. MRP1-CD28 bi-specific oligonucleotide aptamers: Target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget 2016, 7, 23182–23196. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Small, E.; Demkow, T.; Gerritsen, W.R.; Rolland, F.; Hoskin, P.; Smith, D.C.; Parker, C.; Chondros, D.; Ma, J.; Hege, K. A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel versus docetaxel plus prednisone in symptomatic, castration-resistant prostate cancer (CRPC). In Proceedings of the 2009 Genitourinary Cancers Symposium, Orlando, FL, USA, 26–28 February 2009; American Society of Clinical Oncology: Alexandria, VA, USA, 2009. [Google Scholar]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to C-4 blockade in melanoma. New Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrors, B.; Vascotto, F.; Castle, J.C.; et al. Mutant mhc class П epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Povsic, T.J.; Vavalle, J.P.; Aberle, L.H.; Kasprzak, J.D.; Cohen, M.G.; Mehran, R.; Bode, C.; Buller, C.E.; Montalescot, G.; Cornel, J.H.; et al. A phase 2, randomized, partially blinded, active-controlled study assessing the efficacy and safety of variable anticoagulation reversal using the reg1 system in patients with acute coronary syndromes: Results of the radar trial. Eur. Heart J. 2013, 34, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Ganson, N.J.; Povsic, T.J.; Sullenger, B.A.; Alexander, J.H.; Zelenkofske, S.L.; Sailstad, J.M.; Rusconi, C.P.; Hershfield, M.S. Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a pegylated rna aptamer. J. Allergy Clin. Immunol. 2016, 137, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
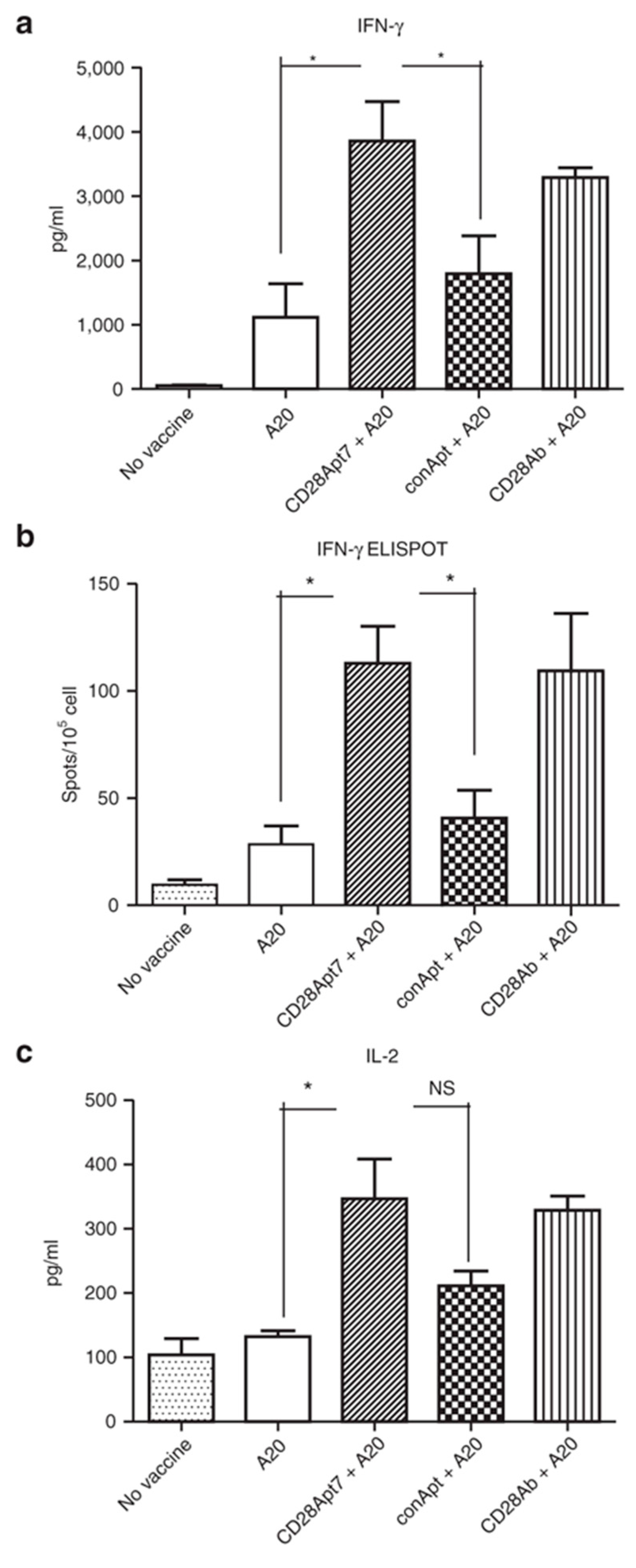

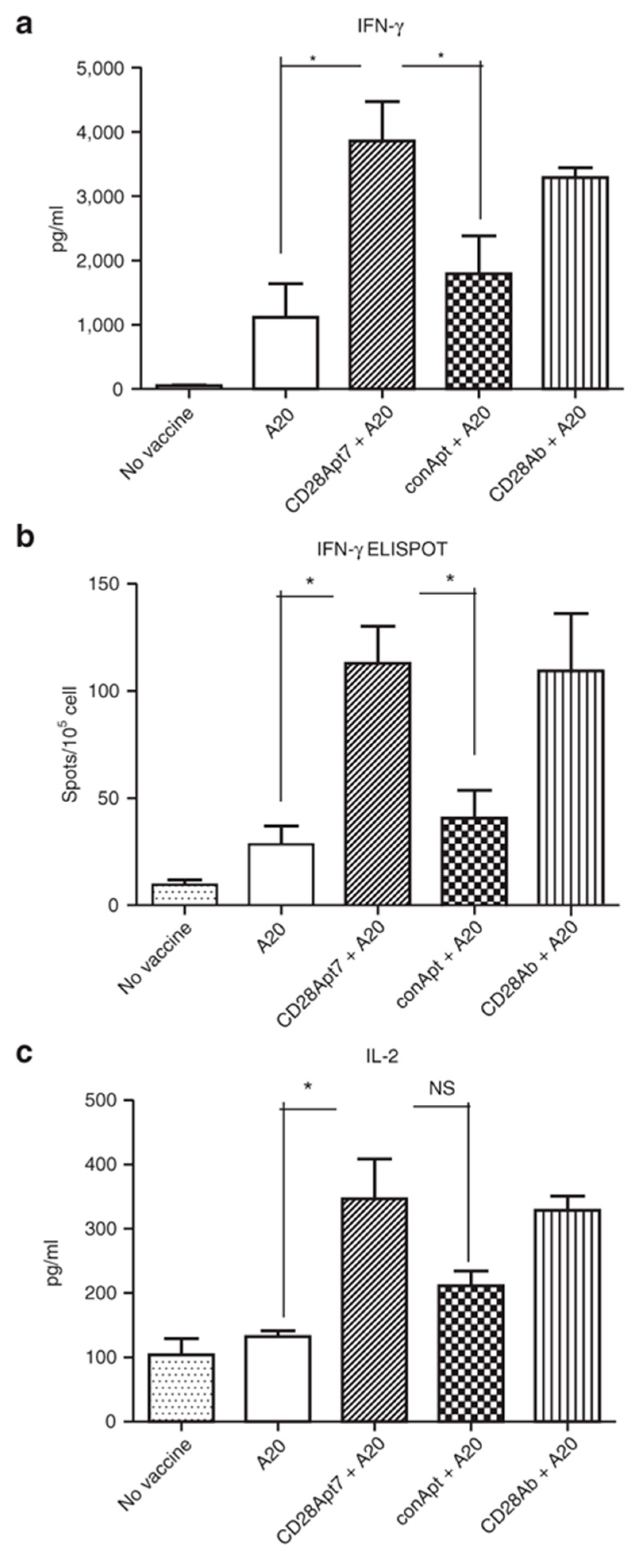{kind=link}
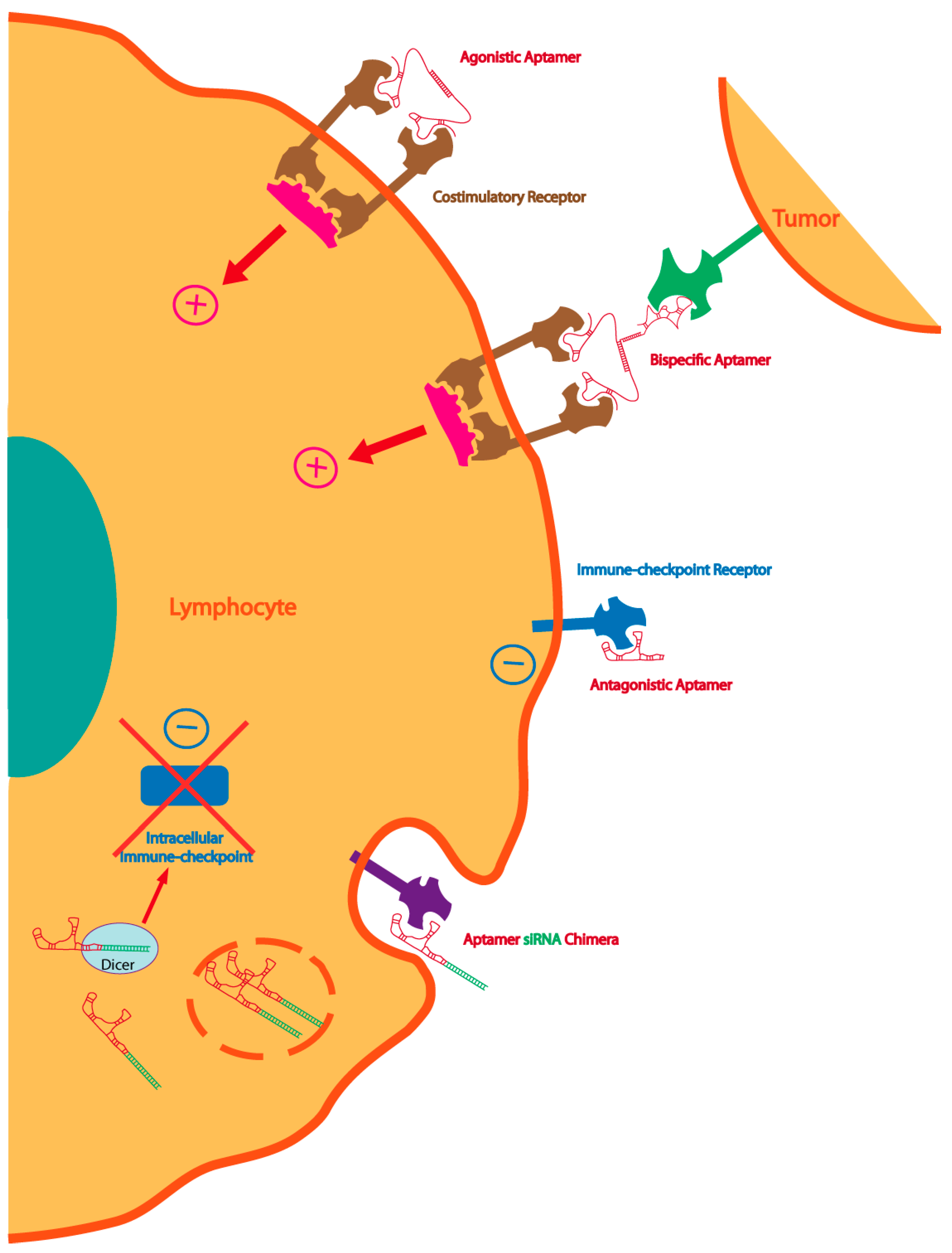
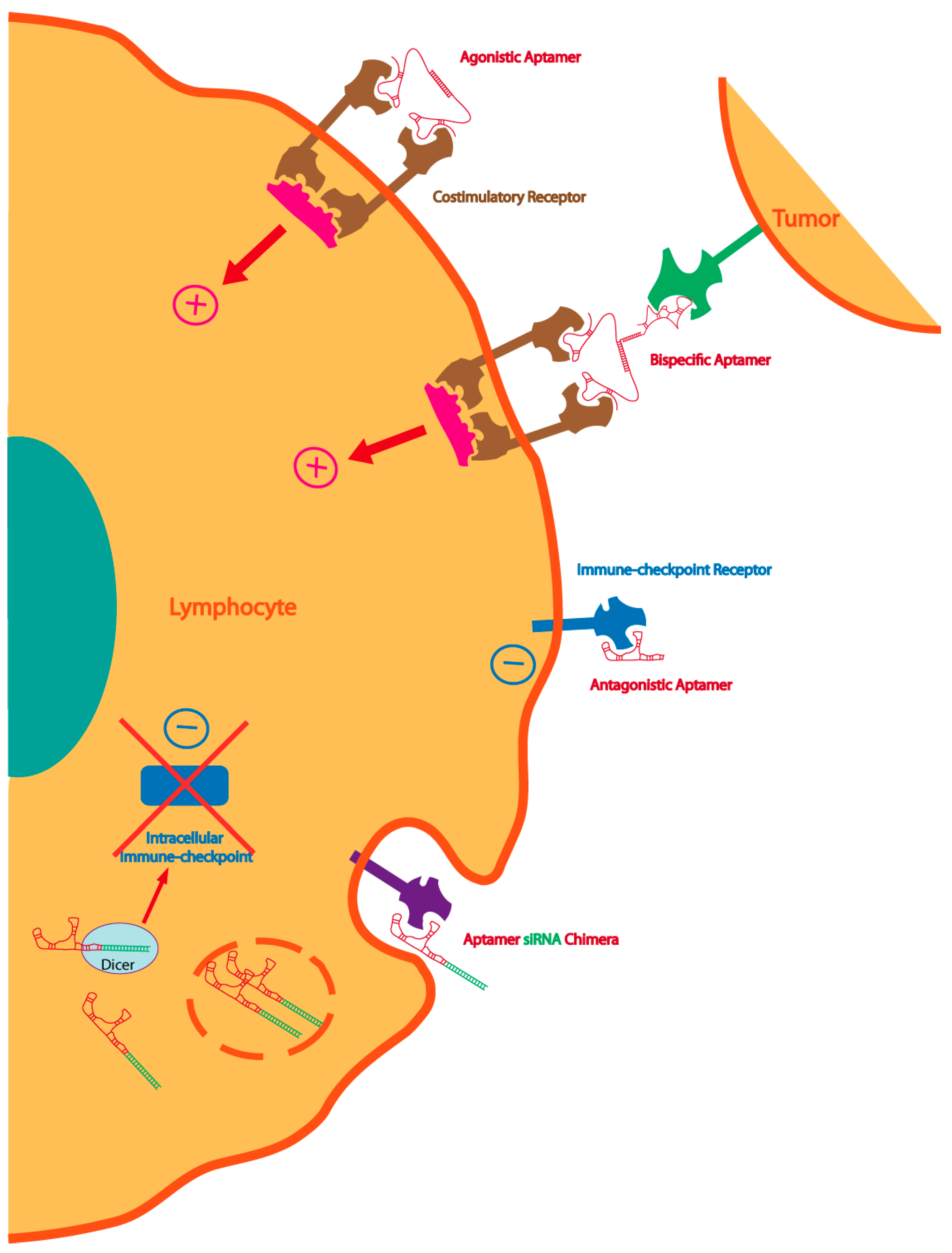{kind=link}
| Aptamer | Application in Cancer Immunotherapy | Type of Tumor |
|---|---|---|
| CTLA-4 | Immune-checkpoint blockade [36] | Melanoma |
| Targeting STAT3 siRNA [53] | Lymphoma, Colon Cancer, Kidney Cancer, Fibrosarcoma | |
| PD1 | Immune-checkpoint blockade [37] | Colon Cancer |
| TIM3 | Immune-checkpoint blockade [39] | Colon Cancer |
| IL10R | Immune-checkpoint blockade [40] | Colon Cancer |
| 4-1BB | Costimulatory receptor agonist [56] | Mastocytoma |
| Targeting costimulation to the tumor [64,65] | Melanoma, Colon Cancer, Breast Cancer, Oncogene-induced high-grade Glioma, MCA Fibrosarcomas | |
| OX40 | Costimulatory receptor agonist [59] | Melanoma |
| CD28 | Costimulatory receptor agonist [60] | Lymphoma |
| Targeting costimulation to the tumor [66] | Melanoma | |
| CD40 | Stimulatory receptor agonist [61] | Lymphoma |
© 2016 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastor, F. Aptamers: A New Technological Platform in Cancer Immunotherapy. Pharmaceuticals 2016, 9, 64. https://doi.org/10.3390/ph9040064
Pastor F. Aptamers: A New Technological Platform in Cancer Immunotherapy. Pharmaceuticals. 2016; 9(4):64. https://doi.org/10.3390/ph9040064
Chicago/Turabian StylePastor, Fernando. 2016. "Aptamers: A New Technological Platform in Cancer Immunotherapy" Pharmaceuticals 9, no. 4: 64. https://doi.org/10.3390/ph9040064
APA StylePastor, F. (2016). Aptamers: A New Technological Platform in Cancer Immunotherapy. Pharmaceuticals, 9(4), 64. https://doi.org/10.3390/ph9040064