
Use of Capsaicin to Treat Pain: Mechanistic and Therapeutic Considerations


{kind=link}
Abstract
:1. Introduction
2. Therapeutic Uses of Capsaicin
3. Functional and Histological Effects
3.1. TRPV1-Expressing Nociceptors
3.2. Variations in Acute Pungency of Capsaicin
3.3. Pungency and Therapeutic Effect?
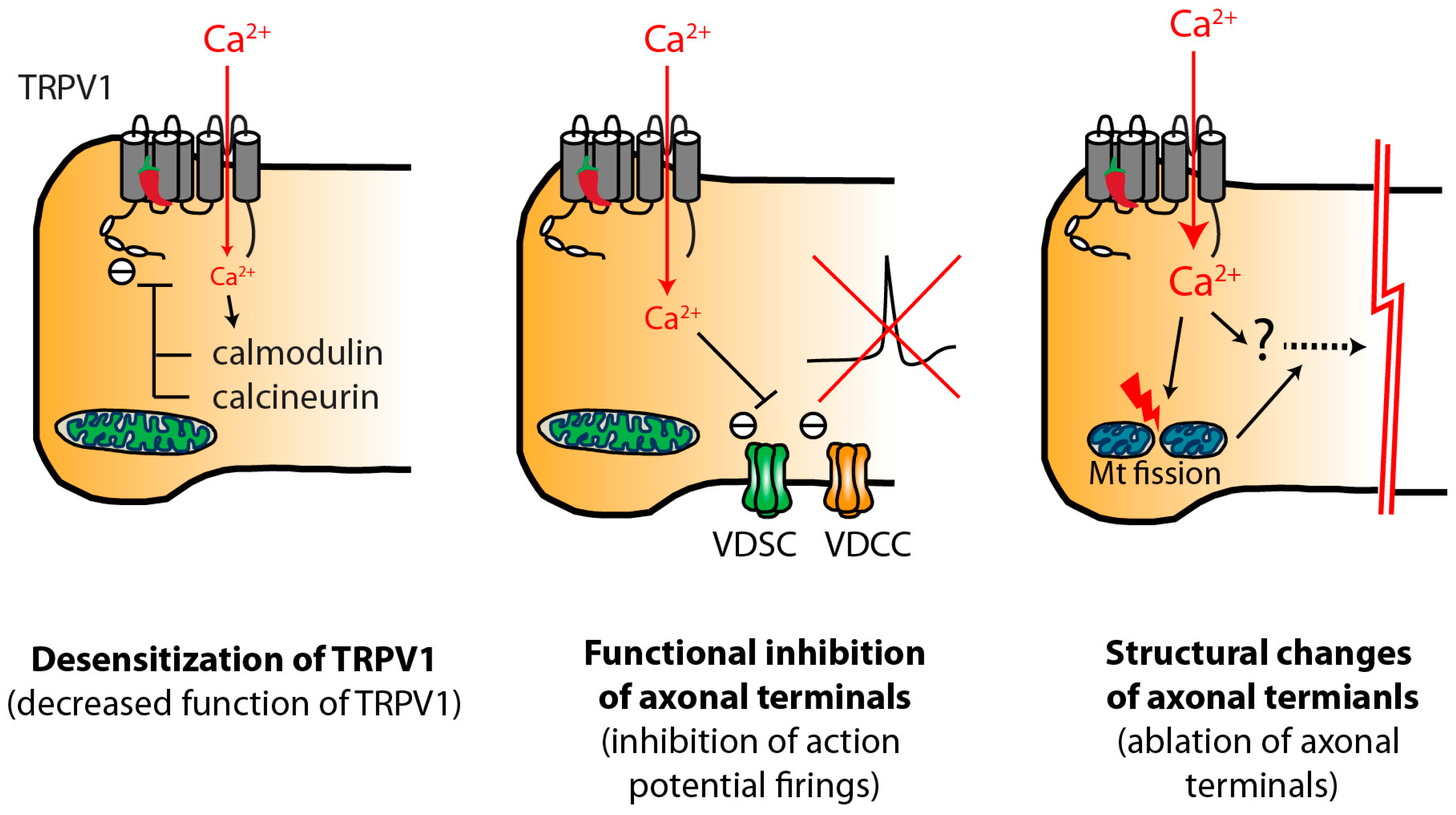
3.4. Transient Analgesia and Decreased Function of TRPV1 Mechanisms
3.5. Long Acting Effects of Capsaicin
4. Potential Mechanisms of Vanilloid-Induced Chemical Ablation of Nociceptor Terminals
5. Clinical Correlates
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [PubMed]
- Chaiyasit, K.; Khovidhunkit, W.; Wittayalertpanya, S. Pharmacokinetic and the effect of capsaicin in capsicum frutescens on decreasing plasma glucose level. J. Med. Assoc. Thai. 2009, 92, 108–113. [Google Scholar] [PubMed]
- Kearney, T.; Hiatt, P.; Birdsall, E.; Smollin, C. Pepper spray injury severity: Ten-year case experience of a poison control system. Prehosp. Emerg. Care 2014, 18, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Spiteller, D. Capsaicin: Tailored chemical defence against unwanted “frugivores”. Chembiochem 2009, 10, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Szolcsanyi, J.; Sann, H.; Pierau, F.K. Nociception in pigeons is not impaired by capsaicin. Pain 1986, 27, 247–260. [Google Scholar] [CrossRef]
- Andersson, J.L.; Lilja, A.; Hartvig, P.; Langstrom, B.; Gordh, T.; Handwerker, H.; Torebjork, E. Somatotopic organization along the central sulcus, for pain localization in humans, as revealed by positron emission tomography. Exp. Brain Res. 1997, 117, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Iadarola, M.J.; Berman, K.F.; Zeffiro, T.A.; Byas-Smith, M.G.; Gracely, R.H.; Max, M.B.; Bennett, G.J. Neural activation during acute capsaicin-evoked pain and allodynia assessed with pet. Brain 1998, 121 Pt 5, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Baron, Y.; Disbrow, E.; Roberts, T.P. Brain processing of capsaicin-induced secondary hyperalgesia: A functional mri study. Neurology 1999, 53, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Szolcsanyi, J.; Pinter, E. Transient receptor potential vanilloid 1 as a therapeutic target in analgesia. Expert Opin. Ther. Targets 2013, 17, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A.; Blumberg, P.M. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol. Rev. 1999, 51, 159–212. [Google Scholar] [PubMed]
- Jancso, G.; Kiraly, E.; Jancso-Gabor, A. Pharmacologically induced selective degeneration of chemosensitive primary sensory neurones. Nature 1977, 270, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Jancso, G.; Kiraly, E.; Joo, F.; Such, G.; Nagy, A. Selective degeneration by capsaicin of a subpopulation of primary sensory neurons in the adult rat. Neurosci. Lett. 1985, 59, 209–214. [Google Scholar] [CrossRef]
- Simone, D.A.; Nolano, M.; Johnson, T.; Wendelschafer-Crabb, G.; Kennedy, W.R. Intradermal injection of capsaicin in humans produces degeneration and subsequent reinnervation of epidermal nerve fibers: Correlation with sensory function. J. Neurosci. 1998, 18, 8947–8959. [Google Scholar] [PubMed]
- Rage, M.; Van Acker, N.; Facer, P.; Shenoy, R.; Knaapen, M.W.; Timmers, M.; Streffer, J.; Anand, P.; Meert, T.; Plaghki, L. The time course of CO2 laser-evoked responses and of skin nerve fibre markers after topical capsaicin in human volunteers. Clin. Neurophysiol. 2010, 121, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Polydefkis, M.; Hauer, P.; Sheth, S.; Sirdofsky, M.; Griffin, J.W.; McArthur, J.C. The time course of epidermal nerve fibre regeneration: Studies in normal controls and in people with diabetes, with and without neuropathy. Brain 2004, 127, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Robbins, W.R.; Staats, P.S.; Levine, J.; Fields, H.L.; Allen, R.W.; Campbell, J.N.; Pappagallo, M. Treatment of intractable pain with topical large-dose capsaicin: Preliminary report. Anesth. Analg. 1998, 86, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Bley, K. Topical capsaicin for pain management: Therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8% patch. Br. J. Anaesth. 2011, 107, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Sven-Rice, A.; Cole, P.; Tan, T.; Moore, R.A. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 2013, 28, CD007393. [Google Scholar]
- Mitchell, K.; Lebovitz, E.E.; Keller, J.M.; Mannes, A.J.; Nemenov, M.I.; Iadarola, M.J. Nociception and inflammatory hyperalgesia evaluated in rodents using infrared laser stimulation after TRPV1 gene knockout or resiniferatoxin lesion. Pain 2014, 155, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Hamalainen, M.M.; Subieta, A.; Arpey, C.; Brennan, T.J. Differential effect of capsaicin treatment on pain-related behaviors after plantar incision. J. Pain 2009, 10, 637–645. [Google Scholar] [CrossRef] [PubMed]
- King, T.; Qu, C.; Okun, A.; Mercado, R.; Ren, J.; Brion, T.; Lai, J.; Porreca, F. Contribution of afferent pathways to nerve injury-induced spontaneous pain and evoked hypersensitivity. Pain 2011, 152, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Okun, A.; DeFelice, M.; Eyde, N.; Ren, J.; Mercado, R.; King, T.; Porreca, F. Transient inflammation-induced ongoing pain is driven by TRPV1 sensitive afferents. Mol. Pain 2011, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Helyes, Z.; Sandor, K.; Borbely, E.; Tekus, V.; Pinter, E.; Elekes, K.; Toth, D.M.; Szolcsanyi, J.; McDougall, J.J. Involvement of transient receptor potential vanilloid 1 receptors in protease-activated receptor-2-induced joint inflammation and nociception. Eur. J. Pain 2010, 14, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Honore, P.; Chandran, P.; Hernandez, G.; Gauvin, D.M.; Mikusa, J.P.; Zhong, C.; Joshi, S.K.; Ghilardi, J.R.; Sevcik, M.A.; Fryer, R.M.; et al. Repeated dosing of ABT-102, a potent and selective TRPV1 antagonist, enhances TRPV1-mediated analgesic activity in rodents, but attenuates antagonist-induced hyperthermia. Pain 2009, 142, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.J.; McQueen, D.S.; Thomson, D.; Gauldie, S.D.; Wilson, A.W.; Salter, D.M.; Chessell, I.P. Attenuation of experimental arthritis in TRPV1R knockout mice. Exp. Mol. Pathol. 2006, 81, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Chapman, R.J.; Woodhams, S.; Sagar, D.R.; Turner, J.; Burston, J.J.; Bullock, C.; Paton, K.; Huang, J.; Wong, A.; et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann. Rheum. Dis. 2015, 74, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.C., 3rd; Xu, L.; Gebhart, G.F. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J. Neurosci. 2005, 25, 10981–10989. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Saloman, J.L.; Weiland, G.; Auh, Q.S.; Chung, M.K.; Ro, J.Y. Functional interactions between NMDA receptors and TRPV1 in trigeminal sensory neurons mediate mechanical hyperalgesia in the rat masseter muscle. Pain 2012, 153, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Katanosaka, K.; Murase, S.; Kashio, M.; Tominaga, M.; Mizumura, K. TRPV1 and TRPV4 play pivotal roles in delayed onset muscle soreness. PLoS ONE 2013, 8, e65751. [Google Scholar] [CrossRef] [PubMed]
- Kissin, E.Y.; Freitas, C.F.; Kissin, I. The effects of intraarticular resiniferatoxin in experimental knee-joint arthritis. Anesth. Analg. 2005, 101, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, E.H.; Lee, K.S.; Lee, K.; Park, S.H.; Na, S.H.; Ko, C.; Kim, J.; Yooon, Y.W. The effects of intra-articular resiniferatoxin on monosodium iodoacetate-induced osteoarthritic pain in rats. Korean J. Physiol. Pharmacol. 2016, 20, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.; Mahowald, M.L.; Frizelle, S.P.; Dorman, C.W.; Funkenbusch, S.C.; Krug, H.E. The effect of intra-articular vanilloid receptor agonists on pain behavior measures in a murine model of acute monoarthritis. J. Pain Res. 2016, 9, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Jancso, G.; Kiraly, E.; Jancso-Gabor, A. Direct evidence for an axonal site of action of capsaicin. Naunyn Schmiedebergs Arch. Pharmacol. 1980, 313, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Neubert, J.K.; Mannes, A.J.; Karai, L.J.; Jenkins, A.C.; Zawatski, L.; Abu-Asab, M.; Iadarola, M.J. Perineural resiniferatoxin selectively inhibits inflammatory hyperalgesia. Mol. Pain 2008, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kissin, I.; Bright, C.A.; Bradley, E.L., Jr. Selective and long-lasting neural blockade with resiniferatoxin prevents inflammatory pain hypersensitivity. Anesth. Analg. 2002, 94, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Kissin, I.; Davison, N.; Bradley, E.L., Jr. Perineural resiniferatoxin prevents hyperalgesia in a rat model of postoperative pain. Anesth. Analg. 2005, 100, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Szabo, T.; Olah, Z.; Iadarola, M.J.; Blumberg, P.M. Epidural resiniferatoxin induced prolonged regional analgesia to pain. Brain Res. 1999, 840, 92–98. [Google Scholar] [CrossRef]
- Eimerl, D.; Papir-Kricheli, D. Epidural capsaicin produces prolonged segmental analgesia in the rat. Exp. Neurol. 1987, 97, 169–178. [Google Scholar] [CrossRef]
- Yaksh, T.L.; Farb, D.H.; Leeman, S.E.; Jessell, T.M. Intrathecal capsaicin depletes substance P in the rat spinal cord and produces prolonged thermal analgesia. Science 1979, 206, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, M.; Bosgraaf, C.A.; Premkumar, L.S. Preservation of acute pain and efferent functions following intrathecal resiniferatoxin-induced analgesia in rats. J. Pain 2011, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.C.; Agnello, K.; Iadarola, M.J. Intrathecal resiniferatoxin in a dog model: Efficacy in bone cancer pain. Pain 2015, 156, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.C.; Iadarola, M.J.; Perkowski, S.Z.; Erin, H.; Shofer, F.; Laszlo, K.J.; Olah, Z.; Mannes, A.J. Physiologic and antinociceptive effects of intrathecal resiniferatoxin in a canine bone cancer model. Anesthesiology 2005, 103, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Brederson, J.D.; Kym, P.R.; Szallasi, A. Targeting TRP channels for pain relief. Eur. J. Pharmacol. 2013, 716, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Aasvang, E.K.; Hansen, J.B.; Malmstrom, J.; Asmussen, T.; Gennevois, D.; Struys, M.M.; Kehlet, H. The effect of wound instillation of a novel purified capsaicin formulation on postherniotomy pain: A double-blind, randomized, placebo-controlled study. Anesth. Analg. 2008, 107, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.M.; Diamond, E.; Schmidt, W.K.; Kelly, M.; Allen, R.; Houghton, W.; Brady, K.L.; Campbell, J.N. A randomized, double blind, placebo controlled trial of injected capsaicin for pain in morton’s neuroma. Pain 2016, 157, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Mou, J.; Paillard, F.; Turnbull, B.; Trudeau, J.; Stoker, M.; Katz, N.P. Efficacy of qutenza® (capsaicin) 8% patch for neuropathic pain: A meta-analysis of the qutenza clinical trials database. Pain 2013, 154, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Ringkamp, M.; Raja, S.N.; Campbell, J.N.; Meyer, R.A. Peripheral mechanisms of cutaneous nociception. In Wall & Melzack’s Textbook of Pain, 6th ed.; McMahon, S.B., Koltzenburg, M., Tracey, I., Turk, D.C., Eds.; Elsevier Health Sciences: Philadelphia, PA, USA, 2013; pp. 1–30. [Google Scholar]
- Campbell, J.N.; LaMotte, R.H. Latency to detection of first pain. Brain Res. 1983, 266, 203–208. [Google Scholar] [CrossRef]
- LaMotte, R.H.; Lundberg, L.E.; Torebjork, H.E. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J. Physiol. 1992, 448, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, M.; Schmid, R.; Handwerker, H.O.; Torebjork, H.E. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain 2000, 123 Pt 3, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Ringkamp, M.; Peng, Y.B.; Wu, G.; Hartke, T.V.; Campbell, J.N.; Meyer, R.A. Capsaicin responses in heat-sensitive and heat-insensitive A-fiber nociceptors. J. Neurosci. 2001, 21, 4460–4468. [Google Scholar] [PubMed]
- Snider, W.D.; McMahon, S.B. Tackling pain at the source: New ideas about nociceptors. Neuron 1998, 20, 629–632. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Helme, R.D.; McKernan, S. Neurogenic flare responses following topical application of capsaicin in humans. Ann. Neurol. 1985, 18, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Simone, D.A.; Baumann, T.K.; LaMotte, R.H. Dose-dependent pain and mechanical hyperalgesia in humans after intradermal injection of capsaicin. Pain 1989, 38, 99–107. [Google Scholar] [CrossRef]
- Carpenter, S.E.; Lynn, B. Vascular and sensory responses of human skin to mild injury after topical treatment with capsaicin. Br. J. Pharmacol. 1981, 73, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.R.; Nicol, G.D.; Vasko, M.R. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 1996, 712, 265–273. [Google Scholar] [CrossRef]
- Nemeth, J.; Helyes, Z.; Oroszi, G.; Jakab, B.; Pinter, E.; Szilvassy, Z.; Szolcsanyi, J. Role of voltage-gated cation channels and axon reflexes in the release of sensory neuropeptides by capsaicin from isolated rat trachea. Eur. J. Pharmacol. 2003, 458, 313–318. [Google Scholar] [CrossRef]
- LaMotte, R.H.; Shain, C.N.; Simone, D.A.; Tsai, E.F. Neurogenic hyperalgesia: Psychophysical studies of underlying mechanisms. J. Neurophysiol. 1991, 66, 190–211. [Google Scholar] [PubMed]
- Henrich, F.; Magerl, W.; Klein, T.; Greffrath, W.; Treede, R.D. Capsaicin-sensitive C- and A-fibre nociceptors control long-term potentiation-like pain amplification in humans. Brain 2015, 138 Pt 9, 2505–2520. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.K.; Simone, D.A.; Shain, C.N.; LaMotte, R.H. Neurogenic hyperalgesia: The search for the primary cutaneous afferent fibers that contribute to capsaicin-induced pain and hyperalgesia. J. Neurophysiol. 1991, 66, 212–227. [Google Scholar] [PubMed]
- Campbell, C.M.; Edwards, R.R.; Carmona, C.; Uhart, M.; Wand, G.; Carteret, A.; Kim, Y.K.; Frost, J.; Campbell, J.N. Polymorphisms in the GTP cyclohydrolase gene (GCH1) are associated with ratings of capsaicin pain. Pain 2009, 141, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Belfer, I.; Segall, S.K.; Lariviere, W.R.; Smith, S.B.; Dai, F.; Slade, G.D.; Rashid, N.U.; Mogil, J.S.; Campbell, C.M.; Edwards, R.R.; et al. Pain modality- and sex-specific effects of comt genetic functional variants. Pain 2013, 154, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Dimova, V.; Oertel, B.G.; Kabakci, G.; Zimmermann, M.; Hermens, H.; Lautenbacher, S.; Ultsch, A.; Lotsch, J. A more pessimistic life orientation is associated with experimental inducibility of a neuropathy-like pain pattern in healthy individuals. J. Pain 2015, 16, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Freed, K.A.; Blangero, J.; Howard, T.; Johnson, M.P.; Curran, J.E.; Garcia, Y.R.; Lan, H.C.; Abboud, H.E.; Moses, E.K. The 57 kb deletion in cystinosis patients extends into TRPV1 causing dysregulation of transcription in peripheral blood mononuclear cells. J. Med. Genet. 2011, 48, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Buntinx, L.; Voets, T.; Morlion, B.; Vangeel, L.; Janssen, M.; Cornelissen, E.; Vriens, J.; de Hoon, J.; Levtchenko, E. TRPV1 dysfunction in cystinosis patients harboring the homozygous 57 kb deletion. Sci. Rep. 2016, 6, 35395. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tian, W.; Fu, Y.; Oyama, T.T.; Anderson, S.; Cohen, D.M. Functional effects of nonsynonymous polymorphisms in the human TRPV1 gene. Am. J. Physiol. Renal. Physiol. 2007, 293, F1865–F1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Joseph, J.; Diatchenko, L.; Ro, J.Y.; Chung, M.K. Agonist-dependence of functional properties for common nonsynonymous variants of human transient receptor potential vanilloid 1. Pain 2016, 157, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Khairatkar-Joshi, N.; Szallasi, A. TRPV1 antagonists: The challenges for therapeutic targeting. Trends Mol. Med. 2009, 15, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Bates, B.D.; Mitchell, K.; Keller, J.M.; Chan, C.C.; Swaim, W.D.; Yaskovich, R.; Mannes, A.J.; Iadarola, M.J. Prolonged analgesic response of cornea to topical resiniferatoxin, a potent TRPV1 agonist. Pain 2010, 149, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Knolle, E.; Zadrazil, M.; Kovacs, G.G.; Medwed, S.; Scharbert, G.; Schemper, M. Comparison of cooling and emla to reduce the burning pain during capsaicin 8% patch application: A randomized, double-blind, placebo-controlled study. Pain 2013, 154, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.; Fischer, M.J.; Rehner, D.; Kienel, S.; Kistner, K.; Sauer, S.K.; Gavva, N.R.; Reeh, P.W.; Nau, C. The vanilloid receptor TRPV1 is activated and sensitized by local anesthetics in rodent sensory neurons. J. Clin. Investig. 2008, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Wang, S. Cold suppresses agonist-induced activation of TRPV1. J. Dent. Res. 2011, 90, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Leffler, A.; Babes, A.; Cendan, C.M.; Carr, R.W.; Kobayashi, J.; Nau, C.; Wood, J.N.; Reeh, P.W. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 2007, 447, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, A.; Murray, A.N.; Mathur, J.; Earley, T.J.; Petrus, M.J.; Patapoutian, A. TRPM8 is required for cold sensation in mice. Neuron 2007, 54, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Del Camino, D.; Murphy, S.; Heiry, M.; Barrett, L.B.; Earley, T.J.; Cook, C.A.; Petrus, M.J.; Zhao, M.; D'Amours, M.; Deering, N.; et al. TRPA1 contributes to cold hypersensitivity. J. Neurosci. 2010, 30, 15165–15174. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Uta, D.; Furue, H.; Tominaga, M. Pain-enhancing mechanism through interaction between TRPV1 and anoctamin 1 in sensory neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 5213–5218. [Google Scholar] [CrossRef] [PubMed]
- Deba, F.; Bessac, B.F. Anoctamin-1 Cl− channels in nociception: Activation by an N-aroylaminothiazole and capsaicin and inhibition by T16A[inh]-A01. Mol. Pain 2015, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Simone, D.A.; Ochoa, J. Early and late effects of prolonged topical capsaicin on cutaneous sensibility and neurogenic vasodilatation in humans. Pain 1991, 47, 285–294. [Google Scholar] [CrossRef]
- Neubert, J.K.; Karai, L.; Jun, J.H.; Kim, H.S.; Olah, Z.; Iadarola, M.J. Peripherally induced resiniferatoxin analgesia. Pain 2003, 104, 219–228. [Google Scholar] [CrossRef]
- Ma, X.L.; Zhang, F.X.; Dong, F.; Bao, L.; Zhang, X. Experimental evidence for alleviating nociceptive hypersensitivity by single application of capsaicin. Mol. Pain 2015, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Baranowski, R.; Lynn, B.; Pini, A. The effects of locally applied capsaicin on conduction in cutaneous nerves in four mammalian species. Br. J. Pharmacol. 1986, 89, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.M.; Lee, K.H.; Hori, Y.; Willis, W.D. Effects of capsaicin applied to a peripheral nerve on the responses of primate spinothalamic tract cells. Brain Res. 1985, 329, 27–38. [Google Scholar] [CrossRef]
- Petsche, U.; Fleischer, E.; Lembeck, F.; Handwerker, H.O. The effect of capsaicin application to a peripheral nerve on impulse conduction in functionally identified afferent nerve fibres. Brain Res. 1983, 265, 233–240. [Google Scholar] [CrossRef]
- Beydoun, A.; Dyke, D.B.; Morrow, T.J.; Casey, K.L. Topical capsaicin selectively attenuates heat pain and A delta fiber-mediated laser-evoked potentials. Pain 1996, 65, 189–196. [Google Scholar] [CrossRef]
- Mitchell, K.; Bates, B.D.; Keller, J.M.; Lopez, M.; Scholl, L.; Navarro, J.; Madian, N.; Haspel, G.; Nemenov, M.I.; Iadarola, M.J. Ablation of rat TRPV1-expressing Adelta/C-fibers with resiniferatoxin: Analysis of withdrawal behaviors, recovery of function and molecular correlates. Mol. Pain 2010, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Dray, A.; Bettaney, J.; Forster, P. Actions of capsaicin on peripheral nociceptors of the neonatal rat spinal cord-tail in vitro: Dependence of extracellular ions and independence of second messengers. Br. J. Pharmacol. 1990, 101, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.B.; Ringkamp, M.; Meyer, R.A.; Campbell, J.N. Fatigue and paradoxical enhancement of heat response in C-fiber nociceptors from cross-modal excitation. J. Neurosci. 2003, 23, 4766–4774. [Google Scholar] [PubMed]
- LaMotte, R.H.; Campbell, J.N. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgments of thermal pain. J. Neurophysiol. 1978, 41, 509–528. [Google Scholar] [PubMed]
- Slugg, R.M.; Meyer, R.A.; Campbell, J.N. Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. J. Neurophysiol. 2000, 83, 2179–2191. [Google Scholar] [PubMed]
- Joseph, J.; Wang, S.; Lee, J.; Ro, J.Y.; Chung, M.K. Carboxyl-terminal domain of transient receptor potential vanilloid 1 contains distinct segments differentially involved in capsaicin- and heat-induced desensitization. J. Biol. Chem. 2013, 288, 35690–35702. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, L.; Novakova-Tousova, K.; Benedikt, J.; Samad, A.; Touska, F.; Vlachova, V. Calcium-dependent desensitization of vanilloid receptor TRPV1: A mechanism possibly involved in analgesia induced by topical application of capsaicin. Physiol. Res. 2008, 57 (Suppl. S3), 59–68. [Google Scholar]
- Liu, L.; Oortgiesen, M.; Li, L.; Simon, S.A. Capsaicin inhibits activation of voltage-gated sodium currents in capsaicin-sensitive trigeminal ganglion neurons. J. Neurophysiol. 2001, 85, 745–758. [Google Scholar] [PubMed]
- Su, X.; Wachtel, R.E.; Gebhart, G.F. Capsaicin sensitivity and voltage-gated sodium currents in colon sensory neurons from rat dorsal root ganglia. Am. J. Physiol. 1999, 277, G1180–G1188. [Google Scholar] [PubMed]
- Onizuka, S.; Yonaha, T.; Tamura, R.; Hosokawa, N.; Kawasaki, Y.; Kashiwada, M.; Shirasaka, T.; Tsuneyoshi, I. Capsaicin indirectly suppresses voltage-gated Na+ currents through TRPV1 in rat dorsal root ganglion neurons. Anesth. Analg. 2011, 112, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Z.; Chen, S.R.; Pan, H.L. Transient receptor potential vanilloid type 1 activation down-regulates voltage-gated calcium channels through calcium-dependent calcineurin in sensory neurons. J. Biol. Chem. 2005, 280, 18142–18151. [Google Scholar] [CrossRef] [PubMed]
- Caudle, R.M.; Karai, L.; Mena, N.; Cooper, B.Y.; Mannes, A.J.; Perez, F.M.; Iadarola, M.J.; Olah, Z. Resiniferatoxin-induced loss of plasma membrane in vanilloid receptor expressing cells. Neurotoxicology 2003, 24, 895–908. [Google Scholar] [CrossRef]
- Sanz-Salvador, L.; Andres-Borderia, A.; Ferrer-Montiel, A.; Planells-Cases, R. Agonist- and Ca2+-dependent desensitization of TRPV1 channel targets the receptor to lysosomes for degradation. J. Biol. Chem. 2012, 287, 19462–19471. [Google Scholar] [CrossRef] [PubMed]
- Dickenson, A.; Ashwood, N.; Sullivan, A.F.; James, I.; Dray, A. Antinociception produced by capsaicin: Spinal or peripheral mechanism? Eur. J. Pharmacol. 1990, 187, 225–233. [Google Scholar] [CrossRef]
- Dickenson, A.H.; Dray, A. Selective antagonism of capsaicin by capsazepine: Evidence for a spinal receptor site in capsaicin-induced antinociception. Br. J. Pharmacol. 1991, 104, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Micevych, P.E.; Yaksh, T.L.; Szolcsanyi, J. Effect of intrathecal capsaicin analogues on the immunofluorescence of peptides and serotonin in the dorsal horn in rats. Neuroscience 1983, 8, 123–131. [Google Scholar] [CrossRef]
- Miller, M.S.; Buck, S.H.; Sipes, I.G.; Burks, T.F. Capsaicinoid-induced local and systemic antinociception without substance P depletion. Brain Res. 1982, 244, 193–197. [Google Scholar] [CrossRef]
- Pinter, E.; Helyes, Z.; Szolcsanyi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol. Ther. 2006, 112, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Szolcsanyi, J.; Bolcskei, K.; Szabo, A.; Pinter, E.; Petho, G.; Elekes, K.; Borzsei, R.; Almasi, R.; Szuts, T.; Keri, G.; et al. Analgesic effect of TT-232, a heptapeptide somatostatin analogue, in acute pain models of the rat and the mouse and in streptozotocin-induced diabetic mechanical allodynia. Eur. J. Pharmacol. 2004, 498, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Helyes, Z.; Pinter, E.; Sandor, K.; Elekes, K.; Banvolgyi, A.; Keszthelyi, D.; Szoke, E.; Toth, D.M.; Sandor, Z.; Kereskai, L.; et al. Impaired defense mechanism against inflammation, hyperalgesia, and airway hyperreactivity in somatostatin 4 receptor gene-deleted mice. Proc. Natl. Acad. Sci. USA 2009, 106, 13088–13093. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Farkas-Szallasi, T.; Lundberg, J.M.; Hokfelt, T.; Wiesenfeld-Hallin, Z.; Szallasi, A. Effects of the capsaicin analogue resiniferatoxin on spinal nociceptive mechanisms in the rat: Behavioral, electrophysiological and in situ hybridization studies. Brain Res. 1997, 752, 52–60. [Google Scholar] [CrossRef]
- Karai, L.; Brown, D.C.; Mannes, A.J.; Connelly, S.T.; Brown, J.; Gandal, M.; Wellisch, O.M.; Neubert, J.K.; Olah, Z.; Iadarola, M.J. Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J. Clin. Investig. 2004, 113, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Premkumar, L.S. Ablation and regeneration of peripheral and central TRPV1 expressing nerve terminals and the consequence of nociception. Open Pain J. 2015, 8, 1–9. [Google Scholar] [CrossRef]
- Mou, J.; Paillard, F.; Turnbull, B.; Trudeau, J.; Stoker, M.; Katz, N.P. Qutenza (capsaicin) 8% patch onset and duration of response and effects of multiple treatments in neuropathic pain patients. Clin. J. Pain 2014, 30, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Szolcsanyi, J.; Jancso-Gabor, A.; Joo, F. Functional and fine structural characteristics of the sensory neuron blocking effect of capsaicin. Naunyn Schmiedebergs Arch. Pharmacol. 1975, 287, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Klein, C.M.; Coggeshall, R.E. The receptive part of the primary afferent axon is most vulnerable to systemic capsaicin in adult rats. Brain Res. 1990, 511, 222–226. [Google Scholar] [CrossRef]
- Jancso, G.; Lawson, S.N. Transganglionic degeneration of capsaicin-sensitive C-fiber primary afferent terminals. Neuroscience 1990, 39, 501–511. [Google Scholar] [CrossRef]
- Otten, U.; Lorez, H.P.; Businger, F. Nerve growth factor antagonizes the neurotoxic action of capsaicin on primary sensory neurones. Nature 1983, 301, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Hikawa, N.; Kusakabe, T.; Kano, M.; Bandou, Y.; Gotoh, H.; Takenaka, T. Mechanism of inhibitory action of capsaicin on particulate axoplasmic transport in sensory neurons in culture. J. Neurobiol. 1993, 24, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Lawson, S.N. The morphological consequences of neonatal treatment with capsaicin on primary afferent neurones in adult rats. Acta Physiol. Hung. 1987, 69, 315–321. [Google Scholar] [PubMed]
- Sugimoto, T.; Xiao, C.; Ichikawa, H. Neonatal primary neuronal death induced by capsaicin and axotomy involves an apoptotic mechanism. Brain Res. 1998, 807, 147–154. [Google Scholar] [CrossRef]
- Hiura, A.; Nakae, Y.; Nakagawa, H. Cell death of primary afferent nerve cells in neonatal mice treated with capsaicin. Anat. Sci. Int. 2002, 77, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.W.; Ichikawa, H.; Fujita, M.; Yamaai, T.; Mukae, K.; Nomura, K.; Sugimoto, T. Involvement of caspase cascade in capsaicin-induced apoptosis of dorsal root ganglion neurons. Brain Res. 2005, 1056, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Hiura, A.; Ishizuka, H. Changes in features of degenerating primary sensory neurons with time after capsaicin treatment. Acta Neuropathol. 1989, 78, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Regan, R.F.; Panter, S.S.; Witz, A.; Tilly, J.L.; Giffard, R.G. Ultrastructure of excitotoxic neuronal death in murine cortical culture. Brain Res. 1995, 705, 188–198. [Google Scholar] [CrossRef]
- Jancso, G.; Karcsu, S.; Kiraly, E.; Szebeni, A.; Toth, L.; Bacsy, E.; Joo, F.; Parducz, A. Neurotoxin induced nerve cell degeneration: Possible involvement of calcium. Brain Res. 1984, 295, 211–216. [Google Scholar] [CrossRef]
- Marsh, S.J.; Stansfeld, C.E.; Brown, D.A.; Davey, R.; McCarthy, D. The mechanism of action of capsaicin on sensory C-type neurons and their axons in vitro. Neuroscience 1987, 23, 275–289. [Google Scholar] [CrossRef]
- Wood, J.N.; Winter, J.; James, I.F.; Rang, H.P.; Yeats, J.; Bevan, S. Capsaicin-induced ion fluxes in dorsal root ganglion cells in culture. J. Neurosci. 1988, 8, 3208–3220. [Google Scholar] [PubMed]
- Chard, P.S.; Bleakman, D.; Savidge, J.R.; Miller, R.J. Capsaicin-induced neurotoxicity in cultured dorsal root ganglion neurons: Involvement of calcium-activated proteases. Neuroscience 1995, 65, 1099–1108. [Google Scholar] [CrossRef]
- Olah, Z.; Szabo, T.; Karai, L.; Hough, C.; Fields, R.D.; Caudle, R.M.; Blumberg, P.M.; Iadarola, M.J. Ligand-induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J. Biol. Chem. 2001, 276, 11021–11030. [Google Scholar] [CrossRef] [PubMed]
- Karai, L.J.; Russell, J.T.; Iadarola, M.J.; Olah, Z. Vanilloid receptor 1 regulates multiple calcium compartments and contributes to Ca2+-induced Ca2+ release in sensory neurons. J. Biol. Chem. 2004, 279, 16377–16387. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Sandin, S.; Rodriguez-Garcia, A.; Alonso, M.T.; Garcia-Sancho, J. The endoplasmic reticulum of dorsal root ganglion neurons contains functional TRPV1 channels. J. Biol. Chem. 2009, 284, 32591–32601. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.; Wend, P.; Purfurst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kee, K.H.; Suh, C.H.; Lim, S.C.; Oh, S.H. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology 2009, 264, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.M.; Basbaum, A.I. Differential ATF3 expression in dorsal root ganglion neurons reveals the profile of primary afferents engaged by diverse noxious chemical stimuli. Pain 2010, 150, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Usachev, Y.M.; DeMarco, S.J.; Campbell, C.; Strehler, E.E.; Thayer, S.A. Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron 2002, 33, 113–122. [Google Scholar] [CrossRef]
- Gover, T.D.; Moreira, T.H.; Kao, J.P.; Weinreich, D. Calcium homeostasis in trigeminal ganglion cell bodies. Cell Calcium 2007, 41, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Shutov, L.P.; Kim, M.S.; Houlihan, P.R.; Medvedeva, Y.V.; Usachev, Y.M. Mitochondria and plasma membrane Ca2+-ATPase control presynaptic Ca2+ clearance in capsaicin-sensitive rat sensory neurons. J. Physiol. 2013, 591, 2443–2462. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.V.; Kim, M.S.; Usachev, Y.M. Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J. Neurosci. 2008, 28, 5295–5311. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, D.Y.; Chung, E.S.; Oh, U.T.; Kim, S.U.; Jin, B.K. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 2005, 25, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.Y.; Shin, J.; Kim, B.M.; Wang, M.H.; Jang, J.H.; Surh, Y.J.; Oh, U. Essential role of mitochondrial permeability transition in vanilloid receptor 1-dependent cell death of sensory neurons. Mol. Cell. Neurosci. 2003, 24, 57–68. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J. Neurosci. 2004, 24, 10878–10887. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, K.Y.; Lu, Y.; Wang, J.; Cui, L.; Kim, S.J.; Chung, J.M.; Chung, K. Mitochondrial Ca2+ uptake is essential for synaptic plasticity in pain. J. Neurosci. 2011, 31, 12982–12991. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.S.; Kim, H.Y.; Wang, J.; Lee, I.; Klann, E.; Chung, J.M.; Chung, K. Persistent pain is dependent on spinal mitochondrial antioxidant levels. J. Neurosci. 2009, 29, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53 (Suppl. S1), 96–102. [Google Scholar] [CrossRef]
- Dedov, V.N.; Mandadi, S.; Armati, P.J.; Verkhratsky, A. Capsaicin-induced depolarisation of mitochondria in dorsal root ganglion neurons is enhanced by vanilloid receptors. Neuroscience 2001, 103, 219–226. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.; Ohno, N.; Hsieh, Y.L.; Mahad, D.J.; Kikuchi, S.; Komuro, H.; Hsieh, S.T.; Trapp, B.D. Mitochondrial fission augments capsaicin-induced axonal degeneration. Acta Neuropathol. 2015, 129, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Movahed, P.; Hinman, A.; Axelsson, H.E.; Sterner, O.; Högestätt, E.D.; Julius, D.; Jordt, S.E.; Zygmunt, P.M. Pungent products from garlic activate the sensory ion channel TRPA1. Proc. Natl. Acad. Sci. USA 2005, 102, 12248–12252. [Google Scholar] [CrossRef] [PubMed]
- Iadarola, M.J.; Gonnella, G.L. Resiniferatoxin for pain treatment: An interventional approach to personalized pain medicine. Open Pain J. 2013, 6, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, J.R.; Rohrich, H.; Lindsay, T.H.; Sevcik, M.A.; Schwei, M.J.; Kubota, K.; Halvorson, K.G.; Poblete, J.; Chaplan, S.R.; Dubin, A.E.; et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J. Neurosci. 2005, 25, 3126–3131. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Winston, J.H.; Sarna, S.K. Neurological and cellular regulation of visceral hypersensitivity induced by chronic stress and colonic inflammation in rats. Neuroscience 2013, 248, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cai, J.; Han, Y.; Xiao, X.; Meng, X.L.; Su, L.; Liu, F.Y.; Xing, G.G.; Wan, Y. Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur. J. Pain 2014, 18, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.J.; Bevan, S.; Wotherspoon, G.; Gentry, C.; Fox, A.; Winter, J. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur. J. Neurosci. 2001, 13, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Chu, Y.; Han, L.; Li, M.; Li, Z.; Lavinka, P.C.; Sun, S.; Tang, Z.; Park, K.; Caterina, M.J.; et al. Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 2014, 81, 873–887. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, M.-K.; Campbell, J.N. Use of Capsaicin to Treat Pain: Mechanistic and Therapeutic Considerations. Pharmaceuticals 2016, 9, 66. https://doi.org/10.3390/ph9040066
Chung M-K, Campbell JN. Use of Capsaicin to Treat Pain: Mechanistic and Therapeutic Considerations. Pharmaceuticals. 2016; 9(4):66. https://doi.org/10.3390/ph9040066
Chicago/Turabian StyleChung, Man-Kyo, and James N. Campbell. 2016. "Use of Capsaicin to Treat Pain: Mechanistic and Therapeutic Considerations" Pharmaceuticals 9, no. 4: 66. https://doi.org/10.3390/ph9040066
APA StyleChung, M.-K., & Campbell, J. N. (2016). Use of Capsaicin to Treat Pain: Mechanistic and Therapeutic Considerations. Pharmaceuticals, 9(4), 66. https://doi.org/10.3390/ph9040066